

Background: Met(E11) mutation in Hb heme pocket undergoes modification to Asp specifically in γ and β subunits.
Results: Asp conversion increased in β/γ chains with increasing peroxide and was seen after autoxidation of mutant hemoglobin crystals.
Conclusion: Asp is formed by an oxidative mechanism involving ferryl heme complexes.
Significance: Subunit-specific modifications aid in the interpretation of hemoglobinopathies and design of oxidatively stable hemoglobins.
Keywords: Aspartate (Aspartic Acid), Heme, Hemoglobin, Mutant, Post-translational Modification (PTM), Hemoglobinopathies, Oxidation of Methionine
Abstract
A pathogenic V67M mutation occurs at the E11 helical position within the heme pockets of variant human fetal and adult hemoglobins (Hb). Subsequent post-translational modification of Met to Asp was reported in γ subunits of human fetal Hb Toms River (γ67(E11)Val → Met) and β subunits of adult Hb (HbA) Bristol-Alesha (β67(E11)Val → Met) that were associated with hemolytic anemia. Using kinetic, proteomic, and crystal structural analysis, we were able to show that the Met → Asp transformation involves heme cycling through its oxoferryl state in the recombinant versions of both proteins. The conversion to Met and Asp enhanced the spontaneous autoxidation of the mutants relative to wild-type HbA and human fetal Hb, and the levels of Asp were elevated with increasing levels of hydrogen peroxide (H2O2). Using H218O2, we verified incorporation of 18O into the Asp carboxyl side chain confirming the role of H2O2 in the oxidation of the Met side chain. Under similar experimental conditions, there was no conversion to Asp at the αMet(E11) position in the corresponding HbA Evans (α62(E11)Val → Met). The crystal structures of the three recombinant Met(E11) mutants revealed similar thioether side chain orientations. However, as in the solution experiments, autoxidation of the Hb mutant crystals leads to electron density maps indicative of Asp(E11) formation in β subunits but not in α subunits. This novel post-translational modification highlights the nonequivalence of human Hb α, β, and γ subunits with respect to redox reactivity and may have direct implications to α/β hemoglobinopathies and design of oxidatively stable Hb-based oxygen therapeutics.
Introduction
Human fetal hemoglobin (HbF)3 and adult hemoglobin (HbA) are tetrameric heme proteins, which serve as the major transporters of oxygen (O2) in fetal and adult life, respectively (1). These proteins consist of two globular α subunits and two globular non-α subunits. The non-α subunits in HbA and their homologs in HbF are β and γ subunits, respectively. These subunits are held together through noncovalent interactions, and each bears a single ferroprotoporphyrin IX (heme) prosthetic group that confers O2 binding capacity (1). Post-translational modifications (PTMs) represent an important biological mechanism for modulating both protein function and increasing the overall functional diversity of the cellular proteome (1). These PTMs involve enzymatic and nonenzymatic modifications of amino acid side chains, including phosphorylation, methylation, acetylation, and glycosylation (2). Some of the commonly known post-translational changes in Hb, such as elevated levels of glycosylation in diabetes and carbamoylation due to elevated serum bicarbonate, are due to metabolic defects, and others are due to mutations that lead to covalent PTMs that can alter the stability of HbA or HbF in specific hemoglobinopathies (1).
Globin gene mutations often result in amino acid substitutions that alter Hb function or stability through diverse mechanisms. Hb variants have been identified where the primary sequence is changed at the substituted amino acid position by a process previously termed a silent PTM (3). In such a scenario, the substituted amino acid is post-translationally modified to another residue due to its proximity to either catalytic amino acid side chains or the reactive heme group. The resulting amino acid is equivalent to a single amino acid substitution in the protein's primary sequence; however, a disparity exists between the substituted amino acid and its codon (4–7). These modifications can be difficult to detect, but previous protein and DNA analyses identified one such modification in a human variant, Hb Bristol-Alesha ((β67(E11)Val → Met), referred to hereafter as βV67M (3, 8–12).
Hb Bristol-Alesha was originally identified in an adult British patient and two unrelated Japanese subjects who suffered from idiopathic Heinz body hemolytic anemia (9–11). These variant Hbs were initially found by protein sequencing to possess a single Val → Asp substitution at position 67 in β subunits, which is at the 11th amino acid along the E-helix (E11 helical position) and adjacent to the iron and bound ligands in the distal heme pocket. A reinvestigation of the British patient using both DNA and protein analysis showed that the original characterization of HbA Bristol-Alesha as βV67D was incorrect; the actual Hb gene mutation was in fact βV67M. Furthermore, the patient's hemolysate was found to contain both βV67M and βV67D. A structurally analogous Val → Met mutation was also reported in Hb α subunits (i.e. HbA Evans (α62(E11)Val → Met), referred to hereafter as αV62M; however, the conversion to Asp was not identified in the patient's blood (13, 14).
A new γ Hb variant, named “Hb Toms River,” was recently discovered in a neonate with transient cyanosis and anemia (15). The baby was heterozygous for a γ-globin (HBG2) missense mutation that resulted in a positionally equivalent Val → Met substitution at amino acid 67 (E11) in the fetal subunit. The recombinant analog HbF (γ67(E11)Val → Met), referred to hereafter as γV67M, was reported to exhibit decreased rates of O2 binding as compared with wild-type HbF, providing an explanation for the cyanosis observed in the affected patient (15). Preliminary mass spectrometric studies of patient hemolysate suggested a mixture of variant γ-globins containing either Met or Asp at position E11. The hemolysate from a neonatal control exhibited only Val at the E11 position.
To date, no structural studies have been performed with HbF chains containing γMet-67 or γAsp-67, and the mechanism for this PTM is unknown. However, the presence of both isoforms in the γV67M hemolysate was supported by the previous HbA Bristol-Alesha (βV67M) studies. It was suggested that Met → Asp conversion is likely driven by an oxidative reaction based on the proximity of the Met(E11) side chain to bound O2 and the heme iron within the distal pocket (15). In such a scenario, post-translational conversion to Asp would likely destabilize the molecule by introducing a polar residue and perhaps a negative charge in the middle of the hydrophobic heme pocket causing precipitation, Heinz bodies, and hemolytic anemia. In the case of HbA Evan(αV62M), where conversion to Asp-62(E11) is not observed, the protein is stable, and little anemia was observed. Thus, these phenotypic differences appear to reflect markedly different rates of Met(E11) → Asp conversion in the variant globin chains (16).
In this study, we used one-dimensional liquid chromatography-tandem mass spectrometry (LC-MS/MS) to confirm the presence of γV67M and γV67D variants in hemolysate from the patient with HbF Toms River. Using recombinant HbF γV67M, we then integrated kinetic and quantitative proteomic approaches to gain mechanistic insight into how γMet-67 in the heme pocket undergoes a post-translational modification to Asp and by what mechanism. The results from these studies establish for the first time a linkage between Met(E11) → Asp conversion and accelerated autoxidation. The latter process generates superoxide (O2⨪), which rapidly dismutates into hydrogen peroxide (H2O2) and facilitates oxidation of the methionine side chain and conversion to Asp. Proof that H2O2 is the causative agent comes from the observation that simple bolus addition of H2O2 greatly enhances formation of Asp in β and γ subunits.
We then compared the impact of positionally equivalent E11 methionine mutations in recombinant HbA βV67M and αV62M variants. Met → Asp conversion was identified in the β(E11) but not in the α(E11) variants under similar experimental conditions. The initial crystal structures of the three recombinant Met(E11) HbCO forms show similar active site thioether side chain orientations, with the γ side chain carbon atoms all being roughly 5.0–5.3 Å from the heme iron. However, in agreement with solution experiments, autoxidation of the crystals revealed electron density maps indicative of Asp(E11) formation in the active site of the HbA β V67M mutant but not in the active site of the HbA αV62M variant. These findings demonstrate the unique redox reactivity of the γ and β heme iron atoms, which leads to post-translational oxidative conversion of amino acid side chains within these subunits but not within the α subunits of human Hb.
EXPERIMENTAL PROCEDURES
Reagents
Chromatography media, columns, and equipment were obtained from GE Healthcare and Sigma. Catalase was purchased from Sigma, and superoxide dismutase was purchased from MP Biomedical, LLC (Santa Ana, CA). 18O-Labeled reagents were obtained from Cambridge Isotope Laboratories (Andover, MA).
Protein Production, Purification, and Handling
Human HbA was purified as described (17) from 50-ml aliquots of whole blood obtained from the Division of Transfusion Medicine, National Institutes of Health, with Institutional Review Board approval. Further purification of HbA to remove catalase and other erythrocyte proteins was done using an ÄKTA FPLC system and an XK 50/100 column packed with Sephacryl-200 chromatography media (GE Healthcare). Catalase detection was performed using spectrophotometric methods as described elsewhere (18). Recombinant Hbs (rHbs) were produced in Escherichia coli using the pHE2 and pHE9 expression vectors, provided by C. Ho and T.-J. Shen (Carnegie Mellon University, Pittsburgh, PA) (19, 20). Expression and purification were performed using established methods (21), except use of the cation exchanger column was omitted. Mutant plasmids were constructed using a QuikChange site-directed mutagenesis kit (Agilent, Santa Clara, CA) and the following mutagenesis primers: HbA Evans (αV62M), 5′-CAC GGT AAA AAA ATG GCT GAT GCT CTG AC-3′ and 5′-GT CAG AGC ATC AGC CAT TTT TTT ACC GTG-3′; HbA Bristol (βV67M), 5′-C CAT GGT AAA AAA ATG CTG GGT GCT TT C-3′ and 5′-GAA AGC ACC CAG CAT TTT TTT ACC ATG G-3′; and HbF Toms River (γV67M), 5′-GTC AAG GCA CAT GGC AAG AAG ATG CTG ACT TCC TTG GGA GAT GCC-3′ and 5′-GGC ATC TCC CAA GGA AGT CAG CAT CTT CTT GCC ATG TGC CTT GAC-3′. Hbs were stored in a carbon monoxide (CO)-liganded form at −80 °C until used.
