

Abstract
The direct conversion of aliphatic carboxylic acids to the corresponding alkyl fluorides has been achieved via visible light-promoted photoredox catalysis. This operationally simple, redox-neutral fluorination method is amenable to a wide variety of carboxylic acids. Photon-induced oxidation of carboxylates leads to the formation of carboxyl radicals, which upon rapid CO2-extrusion and F• transfer from a fluorinating reagent yield the desired fluoroalkanes with high efficiency. Experimental evidence indicates that an oxidative quenching pathway is operable in this broadly applicable fluorination protocol.
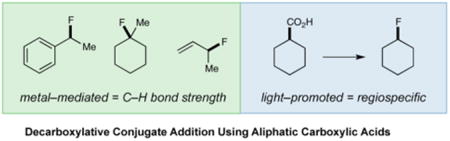
The capacity of fluorine atoms to engender a variety of useful properties in pharmaceuticals, agrochemicals, and performance materials has driven significant research efforts toward the invention of novel fluorination reactions.1–3 Over the past two decades, significant progress has been made toward the production of sp2 C–F bonds;4 however, catalytic methods for sp3 C–F formation have only recently become available.5–8 In particular, metal-mediated radical C–H abstraction/fluorination protocols have been developed to form tertiary aliphatic,6 benzylic,7 as well as allylic8 C–F centers, a strategy that is founded upon the selective functionalization of weak C–H bonds. Despite these important advances, the development of a general sp3 C–F bond-forming platform that is (i) highly regiospecific, (ii) bond strength independent, (iii) operationally simple, and (iv) able to employ readily available, inexpensive starting materials, remains a challenging goal.
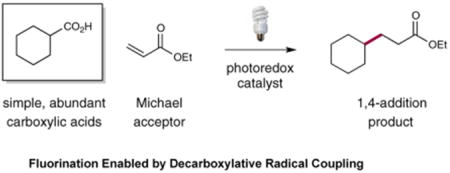
Harnessing visible light as a safe, renewable, and inexpensive source of chemical energy to facilitate the construction of complex organic molecules has emerged recently as a powerful theme in organic chemistry.9 In this context, our group has recently introduced a visible light-mediated alkylation of α,β-unsaturated carbonyl compounds with alkyl radicals, generated via a CO2-extrusion mechanism involving carboxylic acids (eq 1).10 Based on these findings, we wondered if a similar photon-induced decarboxylation strategy might be employed as a general platform for the construction of C–F bonds. Precedent for this transformation has been demonstrated via the silver-mediated Hunsdiecker reaction11 and the work of Sammis to achieve a light-promoted decarboxylative fluorination of 2-aryloxyacetic acids to generate α-oxyfluoro motifs.12 Despite these seminal studies, a general light-mediated strategy for the fluorination of a wide range of aliphatic carboxylic acids has not yet been reported. In this communication, we further advance the visible light activation concept to describe the first broadly applicable
 |
(Eq 1) |
 |
(Eq 2) |
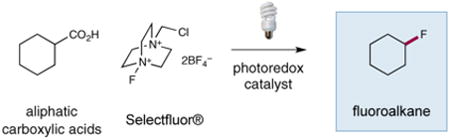
protocol for the photon-mediated decarboxylative fluorination of sp3-carbon-bearing carboxylic acids, using a blue LED light source and a commercial photocatalyst (eq 2).
