

Abstract
CFA/I pili are representatives of a large family of related pili that mediate the adherence of enterotoxigenic Escherichia coli to intestinal epithelial cells. They are assembled via the alternate chaperone-usher pathway and consist of two subunits, CfaB, which makes up the pilus shaft and a single pilus tip-associated subunit, CfaE. The current model of pilus-mediated adherence proposes that CFA/I has two distinct binding activities; the CfaE subunit is responsible for binding to receptors of unknown structure on erythrocyte and intestinal epithelial cell surfaces, while CfaB binds to various glycosphingolipids, including asialo-GM1. In this report, we present two independent lines of evidence that, contrary to the existing model, CfaB does not bind to asialo-GM1 independently of CfaE. Neither purified CfaB subunits nor CfaB assembled into pili bind to asialo-GM1. Instead, we demonstrate that binding activity toward asialo-GM1 resides in CfaE and this is essential for pilus binding to Caco-2 intestinal epithelial cells. We conclude that the binding activities of CFA/I pili for asialo-GM1, erythrocytes, and intestinal cells are inseparable, require the same amino acid residues in CfaE, and therefore depend on the same or very similar binding mechanisms.
INTRODUCTION
Enterotoxigenic Escherichia coli (ETEC) infections are a major public health problem around the world, causing an estimated 200 million cases of diarrhea and 380,000 deaths every year, mainly in developing countries (1, 2). ETEC is also the most common cause of traveler's diarrhea in visitors to areas where it is endemic (3, 4) and is a significant cause of diarrhea in military personnel in the field (5, 6). ETEC strains possess two major types of virulence factor: colonization factors and enterotoxins. Colonization factors mediate the attachment of ETEC to receptors on the mucosal epithelium of the small intestine, thereby facilitating bacterial colonization of the intestine. Enterotoxins elicit the secretion of water and electrolytes from the intestine. Both types of virulence factor have been investigated as potential vaccine targets that aim to either inhibit colonization of the intestine or block the effects of toxin (7).
ETEC strains express a wide variety of colonization factors, most of which are pili (fimbriae) (8). The CFA/I pilus was the first colonization factor to be identified in ETEC (9). CFA/I is a member of a large group of related ETEC pili that belong to the α clade of the fimbrial usher protein (FUP) family (8), which substantially overlaps the class 5 pili defined by an earlier classification scheme (10). This pilus family includes the colonization factors CFA/I, CS1, CS2, CS4, CS14, CS17, CS19, and CS5 (8). Our understanding of the assembly, structure, and function of ETEC pili from the α clade of the FUP family is derived mostly from studies of CS1 and CFA/I pili (10–23). Both CS1 and CFA/I pili are assembled via the alternate chaperone-usher pathway and consist of only two pilus subunits (pilins): a major pilin that makes up the pilus shaft and a single pilus tip-associated pilin. The other components of these pilus systems are a periplasmic pilin chaperone and an usher protein, which are essential for pilus assembly. The pilus tip subunits, CooD in the case of CS1 pili and CfaE in the case of CFA/I pili, are responsible for pilus binding to erythrocytes (17, 18, 20). CfaE also mediates the binding of CFA/I to Caco-2 intestinal cells (10) and to cultured small intestinal biopsy tissue (24).
It is thought that the receptor for CFA/I pili may be a glycolipid. Studies of the binding activities of ETEC pili indicate that CFA/I, and a number of other ETEC pili, bind to the glycosphingolipid asialo-GM1 and various other nonacid glycosphingolipids (25, 26). However, whereas binding to erythrocytes and intestinal epithelial cells is mediated by CfaE, it has been reported that the binding activity for glycosphingolipids resides in the major pilin, CfaB (25). Thus, the current model of CFA/I-mediated adherence proposes that the pilus has two distinct binding activities, one for unidentified receptors on erythrocyte and intestinal epithelial cell surfaces and another for asialo-GM1 and various other glycosphingolipids (25).