Spectrophotometry
Autoxidation was measured in the presence and absence of catalase (200 units/ml) and superoxide dismutase (10 μg/ml; >35 units) to compare differences in spontaneous autoxidation between the various mutants and their wild-type forms, i.e. rHbF and rHbA, respectively. All measurements were made at 37 °C by recording optical absorbance at 576 nm of solutions of 60 μm Hb (heme equivalents) in 20 mm potassium phosphate buffer, pH 7.4, at 22 °C. Data were collected every minute for 16 h, with every 30th (Fig. 1A) or 15th (Fig. 1B) data point plotted. Rate constants with standard deviations were obtained by averaging data from at least three different experiments and fitting each time course to a single-exponent expression with an offset as described previously (22). The extinction coefficients used to determine concentrations of Hb solutions were as follows: 15.15 mm−1 cm−1 at 576 nm for HbO2 in heme equivalents (23); 14.95 mm−1 cm−1 at 568.5 nm for HbCO in heme equivalents; 4.4 mm−1 cm−1 at 630 nm for aquo-met-Hb in heme equivalents; and 43.6 m−1cm−1 at 240 nm for H2O2 (24). Optical absorbance experiments were performed using either an Agilent Cary 100 or an Agilent 8453 UV-visible light optical absorbance spectrophotometer (Agilent).
FIGURE 1.

Effect of Met substitution at position 67 in the distal oxygen binding pocket on autoxidation and production of ROS. A, autoxidation time courses for native HbA, recombinant HbF, and recombinant HbF γV67M. B, effects of catalase and superoxide dismutase on autoxidation and co-oxidation of epinephrine. Solid lines in the main panels correspond to the fitted observed rate constants listed in Table 1. The effects of catalase and superoxide dismutase under the same buffer conditions and temperature are depicted in B using open squares and open triangles, respectively.
The formation of ferryl intermediates was investigated at room temperature by manual and rapid mixing using conventional and stopped-flow spectrophotometry methods. Initially, 1.5 mm stock solutions of HbO2 were treated with slight molar excesses of K3[Fe(CN)6] to generate ferric Hb. Removal of K3[Fe(CN)6] was accomplished using a column containing Sephadex G-25 media (Sigma). For manual mixing experiments, the conversion of 35 μm HbF γV67M from ferric to ferryl to sulfheme states was monitored by recording spectra between 450 and 700 nm before and after the successive addition of 2-fold excess H2O2, 200 units/ml catalase, and 2 mm disodium sulfide (Na2S). Absorbance measurements were made using an Agilent 8453 UV-visible light optical absorbance spectrophotometer (Agilent). The extinction coefficient used to determine the concentration of sulfheme was 24 mm−1 cm−1 at 620 nm (25). Stopped-flow measurements were made using an Applied Photophysics SX-18 microvolume stopped-flow spectrophotometer with photodiode array unit (Leatherhead, Surry, UK). The volume of the cell was 20 μl. Shot volumes were between 100 and 200 μl, and mixing was performed using equal volumes of reactant solutions. The path length was set to 10 mm, and the entrance and exit slit widths were set to 5 nm.
Experimental Conditions for Quantitative Proteomics
Quantitative proteomic experiments were performed on a patient hemolysate containing the HbF Toms River mutation to assess the relative amount γAsp-67 and Met-67. Erythrocytes were collected by centrifugation of citrate-anticoagulated blood from the Hb Toms River patient at age 6 days and from a normal control neonate and resuspended in ∼5 volumes of 10 mm HEPES buffer, pH 7.4. Hemolysate was obtained by three cycles of rapid freeze-thaw followed by centrifugation. The supernatant was collected, resuspended in the same buffer, and stored under nitrogen at −80 °C.
The mechanistic basis of Met → Asp conversion in HbF γV67M was examined using the following oxidative and nonoxidative conditions (experiments 1–6). In all experiments, 178 μm (heme) HbF γV67M was incubated overnight in 20 mm phosphate buffer, pH 7.4, at 22 °C. The experimental redox and ligation conditions included the following: control incubations with CO- and O2-bound ferrous HbF γV67M in air-equilibrated phosphate buffer (experiments 1 and 2); to study the effect of increasing peroxide concentration, ferrous HbF γV67M was incubated with 1.0, 2.5, and 5.0 m excesses of H2O2 per heme (experiments 3–5); to study the effect of the ferric to ferryl transition on Met → Asp conversion, ferric HbF γV67M (prepared as described above) was incubated with a 10-fold m excess H2O2 (experiment 6). In another set of experiments, both HbA βV67M and HbA αV62M were subjected to the same conditions outlined in experiments 1, 2, and 5 as described above.
Oxidation of Hemoglobin Solutions with H218O2
The 18O labeling experiments were performed by incubating 178 μm oxyferrous HbF γV67M with 2.5-fold m excesses of H218O2 (98% atom percent excess) as described above. 18O labeling experiments were repeated with ferric HbF γV67M (condition 6) as described above.
Proteolytic Digestion of Hemoglobin Samples
All samples were precipitated with trichloroacetic acid (TCA) using a ratio of 1 volume of TCA to 4 volumes of sample. Samples were incubated for 10 min, washed with acetone, and then dried. The resulting pellets were resuspended, denatured, and reduced in 6 m urea, 20 mm DTT, 100 mm NH4HCO3, pH 8.0, at 60 °C, for 1 h followed by alkylation with 100 mm iodoacetamide for an additional hour at ambient temperature in the dark. Each sample was then diluted to 2 m urea, 100 mm NH4HCO3, pH 8.0, at 37 °C, and digested overnight with trypsin (1:25 enzyme/substrate ratio) at 37 °C. The resultant peptides were desalted using solid phase extraction, dried down, and resuspended in 0.1% formic acid (C18 Omix, Agilent Technologies, Santa Clara, CA) prior to mass spectrometry analysis.
LC-MS/MS Analysis
All samples in this study were analyzed by reverse phase liquid chromatography/mass spectrometry (LC-MS/MS) using an Easy nLC II Proxeon nanoflow HPLC system coupled on-line to a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific). Peptides were additionally desalted on a 2-cm C18 reversed phase (75 μm inner diameter) pre-column, loaded on a 10-cm 75 μm (inner diameter) C18 reversed phase column (both Easy Columns from Thermo Scientific), and then separated with a linear gradient of 0–45% buffer B (100% acetonitrile, 0.1% formic acid) for 60 min at a flow rate of 300 nl/min. Data were acquired using a top 10 method (for 60 min) dynamically choosing the most abundant precursors (scanned at 400–2000 m/z) from the survey scans for higher energy collisional dissociation fragmentation. Dynamic exclusion duration was 60 s. The survey scans were acquired in the Orbitrap analyzer at a resolution of 70,000 and fragment ions with a resolution of 35,000. Higher energy collisional dissociation was performed with normalized collision energy of 30 eV. The AGC target was set at 2E5, and the underfill ratio was set to 6%.
All XCalibur raw files (submitted to PeptideAtlas database repository) were converted to DTA formatted peak lists and merged into MGF files by the software Proteome Discoverer version 1.3 (Thermo Scientific). MGF data files were searched against the Swiss-Prot Human database (release 2014_03; contains 542,782 sequence entries) supplemented with variant Hb sequences discussed in this study and porcine trypsin using the Mascot (version 2.4) search engine (Matrix Sciences, London, UK). Mascot was searched with a precursor mass tolerance of ±10 ppm and fragment ion mass tolerance of ±25 absolute milli-mass units, i.e. 0.001 Da. Search parameters were as follows: trypsin specificity, 1 missed cleavage, carbamidomethylation of cysteine as a static modification, methionine oxidation to sulfoxides (+16 Da), and oxidation to sulfone (+32 Da) as variable modifications. Mascot output files were analyzed using the software MassSieve (26). MassSieve filters were adjusted to only include peptide identifications with Mascot Ion Scores equal to or exceeding their identity scores (corresponds to ≥95% confidence).
Quantitative Proteomics Analysis
For quantitative experiments, peptides listed in Table 1 (containing Asp or Met in the oxidized or unoxidized form) from LC-MS/MS data were analyzed to quantify changes in the HbF γV67M, HbA βV67M, and HbA αV62M variants under different oxidative conditions. Each peptide listed in Table 1 was validated by Mascot identification and retention time reproducibility. All quantitative experiments were performed in triplicate, and standard deviations were obtained by averaging relative abundance data from three different experiments. Briefly, extracted ion chromatograms (XICs) were generated from the most abundant monoisotopic peak of each isotopic profile (representing charged states of each peptide) to calculate the relative abundance of each peptide mass. To construct XICs, Xcalibur (version 2.2) software was used with a designated mass tolerance of 0.01 Da, and mass precision was set to three decimals. For relative quantification, the ratio of each Asp-67 or Met-67 isoform was calculated based on the sum of the XIC peak area from all forms (including all charge states and versions that result from different cleavage sites), which was normalized to be 100%.
TABLE 1.