Design Plan
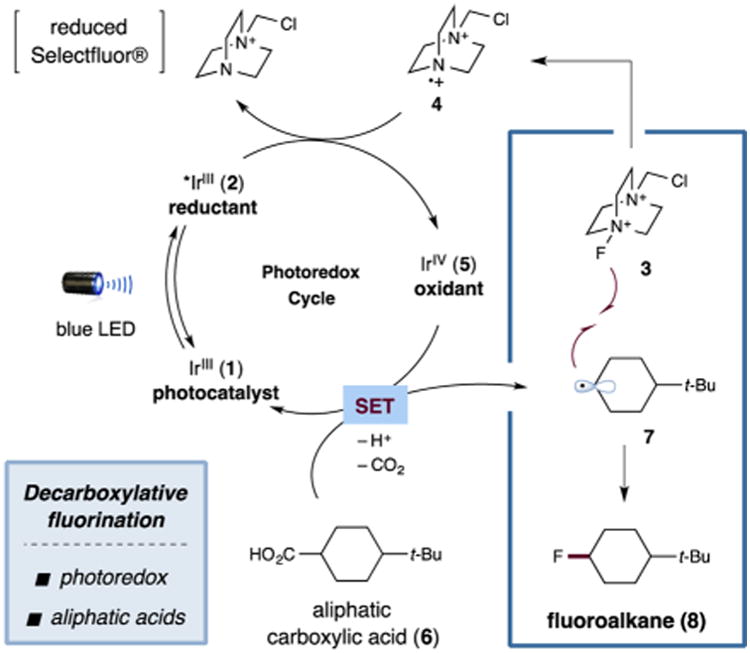
Drawing from the mechanistic insights gained in the course of our decarboxylative alkylation10 and arylation13 methods, we envisioned that a broad range of aliphatic carboxylic acids could be employed as viable precursors to fluoroalkanes. The specific mechanistic details of our proposed visible light-mediated photoredox decarboxylative fluorination are outlined in Scheme 1. Irradiation of heteroleptic iridium(III) photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) with visible light leads to the formation of a long-lived (τ = 2.3 μs)14 excited state, *Ir[dF(CF3)ppy]2(dtbbpy)+ (2), which can undergo oxidative quenching ( vs SCE in CH3CN)14 in the presence of an appropriate electron acceptor. We hypothesized that an initial reduction of a sacrificial quantity of Selectfluor reagent (3; Selectfluor is a trademark of Air Products and Chemicals) ( vs SCE in CH3CN)15 by *Ir(III) 2 via a single electron transfer (SET) process should generate the strongly oxidizing Ir[dF(CF3)ppy]2(dtbbpy)2+ (5). Indeed, the earlier work of Sammis clearly delineated that such a possibility was viable with an electrophilic source of fluorine.12 We further assumed that base-mediated formation of an alkyl carboxylate followed by an SET oxidation ( for hexanoate)16 using the transiently formed Ir(IV) species 5 ( vs SCE in CH3CN)14 would be thermodynamically feasible. This process is envisioned to generate a carboxyl radical, which upon immediate extrusion of CO2 should provide the SOMO species 7.17 Concurrently, reduction of Ir(IV) 5 would regenerate the ground-state photocatalyst 1, thus completing the photoredox cycle. At this stage, direct F-transfer from Selectfluor to the alkyl radical 7 is proposed to forge the desired fluoroalkane bond (8) with concomitant formation of the corresponding Selectfluor radical cation 4. We assume that radical cation 4 would replace Selectfluor in subsequent photoredox cycles as a suitable electron acceptor in the conversion of excited-state *Ir(III) (2) to the requisite Ir[dF(CF3)ppy]2(dtbbpy)2+ (5) species.
Scheme 1. Mechanism for Decarboxylative Fluorination.
Results
We first explored the proposed decarboxylative fluorination reaction in the context of N-benzoyl-4-piperidine-carboxylic acid and Selectfluor (Table 1). Examination of a range of photocatalysts and bases revealed that a combination of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1) and disodium hydrogen phosphate (1 equiv) was superior with respect to reaction efficiency. The use of more oxidizing photocatalysts, such as Ru(phen)32+ or Ru(bpz)32+ (9), resulted in slower or no reaction (entries 1, 3, and 4). Moreover, the use of the Sammis protocol (Ru(bpy)32+ (10) + NaOH, developed for the decarboxylative fluorination of α-oxy acids)12c gave no observable product. Further improvement was achieved via the use of 2 equiv of Na2HPO4 as a base, presumably due to the larger extent of acid deprotonation (entries 5 and 6; 80% vs 90% yield). The critical role of acetonitrile and water as solvent mixture was demonstrated by the absence of any fluorinated product when either of these two solvents was used independently (entries 7 and 8). This observation is likely due to the low solubility of (i) Selectfluor in acetonitrile and (ii) alkyl carboxylic acids in water, whereas the miscible mixture of acetonitrile and water allowed for both substrates to be employed in a homogeneous solution. Lastly, control experiments confirmed the requirement of a photocatalyst, base, and a light source in this new fluorination protocol (entries 9–11).