The initial aim of this study was to elucidate the specific mechanism by which CfaB binds glycosphingolipids. However, during the course of this study, we were unable to demonstrate the direct binding of purified CfaB pilins to asialo-GM1, which served as a model glycosphingolipid. We were also unable to demonstrate CfaB-dependent binding of whole CFA/I pili to asialo-GM1. Instead, we demonstrated that asialo-GM1 binding is associated with CfaE. Purified CfaE binds to asialo-GM1, while purified CfaB has no detectable binding activity. Consistent with these findings, site-directed mutations in CfaE abolished the binding of whole pili to asialo-GM1 and, in addition, abolished the binding of pili to Caco-2 intestinal cells. These same mutations also abolished CFA/I-mediated attachment of piliated bacteria to Caco-2 cells. We therefore conclude that CfaB is unable to bind to asialo-GM1 independently of CfaE and that asialo-GM1 binding is inseparable from CfaE-mediated binding of pili and piliated bacteria to erythrocytes and intestinal cells.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Cultures were grown aerated in Luria-Bertani (LB) broth or agar (27) or 2YT broth (28) at 37°C with antibiotic selection where appropriate. The antibiotics used were ampicillin (Ap) at 100 μg/ml and chloramphenicol (Cm) at 50 μg/ml. E. coli K-12 strains DH5α (29) and DH5α/F′-lacIq (New England BioLabs) were used as general hosts for cloning. E. coli strain BL21(DE3) Rosetta (Novagen) was used for high-level expression under the control of the T7 promoter. MC4100 is a lac deletion-containing E. coli K-12 strain (30) used for expression of the cfa genes under the control of the lac promoter. The plasmids used in this study are listed in Table 1.
TABLE 1.
Plasmids used in this study
| Plasmid | Backbone vector | Relevant characteristic | Reference/source |
|---|---|---|---|
| pHSG576 | Plac-lacZ′ | 33 | |
| pET22b(+) | PT7, lacI | Novagen | |
| pJGX15W | pJRD184 | Ptet-cfaABCE | 34 |
| pEU2124 | pJRD184 | Ptet-cfaABCE1a | 20 |
| pGU1 | pJRD184 | Ptet-cfaABC | This study |
| pGU2 | pHSG576 | Plac-cfaE | This study |
| pGU3 | pHSG576 | Plac-cfaE1a | This study |
| pGU4 | pHSG576 | Plac-cfaE2b | This study |
| pGU5 | pHSG576 | Plac-cfaE3c | This study |
| pGU6 | pET22b(+) | PT7-dsc19cfaB(His)6 | This study |
| pGU7 | pET22b(+) | PT7-dsc19cfaE(His)6 | This study |
R181A mutation in cfaE.
R67A mutation in cfaE.
R67A R181A double mutation in cfaE.
Construction of a cfaE deletion of the CFA/I pilus operon.
The cfaE gene was deleted from the CFA/I pilus operon by amplifying part of pEU2124, excluding cfaE, by inverse PCR with Phusion High Fidelity DNA polymerase (Thermo Fisher Scientific) and primers HSP277 (5′-ACACCAAGTAGTCAAACACTCTAG-3′) and HSP278 (5′-CTATGTAAAAATAAATAAAATTTTATTCAT-3′). The amplicon was phosphorylated with T4 polynucleotide kinase (New England BioLabs) and ligated with T4 DNA ligase (New England BioLabs). DH5α/F′-lacIq was transformed by the ligation reaction, and presumptive clones were identified by PCR. Deletion of the cfaE gene, including the start and stop codons of cfaE, and the fidelity of the remaining cfa operon sequence were confirmed by DNA sequencing. The cfaE deletion construct was given the plasmid designation pGU1 (Table 1).
Construction of cfaE complementation plasmid.
The cfaE gene was PCR amplified from pJGX15W with primers HSP257 (5′-AAAAAGGATCCAAAGGATAAACGATG-3′) and HSP258 (5′-AAAAAGAATTCCTAGAGTGTTTGACTACTAC-3′) and ligated into the BamHI/EcoRI sites of pHSG576 (Table 1). The construct was confirmed by DNA sequencing of the entire cloned insert and designated pGU2 (Table 1).