All targeted peptides including charge state and cleavage variants containing either Met or Asp at position 67 or 62
| Peptides | Amino acid position 67 | (+) Charge state | m/z |
|---|---|---|---|
| γV67 M Toms River | |||
| 67DLTSLGDAIK76 | Asp | 2 | 516.7829 |
| 1 | 1032.5569 | ||
| 67MLTSLGDAIK76 | Met (unoxidized) | 2 | 524.7829 |
| 1 | 1048.5668 | ||
| Met (sulfoxide) | 2 | 532.7879 | |
| 1 | 1064.5653 | ||
| Met (sulfone) | 2 | 540.7815 | |
| 1 | 1080.5606 | ||
| 66KMLTSLGDAIK76 | Met (sulfoxide) | 2 | 596.8342 |
| Met (sulfone) | 2 | 604.832 | |
| βV67 M Bristol-Alesha | |||
| 67DGAFSDGLAHLDNLK76 | Asp | 2 | 562.6215 |
| 67KDGAFSDGLAHLDNLK76 | Asp | 2 | 605.3199 |
| 67MLGAFSDGLAHLDNLK76 | Met (unoxidized) | 2 | 851.4353 |
| 3 | 567.9596 | ||
| Met (sulfoxide) | 2 | 859.4267 | |
| 3 | 573.2903 | ||
| Met (sulfone) | 1 | 578.6223 | |
| βV62 M Evans | |||
| 61MADALTNAVAHVDDMPNALSALSDLHAHKK90 | Met (unoxidized) | 4 | 758.121 |
| 5 | 606.6985 | ||
| Met (sulfoxide) | 4 | 762.1201 | |
| 5 | 609.8976 | ||
| Met (sulfone) | 4 | 766.1195 | |
| 5 | 613.0964 | ||
| 61KMADALTNAVAHVDDMPNALSALSDLHAHKK90 | Met (unoxidized) | 4 | 790.3951 |
| 5 | 632.3174 | ||
| Met (sulfoxide) | 4 | 635.5165 | |
Crystal Structure Determinations for Recombinant Wild-type and Met(E11) Mutants
The CO forms of both HbF wild type and γV67M were crystallized using the batch method from mixtures of concentrated HbCO solutions and 3.4 m sodium/potassium phosphate, pH 7.0. 100–200 μl of this mixture was placed in a sealed, CO-purged Vacutainer, and a drop of toluene was added. The final concentrations were 20 mg/ml protein and 1.95 m precipitant. Crystals were grown over 2–3 days at ambient temperature. Crystals of the recombinant HbA variants were grown as described previously (27) using mixtures of HbCO and 3.4 m sodium/potassium phosphate, pH 6.7. In all cases, crystals were transferred to mother liquor containing 25% sucrose (w/v) immediately before data collection at 100 K. For CO complexes, the mounting solution was saturated with 1 atm of pure carbon monoxide. A cryo-cooling N2 system was used to maintain low temperature (100 K) in the environment of the mounted crystals to reduce radiation damage.
The fetal HbCO γV67M crystals grew in the space group P21 with two tetramers in the asymmetric unit. Because of the small size of the crystals and their poorer diffraction, data for crystals of HbF were collected remotely at the Advanced Photon Source, on the LSCAT beamline 21-1D-D, where the wild-type and mutant HbF crystals diffracted to 1.8 and 2.2 Å, respectively. These data were reduced and scaled using the HKL2000 software.
Data for the mutant adult HbCO crystals were collected using a Rigaku RUH3R rotating anode x-ray generator operated at 50 kV and 90 mA and a Rigaku R-AXIS IV++ image plate detector. Sets of CO crystals of for both adult Met(E11) mutants were taken out of their CO environment and transferred to oxygenated mother liquor. After 4 weeks, the color of these crystals had changed from the cherry-red of the reduced CO forms to the reddish brown of the ferric forms. Then diffraction data were collected from these oxygenated/oxidized crystals as described above. All data were reduced and scaled using the d*TREK software.
The fetal HbCO structures were determined by molecular replacement using the program PHENIX and the previously reported high resolution structure of human HbA (RCSB PDB code 2DN3) as the starting model. After a clear solution was obtained with two tetramers in the asymmetric unit, the appropriate changes were made in the β chains to reflect the correct sequence of amino acids in the γ chain. Crystallographic refinement continued in PHENIX, with several macrocycles that included simulated annealing, bulk solvent correction, and anisotropic scaling of the data, individual coordinate refinement with minimization, and individual isotropic ADP refinement. The CO ligand was introduced into the electron density at this stage, and refinement continued with restraints imposed on the expected Fe-CO geometry. Finally, solvent molecules were included, and combined TLS and individual ADP refinements were carried out.
The adult HbCO structures were solved in PHENIX by direct Fourier synthesis again using the high resolution structure of human HbA (2DN3) as the starting model. The structures of the oxidized proteins (4 and 7 weeks of crystal exposure to O2 at room temperature) were solved using the metHb structure of human HbA (RCSB PDB code 3P5Q) as the starting model. The final refinements were carried out in PHENIX as described above. Map fitting and other manipulations with molecular models were performed using the graphical software COOT. The accession codes for the models, crystal parameters, and statistics of x-ray data collection and refinement are given in Table 2. Crystal structural figures were prepared using the PyMol Molecular Graphics System, version 1.2 (Schrodinger, LLC).
TABLE 2.
Data collection and refinement statistics for the recombinant wild type and mutant HbF and HbA crystal structures
| WT HbFCO | HbCO Toms River α(wt)γ(V67M) | HbCO Bristol-Alesha α(wt)β(V67M) | MetHb Bristol-Alesha α(wt)β(V67M) | HbCO Evans α(V62M)β(wt) | MetHb Evans α(V62M)β(wt) | |
|---|---|---|---|---|---|---|
| PDB entry code | 4MQJ | 4MQK | 4MQG | 4MQI | 4MQC | 4MQH |
| Crystal data | ||||||
| Resolution range | 45.14 to 1.80 Å | 45.1 to 2.24 Å | 51.48 to 1.68 Å | 35.7 to 1.92 Å | 36.9 to 2.2 Å | 50.0 to 2.5 Å |
| Space group | P21 | P21 | P41212 | P41212 | P41212 | P41212 |
| Unit cell parameters (Å, °) | a = 105.65, b = 52.43, c = 106.15, β = 113.84 | a = 106.04, b = 52.23, c = 106.38, β = 114.27 | a = b = 53.43, c = 192.22 | a = b = 53.21, c = 192.48 | a = b = 53.17, c = 192.98 | a = b = 52.93, c = 194.14 |
| Reflections (measured/unique) | 742,084/99,490 | 377,777/51,099 | 246,367/24,312 | 55,639/20,759 | 164,122/14,858 | 92,187/10,243 |
| Completeness | 99.7% (99.2%)a | 99.7% (100%) | 74.0% (17.2%) | 93.5% (96.0%) | 99.7% (99.9%) | 98.0% (94.5%) |
| 〈I〉/σ〈I〉 | 27.9 (5.86) | 20.6 (8.7) | 10.1 (2.3) | 32.1 (2.94) | 8.5 (4.5) | 15.0 (6.0) |
| Redundancy | 7.5 (7.2) | 7.4 (6.7) | 10.1 (3.52) | 2.9 (2.9) | 11.0 (13.0) | 8.9 (7.4) |
| Rmerge | 5.2% (42.0%) | 11.2% (53.7%) | 12.7% (36.2%) | 4.6% (36.0%) | 12.1% (43.7%) | 4.1% (50.0%) |
| Refinement | ||||||
| Resolution range | 45.14 to 1.80 Å | 45.1 to 2.24 Å | 51.48 to 1.65 Å | 35.7 to 1.92 Å | 36.9 to 2.2 Å | 50.0 to 2.5 Å |
| R-factor | 18.0% | 17.7% | 20.3% | 20.1% | 21.4% | 19.8% |
| Rfree | 21.5% | 23.2% | 24.3% | 23.9% | 25.0% | 25.0% |
| Root mean square deviations from ideal values | ||||||
| Bond length | 0.015 Å | 0.008 Å | 0.019 Å | 0.007 Å | 0.023 Å | 0.010 Å |
| Bond angles | 0.93° | 1.092° | 1.068° | 0.988° | 1.184° | 1.182° |
| Residues in most favorable region | 98.0% | 97.0% | 99.0% | 99.0% | 98.0% | 96.0% |
| Residues in additional allowed region | 2.0% | 3.0% | 1.0% | 1.0% | 2.0% | 4.0% |
a Parameters in parentheses are for the outer resolution shell.
RESULTS
Autoxidation and Reactive Oxygen Species Formation
HbO2 undergoes autoxidation, in which the oxygen-bound ferrous (Fe2+) heme iron atom spontaneously oxidizes to the ferric or metHb (Fe3+) state, initially generating a mixture of protonated and anionic superoxide radicals. Autoxidation of Hb is associated with subsequent globin dysfunction and instability due to generation of H2O2 resulting from dismutation of the initial superoxide products (28, 29). Thus, we first surveyed the autoxidation properties of wild-type HbA and HbF and the three recombinant Val → Met(E11) Hb variants (α, β, and γ) by following the formation of metHb via heme spectral changes in the visible wavelength region (Fig. 1A). Estimates of the individual autoxidation rate constants were obtained by fitting the initial phase of the absorbance decreases at 576 nm to single-exponential expressions. As shown in Table 3 and Fig. 1B, catalase reduced the apparent rate of autoxidation of all Hb samples by ∼50%, due presumably to the removal of H2O2, which is generated from release of superoxide and its rapid dismutation at neutral pH. H2O2 can react with HbO2 at rates that are much greater than initial autoxidation, speeding up the overall formation of metHb. Addition of superoxide dismutase by itself has little or no slowing effect showing that it is H2O2 and not O2⨪ that speeds up autoxidation.
TABLE 3.
Observed rate constants for the initial phase of Hb autooxidation in air-equilibrated buffer at pH 7.4, 37 °C
| Sample | kauto | +Catalase | +Superoxide dismutase |
|---|---|---|---|
| h−1 | h−1 | h−1 | |
| HbA | 0.065 ± 0.016 | 0.036 ± 0.014 | 0.041 ± 0.011 |
| HbF | 0.081 ± 0.014 | 0.03 ± 0.0059 | 0.058 ± 0.012 |
| γV67Μ | 0.58 ± 0.047 | 0.32 ± 0.024 | 0.52 ± 0.0076 |
| βV67Μ | 0.81 ± 0.120 | 0.42 ± 0.15 | 0.79 ± 0.16 |
| αV62Μ | 0.58 ± 0.120 | 0.3 ± 0.09 | 0.45 ± 0.06 |
The key results in Table 3 and Fig. 1A are that HbF γV67M autoxidizes almost 8-fold more rapidly than either wild-type HbA or HbF, and similar higher rates of autoxidation are also observed for the HbA βV67M and αV62M variants. Additionally, ferric HbF γV67M is clearly more prone to heme loss and precipitation than the met-forms of wild-type HbA and HbF, as is evidenced by the dramatic increase in solution turbidity toward the end of the experiment, which is characterized by the upward trend in HbF γV67M absorbance at later time points. These data indicate that the γV67M mutation markedly increases the susceptibility of HbF to oxidation and denaturation.