Table 1. Initial Studies and Reaction Optimization.
| ||||
|---|---|---|---|---|
|
| ||||
| entry | photocatalyst | base | solventa | yieldb (%) |
| 1 | Ru(bpz)3(PF6)2 | Na2HPO4 (1 equiv) | CH3CN/H2O | 64 |
| 2 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | CsF (1 equiv) | CH3CN/H2O | 68 |
| 3 | Ru(bpy)3(PF6)2 | NaOH (1 equiv) | CH3CN/H2O | 0 |
| 4 | Ru(phen)3(PF6)2 | Na2HPO4 (1 equiv) | CH3CN/H2O | 0 |
| 5 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | Na2HPO4 (1 equiv) | CH3CN/H2O | 80 |
| 6 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | Na2HPO4 (2 equiv) | CH3CN/H2O | 90 |
| 7 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | Na2HPO4 (2 equiv) | CH3CN | 0 |
| 8 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | Na2HPO4 (2 equiv) | H2O | 0 |
| 9 | none | Na2HPO4 (2 equiv) | CH3CN/H2O | 0 |
| 10 | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | none | CH3CN/H2O | 0 |
| 11c | Ir[dF(CF3)ppy]2 (dtbbpy)PF6 | Na2HPO4 (2 equiv) | CH3CN/H2O | 0 |
Acetonitrile/water 1:1 (v/v) was used as solvent.
Yields determined by 1H NMR using 3,5-bis(trifluoromethyl)bromobenzene as an internal standard.
Reaction performed in the absence of light.
Reaction Scope
Having identified optimal conditions for what we hoped would be a general photocatalytic CO2-extrusion/fluorination protocol, we aimed to define the scope of the carboxylic acid precursor. As shown in Table 2, a wide range of differentially substituted alkyl carboxylates were readily converted to the corresponding alkyl fluorides. It is of note that primary, secondary, and tertiary alkyl carboxylic acids are all well-tolerated, with no observed decrease in efficiency with less-substituted acids. Substrates with a heteroatom in the vicinity of the carboxyl group (α or β) underwent faster CO2-extrusion/fluorination (precursors to 18 and 28–30, 99%, 92%, 90%, and 90% yields, respectively), with reaction times in the 1–3 h range. In addition, 2 equiv of Selectfluor could be used without any decrease in reaction efficiency with these substrates. The same observation was made for benzylic and homobenzylic carboxylic acids (precursors to 14, 17, and 22 (87%, 82%, and 92% yields, respectively), presumably due to stabilization of the transiently formed radical intermediate. It is important to note that unactivated acids were also found to be competent substrates for this fluorination protocol (products 20, 27, and 34 70%, 83%, and 79% yields, respectively). However, when 1,4-phenyldipropionic acid was employed as a substrate, no formation of difluoride 12 was observed, due to the very low solubility of the dicarboxylic acid in the acetonitrile/water mixture. To our delight, when the corresponding preformed disodium salt was employed, the desired difluoride was isolated in 71% yield. Similarly, 4-tert-butylcyclohexanecarboxylic acid provided higher yield of the corresponding fluorocyclohexane 20 when the ratio of the acetonitrile/water medium was adjusted to 3:1. It is interesting to note that while the fluoride 28, derived from ribosic acid, was formed in high yield, the corresponding glucopyranouronic acid derivative did not react under these reaction conditions. We believe that this result can be rationalized by the change in bond strength and oxidation potential of the C–CO2 moiety as it exists in either the axial anomer position (with ribosic acid) or the anomeric equatorial topography (as expected with glucopyranouronic acid). We assume that the axial C–CO2 bond is weaker, and the rate of decarboxylation is faster (in competition with back electron transfer) due to the hyper-conjugative stabilization by the ring oxygen lone pair in the case of the ribosic carboxylic acid system. In contrast, we presume that back electron transfer is competitive with decarboxylation in the case of glucopyranouronic acid, thereby preventing the formation of an α-oxy radical intermediate. Interestingly, no elimination of the fluoride group was observed when this decarboxylative fluorination protocol was used to generate a β-fluoro carbonyl under basic conditions. More specifically, when a 1,4-keto acid substrate was employed, the desired β-fluoroketone 25 was isolated in 96% yield, without observation of the corresponding α,β-unsaturated product. Finally, less reactive substrates, such as unactivated primary (precursors to products 12 and 15) or tertiary carboxylic acids (precursors to 32 and 34) proved to be viable substrates; however, longer reaction times (12–15 h) were required for full conversion of the starting material. This observation is in agreement with the rate of oxidation of the carboxylate (in the case of primary acids) or the rate of formation of the carboxylate (in the case of cyclic tertiary acids), critical steps ahead of the formation of the radical intermediate. Finally, we have performed a series of Stern– Volmer fluorescence quenching studies in an effort to gather evidence with regard to the mechanistic proposal outlined in Scheme 1.19 As revealed in Figure 1, we observed that the emission intensity of the excited state of the heteroleptic catalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 is diminished in the presence of Selectfluor (Figure 1). In contrast, fluorescence quenching was not observed when a solution of sodium 4-tert-butylcyclohexanecarboxylate was exposed to the photoexcited *Ir(III) species. These experiments strongly indicate that the reduction of Selectfluor is likely the initiation point of the photoredox catalytic cycle, as proposed in Scheme 1. Moreover, we presume that the requisite oxidation of the aliphatic carboxylate is performed by the resulting Ir[dF(CF3)ppy]2(dtbbpy)2+ species (also delineated in Scheme 1).