Site-directed mutagenesis of cfaE.
To replace arginine residues at positions 67 and 181 in the CfaE amino acid sequence with alanine, site-directed mutations were introduced into pGU2. To introduce an R181A mutation into cfaE, pGU2 was PCR amplified with primers HSP307 (5′-GCACGATATGATACAACCTATG-3′) and HSP308 (5′-TTTTACATTTAGCTTCAGAACG-3′). HSP307 contains a 5′-terminal alanine codon, GCA, that replaced the AGA arginine codon in the wild-type sequence. The amplicon was phosphorylated and ligated as described previously. Ligated samples were transformed into DH5α/F′-lacIq, and putative clones were confirmed by DNA sequencing. The cfaE gene carrying the R181A mutation was designated cfaE1, and the corresponding plasmid was designated pGU3. Similarly, an R67A mutation was generated by PCR amplification of pGU2 with primers HSP281 (5′-GCAATGAGTTTTTTATGTTTGTCTTCTCAA-3′ and HSP282 (5′-ATCATACAGATGATGGCTTCC-3′). HSP281 contains a 5′-terminal alanine codon, GCA, that replaced the AGG arginine codon in the wild-type CfaE sequence. Clones carrying the cfaE2 mutant allele were confirmed by DNA sequencing and designated pGU4. A double mutation (R67A R181A) was generated by PCR amplification of pGU4 with primers HSP307 and HSP308.
Purification of pili.
LB broth cultures of 800 ml were incubated overnight at 37°C. Bacterial cells were pelleted at 8,000 × g for 10 min and washed in 60 ml of phosphate-buffered saline (PBS) before centrifugation again at 8,000 × g for 10 min. Cell pellets were suspended in 60 ml of PBS and subjected to mechanical shearing for 10 s in a food blender. Bacterial cells were pelleted by centrifugation as described above, and protein in the supernatant was precipitated with ammonium sulfate (10% saturation). The precipitate was centrifuged at 8,200 × g for 10 min, suspended in 4 ml of PBS, and dialyzed against 1 liter of PBS for 18 h, with changes of dialysis buffer every 6 h. Dialyzed pilus preparations were checked for purity by SDS-PAGE separation of proteins that were then visualized with Coomassie blue and immunoblotting with anti-CFA/I antiserum (generated as described below) and stored at 4°C.
Cloning, expression, and purification of in cis donor strand-complemented pilins.
In cis donor strand-complemented CfaB and CfaE, with the same amino acid sequence as previously described (17), were constructed by the addition of a C-terminal extension consisting of a 4-amino-acid flexible hairpin loop, the 19-amino-acid N-terminal sequence of mature CfaB, and a hexahistidine tag. The fusion proteins were given the same designations, dsc19CfaB(His)6 and dsc19CfaB(His)6, used by Poole et al. (17). To construct the dsc19CfaB(His)6 fusion protein, cfaB was PCR amplified from pJGX15W with primers HSP245 (5′-AAAAAACATATGAAATTTAAAAAAACTATTGGTGCAA-3′) and HSP246 (5′-AAAAAACTCGAGTTGCAAAAGATCAATTACAGGATCAACACTAGCTGTTACAGTAATATTTTTCTCTACTTGTTTATTATCGGATCCCAAAGTCATTACAAG-3′). HSP245 contains an introduced NdeI restriction site for cloning (bold) and anneals to 28 bases of the cfaB sequence beginning with the start codon (underlined). HSP246 contains an XhoI restriction site introduced for cloning (bold), a sequence encoding a tetrapeptide linker (DNKQ), the first 19 amino acid residues of CfaB, and a sequence that anneals to the 21 codons of cfaB upstream of the stop codon (underlined). The PCR amplicon was digested with NdeI and XhoI and ligated into the corresponding restriction sites of pET22b (+) (Novagen) downstream of the T7 promoter. This cloning resulted in an in-frame fusion of the extended cfaE insert with the hexahistidine coding sequence of the plasmid vector. The clone was confirmed by double restriction digestion and DNA sequencing and given the designation pGU6 (Table 1).