Peroxide-mediated Ferryl Formation
The pseudoperoxidase reactivity of the Met(E11) Hb variants with H2O2 was examined using UV-visible absorbance spectroscopy and the formation of the ferryl heme species monitored by characteristic changes in the visible spectrum. Fig. 2 shows a typical spectral transition for the reaction of the ferric HbF γV67M mutant (35 μm) with 70 μm H2O2 to form a ferryl species with major peaks at 545 and 585 nm and a flattened region between 600 and 700 nm. To confirm the redox identity of the ferryl state intermediate, Na2S was added to the reaction mixture. Upon the addition of sulfide, the iron in ferryl Hb is reduced to the ferric state, and, at the same time, sulfur is incorporated into the porphyrin ring (see Fig. 2B), which can be monitored by the appearance of an absorbance band at 620 nm (25). Catalase was added prior to sulfide addition to remove any excess H2O2 so that any sulfur addition to the heme group was due only to reaction with ferryl heme complexes. In these experiments, the levels of ferryl Hb in wild-type and mutant HbF were estimated to be 18.6 and 19.3 μm, respectively, based on the reported extinction coefficients for sulfheme (25). Under similar experimental conditions, using Na2S as a derivatizing agent, we calculated the ferryl levels for the reactions of ferric HbA βV67M and HbA αV62M with a 2-fold excess of H2O2 solutions to be similar, 13 and 11.5 μm, respectively (data not shown). The inset in Fig. 2 shows time-resolved spectra for ferryl formation (0–10 min) when ferric HbF γV67M (30 μm) was reacted with 60 μm H2O2 in a stopped-flow spectrophotometer using the same buffer conditions.
FIGURE 2.

Effect of Met substitution at position 67 in the distal oxygen binding pocket on redox ferric to ferryl transitions. Redox ferric to ferryl transition states of recombinant HbF Toms River are shown. A, absorbance spectrum between 450 nm and 700 nm of 35 μm ferric HbF γV67M (black line) after the addition of a 2-fold molar excess of H2O2 (red line). Catalase (200 units/ml) was then added and mixed into the same cuvette, followed by 2 mm Na2S to form sulfheme (blue line). The inset shows time-resolved data in a similar wavelength range derived from the rapid mixing of 30 μm ferric HbF γV67M with 60 μm H2O2 in the same buffer. 40 spectra were recorded over the course of 600 s between 475 nm and 650 nm (25). B, structure of the modified porphyrin generated by the addition of sulfide. This reaction also involves the reduction of a β-carbon double bond in one of the pyrroles in protoporphyrin IX to form a chlorine.
Mass Spectrometry Analysis Confirms Post-translational Conversion of Met to Asp in Patient's Blood and in the Recombinant Protein
We initially used LC-MS survey scans to identify peptides representing γMet-67 (both oxidized sulfoxide and sulfone intermediates) and γAsp-67 variant isoforms from the Toms River patient hemolysate sample (Fig. 3, A and B). The MS/MS spectrum in Fig. 3A (residues 67–76) clearly shows singly charged product ions (b2, b4, b5, and b8) whose mass values are 16 daltons higher than the same fragment ions representing the Asp-67-containing peptide (Fig. 3B). Using the same approach, we also identified γAsp-67 isoforms produced by recombinant HbF γV67M after treatment with H2O2, which was essential for subsequent in vitro quantitative proteomic studies aimed at exploring the mechanism behind this conversion.
FIGURE 3.

LC-MS/MS identification of Asp and Met containing γ67 peptides from Toms River patient's blood. Fragmentation spectra of the singly charged +1 γMet-67 (A) and γAsp-67 (B) tryptic peptides representing residues 67–76 from patient hemolysate. Both spectra show singly charged y and b fragment ions. Displayed above the panels is the primary sequence of the γV67M mutant subunit showing the tryptic peptide (highlighted in red) with position Met-67 (highlighted in blue). * refers to a loss of H2O at serine; ** refers to a loss of H2O at serine and threonine, respectively All charge states of this peptide sequence were the focus of our quantitative proteomic approach.
Oxidation Reactions at the E11 Position in the γ Fetal Hb Subunit Mediates Conversion of Met to Asp
Our kinetic experiments on the spontaneous oxidation of recombinant HbF γV67M were designed to mimic initial oxidative events in patient's blood. They demonstrated that the γV67M mutation resulted in an accelerated rate of autoxidation and concomitant production of H2O2 compared with the wild-type HbF. To explore how these processes contribute to the observed post-translational oxidation of Met → Asp in the patient's blood, we used quantitative proteomics to determine the relative abundance of recombinant γMet-67 and γAsp-67 isoforms under various oxidative reactions (see under “Experimental Procedures”). To quantify isoforms, extracted ion chromatograms (XICs) were generated from the most abundant monoisotopic peak of all Met-67 (thioether, sulfoxide, and sulfone) and Asp-67 peptide isotopic profiles. For example, the most abundant monoisotopic peak (1032.5569 m/z) represented in Fig. 4A for the Asp-67-containing peptide, DLTSLGDAIK, was used to construct the XIC in Fig. 4B. Because the γ67 amino acid could only exist as Met (thiol ether, sulfoxide, and sulfone) or Asp, the percentage of γMet-67 and γAsp-67 isoforms was calculated based on the sum of the XIC peak area from all charged forms of Met-67 and Asp-67 peptides (see Table 1).
FIGURE 4.

Extracted ion chromatogram of γAsp-67 tryptic peptide. XICs were generated from the most abundant monoisotopic peak of each peptide isotopic profile as described under “Experimental Procedures.” A, representative isotopic profile of the singly charged γAsp-67 tryptic peptide, DLTSLGDAIK. B, XIC of the γAsp-67 tryptic peptide (residues 67–76).
The results of these studies are summarized in Table 4 and Fig. 5 and illustrate several key features of the Met → Asp conversion as follows. 1) HbF Toms River from the patient hemolysate contained substantial amounts of oxidized γMet-67 (sulfoxide and sulfone forms, and γAsp-67 isoforms, indicating considerable oxidation in the infant's red blood cells. 2) In air-equilibrated buffer, where autoxidation forms H2O2, both γMet-67 (primarily in oxidized forms) and γAsp-67 peptides were present. 3) Under conditions where CO was liganded to ferrous HbF γV67M, the levels of γAsp-67 peptides were significantly reduced. The CO inhibits binding of O2 and subsequent autoxidation, thereby reducing production of H2O2. However, small amounts of γMet-67 sulfone/sulfoxides levels were detected in the CO-liganded recombinant protein. Although methionine sulfoxide oxidation is a common PTM associated with sample handling, a proportion of these intermediates probably formed during growth and the subsequent production process in host E. coli cells. 4) Treatment of ferrous HbF γV67M with increasing H2O2 increased the ratio of γAsp-67/Met-67 in a dose-related fashion. When 2.5 and 5.0 m excess H2O2 were used, there were 7- and 13-fold increases in the Asp-67/Met-67 ratio relative to the CO-ligand control, respectively. This dose-dependent increase in γAsp-67 levels strongly suggests that oxidative Met → Asp conversion involves a H2O2-driven mechanism in the patient's blood. 5) Finally, when H2O2 was added to ferric HbF γV67M (Fig. 5, condition 6) oxidation of γMet-67 to sulfoxide and sulfone occurred significantly, but little formation of Asp was observed, which suggests that Met → Asp conversion specifically involves oxidation of ferrous HbF γV67M to produce the ferryl state without any porphyrin or protein radicals (see proposed mechanism shown under “Discussion”).
TABLE 4.
Quantitative proteomic data representing oxidative reactions with HbF Toms River (γV67M)
| Reaction conditions | Asp-67 ratio | Met-67 ratio | Met-67 sulfoxide ratio | Met-67 sulfone ratio | Asp-67 fold change compare with condition 1 |
|---|---|---|---|---|---|
| % | % | % | % | ||
| Patient hemolysate | 7.7 ± 1.4 | 6.0 ± 1.9 | 60.5 ± 3.8 | 25.7 ± 3.7 | |
| 1) CO ligand control | 1.2 ± 0.15 | 51.8 ± 9.74 | 33.48 ± 16.44 | 13.58 ± 6.60 | 1 |
| 2) Control (air equilibration buffer) | 5.0 ± 1.0 | 34.5 ± 7.84 | 49.30 ± 9.18 | 11.24 ± 3.96 | 4.2 |
| 3) 1:1 H2O2 | 5.7 ± 0.3 | 7.18 ± 0.69 | 56.55 ± 0.45 | 30.58 ± 0.47 | 4.8 |
| 4) 2.5:1 H2O2 | 8.8 ± 0.8 | 5.94 ± 0.23 | 52.75 ± 2.25 | 32.66 ± 2.61 | 7.3 |
| 5) 5:1 H2O2 | 15.5 ± 4.2 | 4.03 ± 0.65 | 39.44 ± 3.85 | 41.07 ± 2.73 | 12.9 |
| 6) Ferric/5:1 H2O2 | 2.0 ± 0.4 | 1.21 ± 0.06 | 52.83 ± 0.28 | 43.99 ± 0.53 | 1.6 |
FIGURE 5.
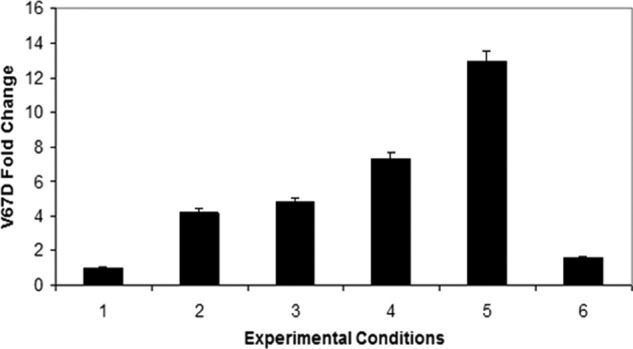
Histogram representing the fold change differences of Asp-67 formation for experimental conditions listed in Table 4. These data indicate Met to Asp conversion increases with increasing H2O2.