Table 2. Decarboxylative Fluorination: Scope of Carboxylic Acidsa.

|
Isolated yields, see Supporting Information for experimental details.
Reaction time 1 h.
Reaction time 3 h.
Reaction time 12 h.
Reaction time 15 h.
Reaction run with Ru(bpz)3(PF6)2 instead of Ir[dF(CF3)ppy]2(dtbbpy)PF6.18
Reaction run using 2 equiv of Selectfluor.
Reaction run in acetonitrile/water 3:1.
Obtained as a single diastereomer.
Obtained as 2.5:1 trans/cis mixture of diastereomers (major diastereomer shown).
Figure 1.
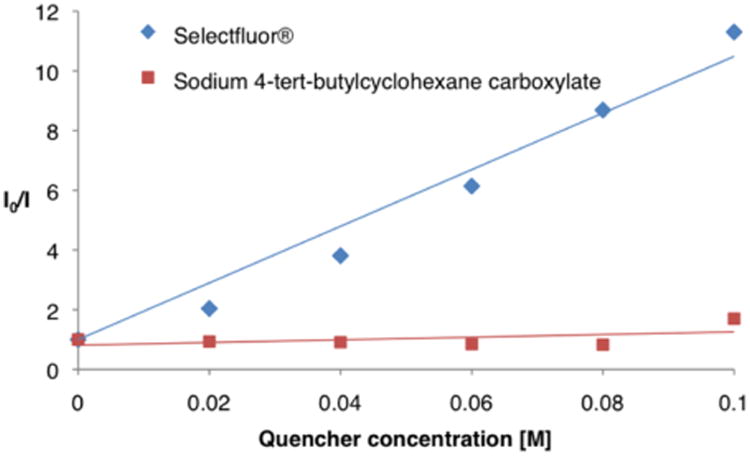
Ir[dF(CF3)ppy]2(dtbbpy)PF6 emission quenching with Selectfluor and sodium 4-tert-butylcyclohexanecarboxylate.
In summary, we have developed a photoredox-assisted decarboxylative fluorination protocol and demonstrated its utility over a wide range of carboxylic acid substrates. In contrast to previously described methods, this redox-neutral reaction does not require activated substrates. In addition, its operational simplicity and mild reaction conditions allow for the synthesis of a diverse collection of valuable fluorinated products. Notably, under these reaction conditions more activated substrates require lower amounts of the electrophilic fluorinating reagent and shorter reaction times. Mechanistic studies have provided evidence supporting an oxidative quenching pathway, in which reduction of the N–F bond of Selectfluor initiates the photoredox cycle prior to the carboxylic acid oxidation/ decarboxylation sequence.
Supplementary Material
Acknowledgments
Financial support was provided by the NIGMS (R01 GM093213-05) and kind gifts from Merck, Amgen, and AbbVie.
Footnotes
Supporting Information: Experimental procedures and spectral data. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.5b02244.
Notes: The authors declare no competing financial interest.
References
- 1.Muller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 2.(a) Furuya T, Kuttruff CA, Ritter T. Curr Opin Drug Disc. 2008;11:803. [PubMed] [Google Scholar]; (b) Neumann CN, Ritter T. Angew Chem, Int Ed. 2015;54:3216. doi: 10.1002/anie.201410288. [DOI] [PubMed] [Google Scholar]
- 3.(a) Hagmann WK. J Med Chem. 2008;51:4359. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]; (b) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Furuya T, Kamlet AS, Ritter T. Nature. 2011;473:470. doi: 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hull KL, Anani WQ, Sanford MS. J Am Chem Soc. 2006;128:7134. doi: 10.1021/ja061943k. [DOI] [PubMed] [Google Scholar]; (c) Watson DA, Su M, Teverovskiy G, Zhang Y, García-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:166. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee HG, Milner PJ, Buchwald SL. J Am Chem Soc. 2014;136:3792. doi: 10.1021/ja5009739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected examples, see: Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826. doi: 10.1021/ja051805f.Kalow JA, Doyle AG. J Am Chem Soc. 2010;132:3268. doi: 10.1021/ja100161d.Katcher MH, Sha A, Doyle AG. J Am Chem Soc. 2011;133:15902. doi: 10.1021/ja206960k.Hollingworth C, Hazari A, Hopkinson MN, Tredwell M, Benedetto E, Huiban M, Gee AD, Brown JM, Gouverneur V. Angew Chem, Int Ed. 2011;50:2613. doi: 10.1002/anie.201007307.Topczewski JJ, Tewson TJ, Nguyen HM. J Am Chem Soc. 2011;133:19318. doi: 10.1021/ja2087213.Barker TJ, Boger DL. J Am Chem Soc. 2012;134:13588. doi: 10.1021/ja3063716.Sladojevich F, Arlow SI, Tang P, Ritter T. J Am Chem Soc. 2013;135:2470. doi: 10.1021/ja3125405.