To construct the dsc19CfaE(His)6 fusion protein, the coding sequence for CfaB in pGU6 was replaced with the coding sequence for CfaE while retaining the sequences encoding the tetrapeptide linker and donor strand-complementing N-terminal sequence of CfaB. To achieve this, a section of pGU6 lacking the CfaB coding sequence was amplified with outward-facing PCR primers HSP259 (5′-TTATTTCTAGAGGGGAATTG-3′, containing the vector-borne XbaI site [bold]) and HSP260 (5′-GATAATAAACAAGTAGAGAAAA-3′). The cfaE gene was PCR amplified with primers HSP261 (5′-AAAATCTAGAGAATGATAAAGGATAAACGATG-3′, containing an XbaI site [bold]) and HSP306 (5′-GAGTGTTTGACTACTTGGTGTGAA-3′). Both amplicons were phosphorylated with T4 polynucleotide kinase (New England BioLabs), digested with XbaI, and ligated. Plasmid clones in which the CfaB coding sequence was replaced with the CfaE coding sequence were confirmed by DNA sequencing and given the designation pGU7 (Table 1).
To express and purify the fusion proteins, BL21(DE3) Rosetta/pGU6 and BL21(DE3) Rosetta/pGU7 were grown with aeration in 1 liter of 2YT broth supplemented with 0.4% (vol/vol) glycerol and appropriate antibiotics at 37°C. At an optical density at 600 nm of 0.4, isopropyl-β-d-thiogalactopyranoside was added to a concentration of 1 mM and cultures were incubated at 16°C for 16 h. Cells were harvested by centrifugation and resuspended in lysis buffer (50 mM Tris-HCl [pH 8], 300 mM NaCl, 10% [vol/vol] glycerol, 300 μg/ml lysozyme) supplemented with protease inhibitor cocktail (Roche) as recommended by the manufacturer. The cell suspension was incubated for enzymatic lysis overnight at 4°C before centrifugation at 104,000 × g for 1 h (Beckman Optima L-100XP ultracentrifuge). The presence of fusion protein was monitored in both the soluble and insoluble fractions by SDS-PAGE and immunoblotting with antihexahistidine antibody (Sigma-Aldrich). Proteins in the soluble fractions were precipitated by the addition of ammonium sulfate to 30% saturation and dialyzed three times, for 6 h each time, against 1-liter changes of Ni-nitrilotriacetic acid binding buffer (sodium phosphate buffer [pH 7.5], 300 mM NaCl, 10% [vol/vol] glycerol, 0.02% [vol/vol] Triton X-100, 25 mM imidazole) supplemented with protease inhibitor cocktail (Roche). Protein solutions were loaded onto a HisTrap column (Sigma) at a rate of 0.5 ml/min with a GE-AKTA Start System. Columns were washed with washing buffer (sodium phosphate buffer [pH 7.5], 500 mM NaCl, 50 mM imidazole, 10% [vol/vol] glycerol), and proteins were eluted with an imidazole concentration gradient of 100 to 500 mM in wash buffer. The purity of eluents was confirmed by SDS-PAGE. Purified fractions were pooled and dialyzed against PBS (pH 7).
Transmission electron microscopy.
Whole bacterial cells and purified pili were prepared, negatively stained with phosphotungstic acid, and examined by transmission electron microscopy as previously described (23).
Production of antiserum.
A New Zealand White rabbit was inoculated subcutaneously with 200 μg of CFA/I pili in complete Freund's adjuvant. Repeat inoculations with 200 μg of CFA/I pili in incomplete Freund's adjuvant were performed on days 30 and 54 after the first inoculation. Serum was collected on day 76.
Pilus and pilus subunit binding assays.