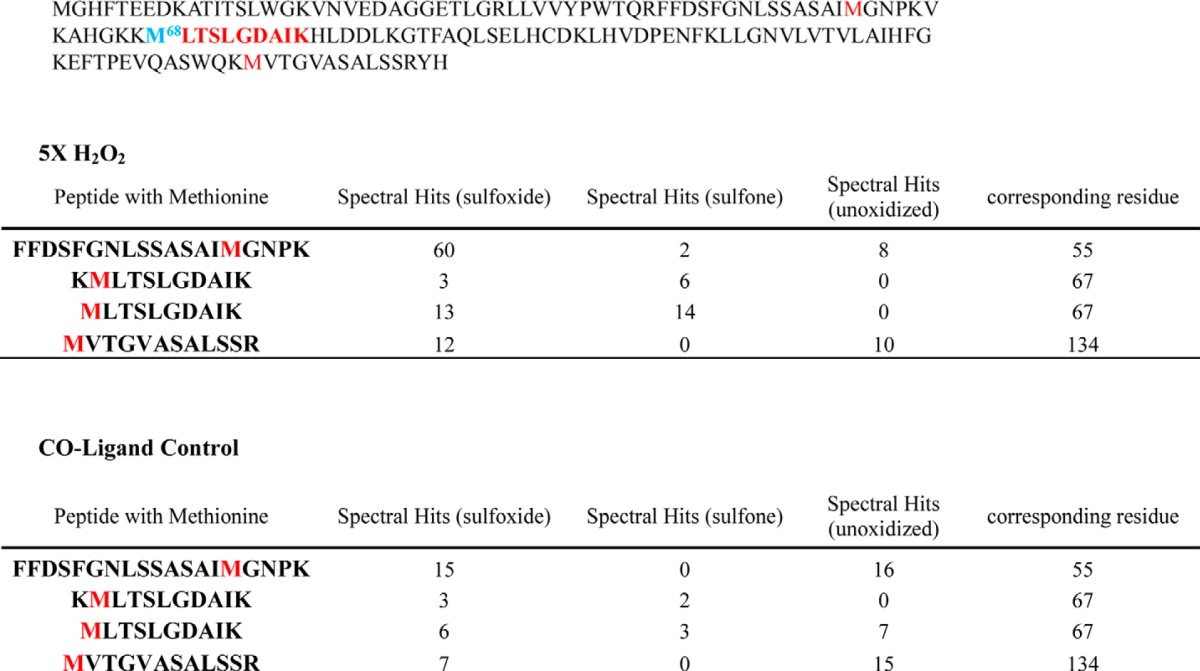
An additional observation from these experiments is that Mascot database searches of LC-MS/MS data revealed that methionine sulfone oxidation occurs almost exclusively on the Met-67-containing peptides (Table 5), for both ferrous and ferric forms of HbF γV67M. This observation supports previous reports that methionine sulfone oxidation is substantially enhanced when the side chain is in close proximity to a metal ion containing group in what is referred to as a caged process (7, 30, 31). The presence of sulfones at the Met-67 position in the HbF variant supports this view because the thiol ether side chain is clearly very close to the reactive heme iron atom as shown in the crystal structure (see below).
TABLE 5.
Number of spectra matched to methionine (spectral hits)-containing peptides (and their charge states) in the γ subunit (Hb Toms River) of H2O2 and control-treated
18O Labeling Confirms That Peroxide Is the Source of Oxygen in the Aspartic Side Chain
To further probe the role of H2O2 in the oxidative Met → Asp conversion, H218O2 (2.5-fold excess relative to heme) was mixed with both ferrous and ferric HbF γV67M. Analyses of LC-MS/MS and full MS data identified oxidized Met-67- and Asp-67-containing peptides as the only peptides that incorporated 18O. Additionally, we identified 18O-labeled γAsp-67 isoforms when H218O2 was mixed with ferrous HbF γV67M but not the ferric form, which correlates with the results from Table 4. The isotopic profile in Fig. 6 illustrates both 16O- and 18O-labeled isotopologous DLTSLGDAIK peptides after H218O2 treatment of ferrous HbF γV67M. The most abundant isotopic peak (M + H)+ of 1036 m/z represents the DLTSLGDAIK peptide with two 18O-labeled carboxyl oxygen molecules. The peak at 1034 m/z represents the same peptide with a mixture of 18O/16O-labeled carboxyl oxygen, and the lowest abundant 1032 m/z peak represents the unlabeled peptide (same m/z as the most abundant isotopic peak in Fig. 4A). This isotopic profile indicates that H218O2 is the source of oxygen in the Asp-67 carboxyl side chain. This result is further substantiated by the MS/MS spectrum showing isotopologous fragment ions of the labeled and unlabeled peptides (Fig. 7). Both data sets unambiguously assign H2O2 as a major source of oxygen atoms incorporated into the Asp carboxyl side chain during the oxidative conversion of Met-67. Furthermore, the isotopic labeling data in Figs. 6 and 7 reveal mono- and di-18O-labeled γAsp-67 species, suggesting that the oxidative conversion involves a complex multistep mechanism (see the proposed mechanism accompanying description under “Discussion”).
FIGURE 6.

Isotopic profile represented by labeled isotopologous peptides after oxidation with H218O2. The most abundant isotopic peak (M + H) of 1036 m/z represents the γAsp-67 tryptic peptide with two 18O-labeled carboxyl oxygen molecules. The 1034.56 m/z peak represents the γAsp-67 tryptic peptide with a mixture of 18O/16O-labeled carboxyl oxygen, and the lowest abundance 1032.56 m/z represents the unlabeled peptide (M + H).
FIGURE 7.

Fragmentation spectra of isotopologous γAsp-67 tryptic peptides after oxidation with H218O2. The precursor isolation width was increased to fragment the labeled and unlabeled isotopologous peptides represented by the isotopic profile in Fig. 6. A, MS/MS spectrum shows the isotopologous fragment ions of both the labeled and unlabeled peptides, respectively. B and C, both zoomed in panels correspond to the rectangular area of the MS/MS spectrum representing isotopologous b8 fragment ions sets. * refers to a loss of H2O at serine; ** refers to a loss of H2O at serine and threonine, respectively.
Oxidative Modification of Met to Asp at the E11 Position Also Occurs in β but Not in α Subunits of Human Hb
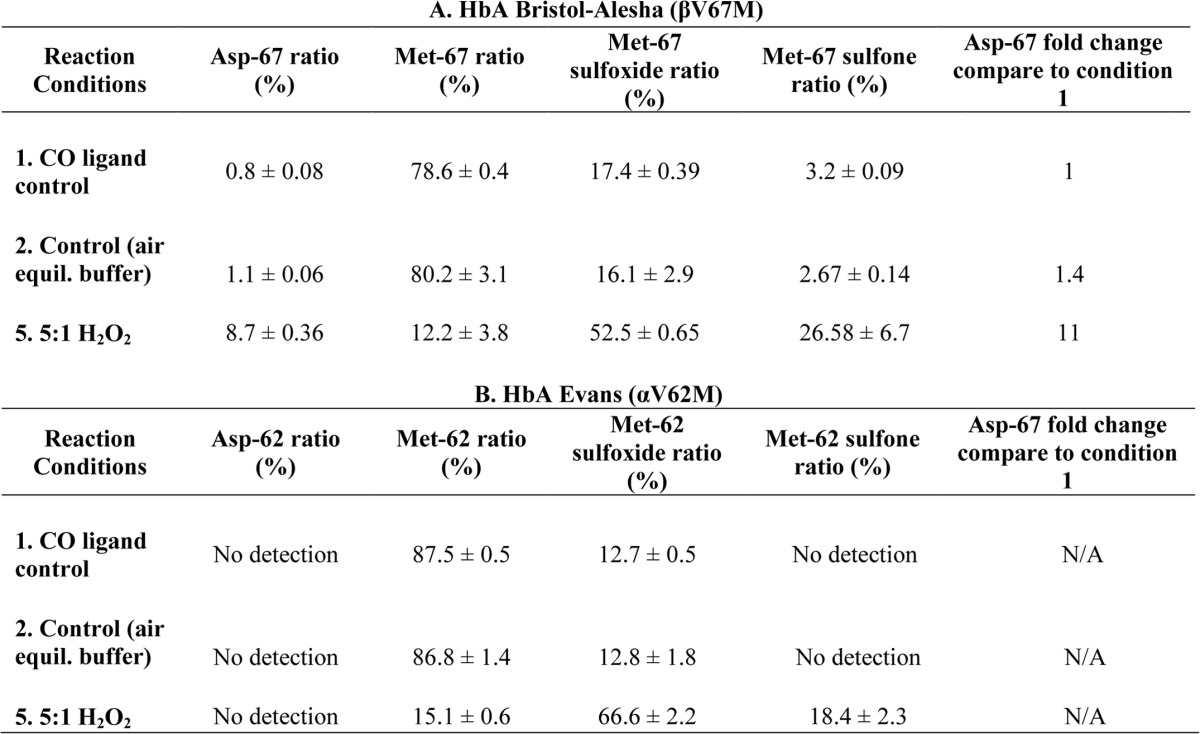
We recently reported that β but not α subunits of human Hb contribute significantly to the oxidative burden of the protein. This is reflected by the accumulation of ferryl intermediates in the β subunit of oxidized Hb (22). To follow-up these studies, we examined the positional equivalent Met(E11) mutations in HbA βV67M and αV62M, respectively. As described for the recombinant Toms River variant, we performed the same quantitative proteomic analyses to quantify Met and Asp levels at positions 67 for βV67M and 62 for the αV62M mutants (experimental conditions summarized in Table 6, A and B). These data show for the first time that oxidative Met(E11) → Asp conversion is subunit-dependent. Molar excess of H2O2/heme facilitated the conversion of Met → Asp in the β subunit at levels similar to those observed in the Met(E11) γ variant but not in mutant α subunits. In fact, we were unable to detect any Asp-62 in recombinant HbA Evans, regardless of conditions. However, in all cases, H2O2 addition did dramatically increase the levels of Met(E11) sulfoxide and sulfone in both subunits. It is noteworthy that untreated βMet(E11) HbA shows measurable amounts of sulfone and higher amounts of sulfoxide than the corresponding αMet(E11) HbA variant.
TABLE 6.