- 6.(a) Liu W, Huang X, Cheng MJ, Nielsen RJ, Goddard WA, III, Groves JT. Science. 2012;337:1322. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]; (b) Bloom S, Knippel JL, Lectka T. Chem Sci. 2014;5:1175. [Google Scholar]
- 7.(a) Liu W, Groves JT. Angew Chem, Int Ed. 2013;52:6024. doi: 10.1002/anie.201301097. [DOI] [PubMed] [Google Scholar]; (b) Bloom S, Pitts CR, Woltornist R, Griswold A, Holl MG, Lectka T. Org Lett. 2013;15:1722. doi: 10.1021/ol400424s. [DOI] [PubMed] [Google Scholar]; (c) Bloom S, Sharber SA, Holl MG, Knippel JL, Lectka T. J Org Chem. 2013;78:11082. doi: 10.1021/jo401796g. [DOI] [PubMed] [Google Scholar]; (d) Amaoka Y, Nagatomo M, Inoue M. Org Lett. 2013;15:2160. doi: 10.1021/ol4006757. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bloom S, Pitts CR, Miller DC, Haselton N, Holl MG, Urheim E, Lectka T. Angew Chem, Int Ed. 2012;51:10580. doi: 10.1002/anie.201203642. [DOI] [PubMed] [Google Scholar]; (b) Braun MG, Doyle AG. J Am Chem Soc. 2013;135:12990. doi: 10.1021/ja407223g. [DOI] [PubMed] [Google Scholar]
- 9.(a) Nicewicz D, MacMillan DWC. Science. 2008;32:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schultz DM, Yoon TP. Science. 2014;343:6174. doi: 10.1126/science.1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527. doi: 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; (d) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]
- 10.Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin F, Wang Z, Li Z, Li C. J Am Chem Soc. 2012;134:10401. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]
- 12.(a) Rueda-Becerril M, Chatlova-Sazepin C, Leung JCT, Okbinoglu T, Kennepohl P, Paquin JF, Sammis GM. J Am Chem Soc. 2012;134:4026. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]; (b) Leung JCT, Chatalova-Sazepin C, West J, Rueda-Becerril M, Paquin JF, Sammis GM. Angew Chem, Int Ed. 2012;51:10804. doi: 10.1002/anie.201206352. [DOI] [PubMed] [Google Scholar]; (c) Rueda-Becerril M, Mahe O, Drouin M, Majewski MB, West JG, Wolf MO, Sammis GM, Paquin JF. J Am Chem Soc. 2014;136:2637. doi: 10.1021/ja412083f. [DOI] [PubMed] [Google Scholar]
- 13.Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowry MS, Goldsmith JL, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712. [Google Scholar]
- 15.(a) Stavber S, Zupan M. Acta Chim Slov. 2005;52:13. [Google Scholar]; (b) Girina GP, Fainzil'berg AA, Feoktistov LG. Russ J Electrochem. 2000;36:162. [Google Scholar]
- 16.Galicia M, Gonzalez FJ. J Electrochem Soc. 2002;149:D46. [Google Scholar]
- 17.Bockman TM, Hubig SM, Kochi JK. J Org Chem. 1997;62:2210. doi: 10.1021/jo9617833. [DOI] [PubMed] [Google Scholar]
- 18.Fluorides 22, 25, and 29 were also obtained using Ir[dF(CF3) ppy]2(dtbbpy)PF6 photocatalyst in 71%, 89%, and 85% yield, respectively. However, we observed slightly increased efficiency with the ruthenium-based photocatalyst Ru(bpz)3(PF6)2 in these three cases.
- 19.See Supporting Information for further details about fluorescence quenching studies.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.