Quantitative proteomic data representing oxidative reactions with HbA Bristol-Alesha βV67M and HbA Evans αV62M
Crystal Structures of the Met(E11) Variants of Human α, β, and γ Subunits
Crystal structures were determined for wild-type HbF, HbF γV67M, HbA βV67M, and HbA αV62M mutants (Fig. 8). As shown in Fig. 8B, the orientations of the Met(E11) side chains in the mutant α, β, and γ subunits are all very similar in the HbCO forms, with the ϵCH3 atoms pointing toward the interior of the subunits. The γCH2 atoms in all three proteins are at similar distances from the iron atom (5.0–5.3 Å). However, when the CO forms of HbA αV62M and βV67M crystals were exposed to oxygen and allowed to slowly oxidize over a few weeks, several interesting changes were observed as follows (Fig. 9, A and B). 1) After 4 weeks, the electron density in the β subunit of the HbA βV67M crystals seems to indicate the appearance of a shorter side chain consistent with Asp at the E11 position (Fig. 9A). The electron density could also be interpreted in terms of mixtures of sulfoxide, sulfone, and an Asp side chain. 2) The ligand electron density at the iron atom in the β subunit of the HbA βV67M structure looks more compatible with a Fe4+=O (oxoferryl) complex than a Fe3+-OH2 (ferric) complex (Fig. 9, A, left panel, and B, upper right). The distance from the iron atom to the center of the ligand electron density is 1.9 Å, which is shorter than the normal Fe-O distance of 2.2–2.3 Å in most aquo-metHb and metMb structures. The “bifurcated” difference in electron density for the E11 side chain and the unusual ligand electron density in the mutant β chain were persistent over two different data sets collected with different crystals. 3) In contrast, after 7 weeks there was little or no change in the electron density for the αE11 side chain in the oxidized HbA αV62M crystals, indicating no shortening of the Met-62 side chain (Fig. 9B, middle panel). 4) In the wild-type β subunit of oxidized HbA αV62M, there also appears to be a ferryl Hb Fe4+=O complex, as judged by the Fe-O bond distance of 1.9 Å (Fig. 9B, bottom right panel), whereas the ligand in the mutant Hb α subunit appears to be a coordinated water molecule, about 2.3 Å away from the iron atom (Fig. 9B, bottom middle panel).
FIGURE 8.

Structures of the active sites of HbF Toms River γV67M, HbA Bristol-Alesha βV67M, and HbA Evans αV62M. A, 2Fo − Fc electron density of γMet(E11) in HbCO Toms River 2Fo − Fc, refined electron density (PDB 4MQK). B, comparisons of Met(E11) side chain orientations in refined models for the CO forms of HbF γV67M (PDB 4MQK), HbA βV67M (PDB 4MQG), and HbA αV62M (PDB 4MQC).
FIGURE 9.

Structures of the oxidized forms of recombinant HbA Bristol-Alesha βV67M and HbA Evans αV62M. A, oxidation of Hb βV67M crystals (HbCO form, PDB 4MQG; metHb form after 4 weeks, PDB 4MQI). The electron density in the middle panel for Bristol-Alesha suggests the appearance of Asp at the E11 position due to oxidation. The ligand electron density in the right-hand panel for the β mutant heme group suggests a ferryl complex with a single coordinated distal atom rather than aquometHb complex with a spherical water oxygen atom about 2.3 Å away from the central iron atom. B, oxidation of αV62M crystals (HbCO form, PDB 4MQC; metHb form after 7 weeks, PDB 4MQH). There was no change in the electron density for the αE11 side chain in the oxidized Hb Evans crystals indicating no modification of the Met-62 residue. However, the coordination in the wild-type met-β active site again suggests a significant fraction of ferryl complex (right-hand panel).
An unambiguous structural interpretation of the iron-ligand electron density in the β subunits of autoxidized HbA Evans and HbA Bristol-Alesha is not possible at the resolution of the diffraction data. The modeled Fe-O distance of 1.9 Å is consistent with previously reported ferryl complexes in peroxidases (32). However, it is also possible that the electron density map could represent a mixture of hexacoordinate Fe3+-OH2 and pentacoordinate Fe3+ complexes (33, 34) that the occupancy of water coordinated to the iron atom in β subunits of metHbA is less than what is observed in α subunits.
In an attempt to address this ambiguity, we redissolved autoxidized HbCO Evans crystals and examined their spectral properties compared with fresh samples that were oxidized by ferricyanide. The autoxidized Hb Evans crystal samples showed evidence for small amounts of ferryl-like spectral species, and addition of N2S led to the appearance of small 620-nm bands indicative of sulfheme (data not shown). However, the amounts of these species were small indicating that not all the β subunits were modified.
Regardless of the exact interpretation, all these structural observations reinforce the idea of a greater propensity of the β subunit to form reactive ferryl species that enhance the conversion of Met(E11) → Asp in β but not α subunits. Fig. 9A, upper panels, shows sample diffraction patterns from the HbA βV67M crystals before, during, and after complete oxidation. There are clear losses in resolution upon oxidation, and no useful data could be collected from crystals that were exposed to air for 7 weeks, presumably due to degradation, heme loss, precipitation, and destruction of the crystals.
Similar oxidation experiments could not be carried out with recombinant fetal HbCO γV67M crystals because of their smaller size and more complex asymmetric unit (two tetramers versus one αβ dimer in HbA crystals). This complexity required use of the Advanced Photon Source, high intensity synchrotron x-ray source, which often leads to radiation-induced damage. In the crystal structure of the CO form of HbF Toms River, only one of the γ subunits showed complete electron density for the Met(E11) side chain (Fig. 8), whereas the other γ subunits showed loss of electron density similar to that seen for the β E11 side chain in oxidized Hb Bristol-Alesha crystals (data not shown). Although high intensity x-ray beams produce hydrated electrons that can reduce metal atoms, these electrons also generate reactive oxygen species, including superoxide and H2O2, which in turn facilitate degradation of the Met(E11) side chain in β subunits.
DISCUSSION
The pseudo-peroxidase activities of Hb have been under intense investigation in vitro, and more recent experimental evidence from animal studies supports the notion that these reactions occur in vivo with some potentially serious consequences (35, 36). H2O2 is known to oxidize ferrous Hb (HbFe2+) and ferric Hb (HbFe3+) to generate higher oxidation states of the protein, including the oxoferryl Hb (HbFe4+ = O2−) intermediate and protein-based cation radicals (+·HbFe4+ = O2−), respectively, as shown in Equations 1 and 2,
Low temperature EPR was used to monitor the amino acid ferryl radicals following H2O2 addition to the ferric form of the mutants (data not shown). A signal was observed at g = 6 and represented high spin ferric heme, and a second signal was observed at g = 2 and represented the tyrosine free radical as described in previous publications (22, 37).
Both the ferryl heme and its associated protein cation radical induce a wide variety of oxidative reactions affecting both the protein itself and nearby molecules due to their high midpoint redox potentials (E1/20 ∼1.0 V) (38). These internal reactions result in modification of the heme, its attachment to nearby amino acids, and irreversible oxidation of amino acids in some “hot spots,” particularly the βCys-93 side-chain (39, 40).
Oxidation-related mechanisms also play a pathophysiological role in persons with unstable Hbs and other related hemoglobinopathies (16). In this study, we report for the first time the mechanistic basis of an oxidation-mediated post-translational conversion of Met to Asp at the E11 position within the heme binding pockets of HbF Toms River (γV67M) and HbA Bristol-Alesha (βV67M), but not in the αMet(E11) analog HbA Evans (αV62M). Our autoxidation experiments, which approximate physiological conditions, show that the Val(E11) → Met mutation accelerates autoxidation of HbO2 roughly 10-fold regardless of whether or not the substitution is in γ, β, or α subunits. In the presence of catalase, small but significant reductions in these rates are observed, confirming the buildup of H2O2 during the autoxidation process (Table 3). Moreover, all three Hbs exhibited pseudoperoxidase activities through formation of classical ferric/ferryl redox intermediates (see Equations 1 and 2) in the presence of H2O2 as exemplified by the UV-visible spectral data (Fig. 2) for Toms River γV67M, and in the case of the ferric forms of these mutants, ferryl protein radicals were detected by EPR spectroscopy (data not shown).
The proteomic results in Tables 4 and 6 indicate that all three variants exhibit substantial increases in Met(E11) sulfoxide and sulfone levels with increasing H2O2. Despite this, Met → Asp conversion was not detected in the HbA αV62M mutant, even following addition of a 5-fold excess of H2O2. Conversely 10–15% of the Met(E11) was converted to Asp in the mutant γ and β subunits. Oxidative Met → Asp conversion also appeared to occur during oxidation of HbA Bristol-Alesha crystals but not in the crystals of the corresponding αMet(E11) Hb Evans variant (Fig. 9). Taken together, these data clearly indicate a difference in redox reactivity between the subunits and suggest that the lack of Asp formation in Hb Evans αV62M may be due to the higher degree of oxidative stability of α subunits relative to that of γ and β subunits in HbF Toms River and HbA Bristol-Alesha, respectively (3, 9–11, 15).
The apparent differences in redox reactivity correlate with our previously reported studies indicating that α subunits form fewer transient ferryl intermediates and accumulate much smaller amounts of detectable protein radicals when exposed to H2O2 in solution (22). This difference appears to be due to rapid autoreduction rates of ferryl α chains, which, in turn, have been attributed in part to the proximity of αTyr-42 to the heme iron atom. This external tyrosine is proposed to act as a redox cofactor that provides an electron transfer route involved in enhancing ferryl heme iron reduction (41, 42). The γ and β subunits appear to lack this electron transfer route and, as reflected by our data, exhibit higher redox potentials. It should be noted that neither human β nor γ subunits (in human Hb) possess an equivalent amino acid residue to αTyr-42. Interestingly, both of these β-like subunits share a common evolutionary ancestry and transcriptional control and thermodynamically exhibit a higher redox potential than α subunits (29).
Our proteomic 18O labeling and crystal structure data also suggest that the Met → Asp conversion involves oxidations catalyzed by the heme group cycling through its ferryl state followed by the net addition of an oxygen atom to the side chain carbon atoms of an oxidized form of the Met(E11). Such an addition is known to occur for sulfoxides, which, in turn, can undergo what is known as Pummerer rearrangement that leads to the elimination of sulfur from the alkyl sulfoxide (43, 44). On this basis, we present a mechanism in Fig. 10 as a working hypothesis consistent with the data presented in this report.
FIGURE 10.

Proposed mechanism of methionine-to-aspartate conversion. The scheme describes major steps involved in the transformation of methionine to aspartate. The first step involves oxidation of the methionine to sulfoxide by hydrogen peroxide via an initial peroxo-complex followed by heterolytic cleavage of the O–O bond generating a ferryl and then a ferric species. The sulfoxide rearranges to a thiol hemiacetal through a Pummerer rearrangement in which an alkyl sulfoxide rearranges to an α-substituted sulfide when treated with acid and then a strong nucleophile, in this case a hydroxide anion. The sulfoxide oxygen atom is converted into a good leaving group by coordinating with the ferric atom acting as strong Lewis acid, which is then followed by reaction with a hydroxide anion or water. The resultant thiol hemiacetal then breaks down into methyl mercaptan and aspartatyl aldehyde. The net result of the rearrangement is reduction of the sulfur atom and oxidation of the α carbon of the amino acid side chain to an aldehyde. The final step involves oxidation of the aldehyde with hydrogen peroxide, presumably by another ferryl species with donation of the oxo oxygen atom, to form aspartate. If the reaction starts with ferric instead of the ferrous heme, there is a tendency for the reaction to proceed to the sulfone, instead of the Pummerer rearrangement as an alternative path leading to limited Asp production. For simplicity of presentation, we use (FeOH)3+ and (FeO)3+ to denote the protonated form of the oxyferryl species (Fe4+=O2−) and the oxyferryl species with associated radical cation (+·HbFe4+=O2−), respectively.
When the reaction commences with the heme in the ferrous state (Fig. 10, step 1), the initial reaction with H2O2 yields an oxoferryl species (HbFe4+=O2−) and is not associated with a cation radical (see Equation 1). This species can be protonated yielding a highly reactive form that can be considered to have some characteristics of its isoelectronic equivalent Fe3+OH•, which may also be written as (FeOH)3+ (45), and is able to add a hydroxyl radical to a nearby side chain. The sulfur atom of methionine, having a lone pair, can react with a hydroxyl radical, lose a proton, and form the sulfoxide observed in our MS data (Fig. 10, step 1 or 2). The heme group is left in the ferric state and can react with H2O2 in further steps.
The subsequent reaction between H2O2 and the ferric heme yields both oxoferryl heme and an associated cation radical (see Equation 2) forming a transient species that is chemically equivalent to compound I of peroxidases and may be written as +·Fe4+=O2− or (FeO)3+. Although this form is extremely short lived in Hbs and Mbs, both the oxidizing equivalents from H2O2 (i.e. the protein or porphyrin radical and the reactive Fe+3OH•) may be transferred to substrates or nearby amino acid side chains. Such transfer occurs in the reduction of ascorbate or in the formation of heme-to-protein crosslinks in these proteins (46). The hydroxyl radical can thus react again with the sulfur atom of the Met(E11) sulfoxide to form the sulfone. Both sulfoxides and sulfones are readily formed when all three Met(E11) Hb variants are reacted with high levels of H2O2 regardless of the initial iron oxidation state.
An acidic environment may cause decomposition of the sulfoxide to form an aldehyde and a methylmercaptan (steps 3–6). This decomposition may occur via a Pummerer rearrangement in which a sulfoxide, when treated with an acid or an anhydride, is converted to an α-substituted sulfide. It proceeds by protonation or esterification of the oxygen atom of the sulfoxide to generate a sulfonium intermediate. Subsequent cleavage of the S–O and C(α)–H bonds results in the release of a proton and formation of a thionium cation. The carbon atom of the sulfenium (thionium) group is then subject to nucleophilic addition in this case by a water or hydroxide anion to generate a thiol hemiacetal. The attacking water or hydroxide could be a product of the labeled H2O2 that was bound to the iron atom or unlabeled from the bulk water. To promote S–OH bond scission with the participation of a single proton in the heme pocket, we propose that the Lewis acid iron of the heme group coordinates with the oxygen atom of the original sulfoxide (Fig. 10, step 2), thus polarizing the S–O bond and facilitating a heterolytic bond cleavage.
Depending on the source of the water nucleophile (i.e. H2O2 or bulk water), which attacks the sulfenium cation, the aldehyde product of step 6 in Fig. 10 will contain either labeled or unlabeled oxygen. There appears to be roughly a 50:50% chance of labeled or unlabeled water or hydroxide attacking the sulfenium ion as we find that a significant amount, ∼50%, of the final aspartate is doubly labeled with 18O. Further oxidation of the labeled aldehyde (Fig. 10, step 7) with the labeled H2O2 gives the insertion of at least one labeled oxygen atom in the final aspartate side chain.
Our MS data (Table 4) suggest that if the whole process starts with ferric heme instead of the ferrous form, there is a greater tendency for the reaction to be driven to the sulfone, instead of the Pummerer rearrangement, which represents a competing side pathway, resulting in very little Asp production. We may account for this by proposing that the positive charge of the radical cation that forms when starting with ferric heme resides, if only transiently, on the sulfoxide residue thus inhibiting coordination of the sulfoxide to the ferric iron, a requirement for entry into the Pummerer rearrangement. H2O2 would not be hindered from approaching the heme, and thus the pathway to sulfone is favored (Fig. 10, step 7).
Additional quantitative studies are required to verify that the Met → Asp conversion of fetal Hb Toms River and adult HbA Bristol-Alesha occurs precisely through this series of reactions. However, the mechanistic steps described in Fig. 10 are consistent with known chemistry and open to experimental test. It should be noted that at each redox step there are competing reactions that have not been made explicit, e.g. migration of radicals away from the heme vicinity that results in the loss of oxidizing equivalents. These additional reactions in turn lead to nonintegral, and indeed condition-dependent, stoichiometries, making interpretation of the product ratios provided in Table 4 difficult, but these problems do not detract from the essential correctness of the scheme.
In addition to the absence of an equivalent amino acid residue αTyr-42, the observed differences in oxidative Met → Asp conversion (between α versus β and γ) may also be due to specific stereochemical features of the distal pocket of each subunit. Indeed, the active sites of β-like subunits in HbA are more accessible to larger ligands and solvent and appear to be more flexible as judged by higher rates of heme dissociation (47, 48). This increased flexibility and solvent accessibility in β subunits could enhance formation of the iron-sulfoxide complex that enhances sulfenium cation formation, facilitates water or hydroxide addition to the sulfenium carbon atom, and accelerates dissociation of the methylmercaptan (Fig. 10, step 3). Regardless of the precise mechanisms responsible for the Met → Asp transition, our data (a) demonstrate a novel post-translational oxidative modification with clinical consequences in Hb variant β and γMet(E11) chains; (b) highlight the marked non-equivalence of α versus β and γ subunits with respect to redox reactivity; (c) re-enforce the conclusion that native Hb β subunits, which bear the burden of oxidative modifications when Hb is challenged with H2O2, must be re-engineered to be more oxidatively stable in Hb-based oxygen therapeutics. From a clinical perspective, our findings show how a PTM can modify a hemoglobinopathy phenotype, in this case by converting a low O2 affinity Hb mutation causing cyanosis (γMet-67) to a destabilizing one associated with hemolytic anemia (γAsp-67).
Similar mutation-associated PTMs could occur in other Hb variants or in other heme-containing proteins, particularly when Met is introduced near a reactive heme iron. Under these circumstances, DNA sequencing, proteomic studies, and biophysical characterizations are required to fully understand the molecular and physical consequences of the mutation.
Acknowledgments
We acknowledge the assistance of Francine Wood in the preparation of HbA. We thank Drs. Paul and Anne Hudrlik of Howard University for the discussion related to the Pummerer arrangement.
This work was supported, in whole or in part, by National Institutes of Health Grant P01-HL110900 from NHLBI (to A. I. A. and J. S. O.). This work was also supported by Welch Grant C-0612 (to J. S. O.) and United States Food and Drug Administration Grants MODSCI 2012 (to A. I. A.), P30 DK090969 (to M. J. W.), and R01 DK61692 (to M. J. W.).
- HbF
- human fetal Hb
- HbA
- adult Hb
- rHb
- recombinant Hb
- PTM
- post-translational modification
- XIC
- extracted ion chromatogram
- PDB
- Protein Data Bank
- metHb
- methemoglobin.
REFERENCES
- 1. Bunn H. F., Forget B. G. (1986) in Hemoglobin: Molecular, Genetic, and Clinical Aspects (Dyson J., ed) W. B. Saunders Co., Philadelphia [Google Scholar]
- 2. Zurbriggen K., Schmugge M., Schmid M., Durka S., Kleinert P., Kuster T., Heizmann C. W., Troxler H. (2005) Analysis of minor hemoglobins by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin. Chem. 51, 989–996 [DOI] [PubMed] [Google Scholar]
- 3. Rees D. C., Rochette J., Schofield C., Green B., Morris M., Parker N. E., Sasaki H., Tanaka A., Ohba Y., Clegg J. B. (1996) A novel silent posttranslational mechanism converts methionine to aspartate in hemoglobin Bristol (β67[E11] Val-Met → Asp). Blood 88, 341–348 [PubMed] [Google Scholar]
- 4. Fucci L., Oliver C. N., Coon M. J., Stadtman E. R. (1983) Inactivation of key metabolic enzymes by mixed-function oxidation reactions: possible implication in protein turnover and ageing. Proc. Natl. Acad. Sci. U.S.A. 80, 1521–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stadtman E. R. (1990) Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences. Free Radic. Biol. Med. 9, 315–325 [DOI] [PubMed] [Google Scholar]
- 6. Levine R. L. (1983) Oxidative modification of glutamine synthetase. II. Characterization of the ascorbate model system. J. Biol. Chem. 258, 11828–11833 [PubMed] [Google Scholar]
- 7. Stadtman E. R. (1992) Protein oxidation and aging. Science 257, 1220–1224 [DOI] [PubMed] [Google Scholar]
- 8. Miyazaki A., Nakanishi T., Kishikawa M., Shimizu A., Ohba Y., Tanaka A., Sasaki H. (1996) Post-translational modification from methionine to aspartic acid residue on a variant hemoglobin, Hb Bristol, a proof by ESI-MS-MS. J. Mass Spectrom. 31, 1311–1313 [DOI] [PubMed] [Google Scholar]
- 9. Steadman J. H., Yates A., Huehns E. R. (1970) Idiopathic heinz body anaemia: Hb-Bristol (β67 (E11) Val→Asp). Br. J. Haematol. 18, 435–446 [DOI] [PubMed] [Google Scholar]
- 10. Sakuragawa M., Ohba Y., Miyaji T., Yamamoto K., Miwa S. (1984) A Japanese boy with hemolytic anemia due to an unstable hemoglobin (Hb Bristol). Nihon Ketsueki Gakkai Zasshi 47, 896–902 [PubMed] [Google Scholar]
- 11. Ohba Y., Matsuoka M., Miyaji T., Shibuya T., Sakuragawa M. (1985) Hemoglobin Bristol or β67(E11) Val→Asp in Japan. Hemoglobin 9, 79–85 [DOI] [PubMed] [Google Scholar]
- 12. Molchanova T. P., Postnikov YuV, Pobedimskaya D. D., Smetanina N. S., Moschan A. A., Kazanetz E. G., Tokarev YuN, Huisman T. H. (1993) Hb Alesha or α2 β(2)67(E11)Val→Met: a new unstable hemoglobin variant identified through sequencing of amplified DNA. Hemoglobin 17, 217–225 [DOI] [PubMed] [Google Scholar]
- 13. Wilson J. B., Webber B. B., Kutlar A., Reese A. L., McKie V. C., Lutcher C. L., Felice A. E., Huisman T. H. (1989) Hb Evans or α(2) 62(E11)Val→Met β(2); an unstable hemoglobin causing a mild hemolytic anemia. Hemoglobin 13, 557–566 [DOI] [PubMed] [Google Scholar]
- 14. Zanotto M. I., Calvo K., Schvartzman G., Deana A., Noguera N. L., Brags I., Milani A. (2010) Hemolytic anemia due to hemoglobin Evans in an Argentinean family. n. Arch. Argentinos de Pediatria 108, 130–133 [DOI] [PubMed] [Google Scholar]
- 15. Crowley M. A., Mollan T. L., Abdulmalik O. Y., Butler A. D., Goodwin E. F., Sarkar A., Stolle C. A., Gow A. J., Olson J. S., Weiss M. J. (2011) A hemoglobin variant associated with neonatal cyanosis and anemia. N. Engl. J. Med. 364, 1837–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thom C. S., Dickson C. F., Gell D. A., Weiss M. J. (2013) Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harbor Perspect. Med. 3, a011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wiedermann B. L., Olson J. S. (1975) Acceleration of tetramer formation by the binding of inositol hexaphosphate to hemoglobin dimers. J. Biol. Chem. 250, 5273–5275 [PubMed] [Google Scholar]
- 18. Aebi H., Packer L. (1984) [13]Catalase in vitro. Methods Enzymol. 105, 121–126 [DOI] [PubMed] [Google Scholar]
- 19. Shen T. J., Ho N. T., Simplaceanu V., Zou M., Green B. N., Tam M. F., Ho C. (1993) Production of unmodified human adult hemoglobin in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 90, 8108–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen T. J., Ho N. T., Zou M., Sun D. P., Cottam P. F., Simplaceanu V., Tam M. F., Bell D. A., Jr., Ho C. (1997) Production of human normal adult and fetal hemoglobins in Escherichia coli. Protein Eng. 10, 1085–1097 [DOI] [PubMed] [Google Scholar]
- 21. Birukou I., Schweers R. L., Olson J.S. (2010) Distal histidine stabilizes bound O2 and acts as a gate for ligand entry in both subunits of adult human hemoglobin. J. Biol. Chem. 285, 8840–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mollan T. L., Banerjee S., Wu G., Parker Siburt C. J., Tsai A.-L., Olson J. S., Weiss M. J., Crumbliss A. L., Alayash A. I. (2013) α-Hemoglobin stabilizing protein (AHSP) markedly decreases the redox potential and reactivity of α subunits of human HbA with hydrogen peroxide. J. Biol. Chem. 288, 4288–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Banerjee R., Alpert Y., Leterrier F., Williams R. J. (1969) Visible absorption and electron spin resonance spectra of the isolated chains of human hemoglobin. Discussion of chain-mediated heme-heme interaction. Biochemistry 8, 2862–2867 [DOI] [PubMed] [Google Scholar]
- 24. Jiang Z.-Y., Woollard A. C., Wolff S. P. (1990) Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 268, 69–71 [DOI] [PubMed] [Google Scholar]
- 25. Berzofsky J. A., Peisach J., Blumberg W. E. (1971) Sulfheme proteins. I. Optical and magnetic properties of sulfmyoglobin and its derivatives. J. Biol. Chem. 246, 3367–3377 [PubMed] [Google Scholar]
- 26. Slotta D. J., McFarland M. A., Markey S. P. (2010) MassSieve: Panning MS/MS peptide data for proteins. Proteomics 10, 3035–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Birukou I., Soman J., Olson J. S. (2011) Blocking the gate to ligand entry in human hemoglobin. J. Biol. Chem. 286, 10515–10529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brantley R. E., Jr., Smerdon S. J., Wilkinson A. J., Singleton E. W., Olson J. S. (1993) The mechanism of autoxidation of myoglobin. J. Biol. Chem. 268, 6995–7010 [PubMed] [Google Scholar]
- 29. Shikama K. (1998) The molecular mechanism of autoxidation for myoglobin and hemoglobin: A venerable puzzle. Chem. Rev. 98, 1357–1374 [DOI] [PubMed] [Google Scholar]
- 30. Stadtman E. R., Levine R. L. (2000) Protein oxidation. Ann. N.Y. Acad. Sci. 899, 191–208 [DOI] [PubMed] [Google Scholar]
- 31. Kowalik-Jankowska T., Rajewska A., Jankowska E., Wiśniewska K., Grzonka Z. (2006) Products of Cu(II)-catalyzed oxidation of the N-terminal fragments of α-synuclein in the presence of hydrogen peroxide. J. Inorg. Biochem. 100, 1623–1631 [DOI] [PubMed] [Google Scholar]
- 32. Berglund G. I., Carlsson G. H., Smith A. T., Szöke H., Henriksen A., Hajdu J. (2002) The catalytic pathway of horseradish peroxidase at high resolution. Nature 417, 463–468 [DOI] [PubMed] [Google Scholar]
- 33. Yi J., Thomas L. M., Richter-Addo G. B. (2011) Structure of human R-state aquomethemoglobin at 2.0 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 647–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hersleth H. P., Dalhus B., Görbitz C. H., Andersson K. K. (2002) An iron hydroxide moiety in the 1.35 Å resolution structure of hydrogen peroxide derived myoglobin compound II at pH 5.2. J. Biol. Inorg. Chem. 7, 299–304 [DOI] [PubMed] [Google Scholar]
- 35. Buehler P. W., Alayash A. I. (2007) Oxidation of hemoglobin: mechanisms of control in vitro and in vivo. Transfusion Alternatives in Transfusion Medicine 9, 204–212 [Google Scholar]
- 36. Butt O. I., Buehler P. W., D'Agnillo F. (2011) Blood-brain barrier disruption and oxidative stress in guinea pig after systemic exposure to modified cell-free hemoglobin. Am. J. Pathol. 178, 1316–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cooper C. E., Schaer D. J., Buehler P. W., Wilson M. T., Reeder B. J., Silkstone G., Svistunenko D. A., Bulow L., Alayash A. I. (2013) Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine β145. Antioxid. Redox Signal. 18, 2264–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonaventura C., Henkens R., Alayash A. I., Banerjee S., Crumbliss A. L. (2013) Molecular controls of the oxygenation and redox reactions of hemoglobin. Antioxid. Redox Signal. 18, 2298–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jia Y., Wood F., Menu P., Faivre B., Caron A., Alayash A. I. (2004) Oxygen binding and oxidation reactions of human hemoglobin conjugated to carboxylate dextran. Biochim. Biophys. Acta 1672, 164–173 [DOI] [PubMed] [Google Scholar]
- 40. Reeder B. J. (2010) The redox activity of hemoglobins: from physiologic functions to pathologic mechanisms. Antioxid. Redox Signal. 13, 1087–1123 [DOI] [PubMed] [Google Scholar]
- 41. Reeder B. J., Grey M., Silaghi-Dumitrescu R.-L., Svistunenko D. A., Bülow L., Cooper C. E., Wilson M. T. (2008) Tyrosine residues as redox cofactors in human hemoglobin. J. Biol. Chem. 283, 30780–30787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reeder B. J., Svistunenko D. A., Cooper C. E., Wilson M. T. (2012) Engineering tyrosine-based electron flow pathways in proteins: the case of aplysia myoglobin. J. Am. Chem. Soc. 134, 7741–7749 [DOI] [PubMed] [Google Scholar]
- 43. Bates D. K., Winters R. T., Sell B. A. (1986) Intramolecular capture of pummerer rearrangement intermediates. 1. Synthesis of pyrrolo[2,1-C][1,4]benzothiazines. J. Heterocyclic Chem. 23, 695–699 [Google Scholar]
- 44. Bates D. K., Winters R. T., Picard J. A. (1992) Intramolecular capture of pummerer rearrangement intermediates. 3. Interrupted pummerer rearrangement-capture of tricoordinate sulfur species generated under pummerer rearrangement conditions. J. Org. Chem. 57, 3094–3097 [Google Scholar]
- 45. Guengerich F. P. (2007) Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J. Biochem. Mol. Toxicol. 21, 163–168 [DOI] [PubMed] [Google Scholar]
- 46. Cooper C. E., Jurd M., Nicholls P., Wankasi M. M., Svistunenko D. A., Wilson M. T. (2005) On the formation, nature, stability and biological relevance of the primary reaction intermediates of myoglobins with hydrogen peroxide. Antioxid. Redox Signal. 21, 3483–3488 [DOI] [PubMed] [Google Scholar]
- 47. Reisberg P. I., Olson J. S. (1980) Rates of isonitrile binding to the isolated α and β subunits of human hemoglobin. J. Biol. Chem. 255, 4151–4158 [PubMed] [Google Scholar]
- 48. Hargrove M. S., Whitaker T., Olson J. S., Vali R. J., Mathews A. J. (1997) Quaternary structure regulates hemin dissociation from human hemoglobin. J. Biol. Chem. 272, 17385–17389 [DOI] [PubMed] [Google Scholar]