Binding of pili and pilus subunits to asialo-GM1 (Sigma-Aldrich) or GM1 (Adipogen Life Sciences) was measured with an enzyme-linked immunosorbent assay (ELISA)-based assay. Microtiter plates (Immulon 1B ELISA plate; Thermo Fisher Scientific) were coated with 100 μl of asialo-GM1 at a 5-μg/ml final concentration. Plates were air dried at 4°C for 12 h, after which they were washed five times with wash buffer (PBS plus 0.5% [wt/vol] bovine serum albumin [BSA]). Wells were blocked with blocking buffer (PBS plus 5% [wt/vol] BSA) overnight at 4°C. Purified pili were subsequently added to asialo-GM1-coated wells and uncoated wells, which served as negative controls. No-pilus control solutions were also added to coated and uncoated wells. Microtiter plates were incubated for 1 h at 37°C and washed five times with wash buffer before the addition of anti-CFA/I antiserum (diluted 1/2,000 in blocking buffer) and incubation for 1 h at room temperature. After five washes to remove unbound antibody, anti-rabbit IgG-alkaline phosphatase conjugate (Santa Cruz) was added at a dilution of 1/5,000 and the mixture was incubated for 1 h at room temperature. After five washes, 100 μl of detection reagent (5 mM diethanolamine, 2 mM NaCl, 2 mM MgCl2, 15 mg of p-nitrophenylphosphate disodium hexahydrate, pH 9.5) was added to each well and the mixture was incubated at 37°C for 45 min. The absorbance of each well was measured at 405 nm on a POLARstar Omega plate reader (BGM Labtech). Absorbance values for no-glycolipid controls were subtracted from corresponding wells containing asialo-GM1. The values for no-pilus control wells were subtracted from the values of test wells.
For assays of pilus binding to Caco-2 cells, cells were grown to 80 to 90% confluence in 96-well tissue culture plates (Falcon) containing RPMI medium supplemented with 20% (vol/vol) fetal bovine serum. All subsequent steps in the Caco-2 binding assay were performed as described for asialo-GM1 binding assays.
Each binding experiment consisted of three technical replicates with the same pilus or protein preparation and three biological replicates with independently derived preparations. Differences between the mean values of biological replicates were analyzed for statistical significance with a one-tailed Student t test.
Assay of bacterial adherence to Caco-2 cells and erythrocytes.
Caco-2 cells were grown to confluence in 24-well tissue culture plates and inoculated with suspensions of MC4100/pGU1/pGU2 (wild-type pili), MC4100/pGU1/pGU5 (mutant pili with R67A and R181A substitutions in CfaE), and MC4100 (pilus negative) at a multiplicity of infection of 100. Bacteria and Caco-2 cells were coincubated in RPMI medium for 2 h at 37°C, washed five times with PBS, and then treated with 0.1% (vol/vol) Triton X-100. The Triton X-100 suspension was serially diluted and plated onto LB Ap Cm agar plates. After overnight incubation at 37°C, the CFU count of each plate was calculated. Assays of technical triplicates of each condition were repeated independently three times, and statistical analysis was performed as described above.
The binding of piliated cells to type A human erythrocytes was determined by the slide test for mannose-resistant hemagglutination as previously described (20).
RESULTS
Expression of CfaE is essential for CFA/I pilus assembly.
To study CfaB-mediated pilus binding to asialo-GM1, we attempted to purify CFA/I pili from an E. coli strain that expresses three of the four genes of the CFA/I operon, cfaA, -B, and -C. This approach was predicated on a prior report that the expression of cfaA, -B, and -C is sufficient for the assembly of pili consisting of the major pilin, CfaB, but devoid of CfaE (25). To construct such a mutant, a defined deletion of the cfaE gene was generated in pEU2124, yielding plasmid pGU1 (Table 1). To develop a quantitative, ELISA-based assay for pilus binding to asialo-GM1, pili were extracted and purified from E. coli strains MC4100/pGU1/pGU2 (cfaABCE) and MC4100/pGU1/pHSG576 (cfaABC). Strain MC4100, which served as a negative control for pilus production, was subjected to the same process as pilus-producing strains. Pili prepared from each strain were examined by SDS-PAGE and immunoblotting with anti-CFA/I antiserum (Fig. 1).
FIG 1.
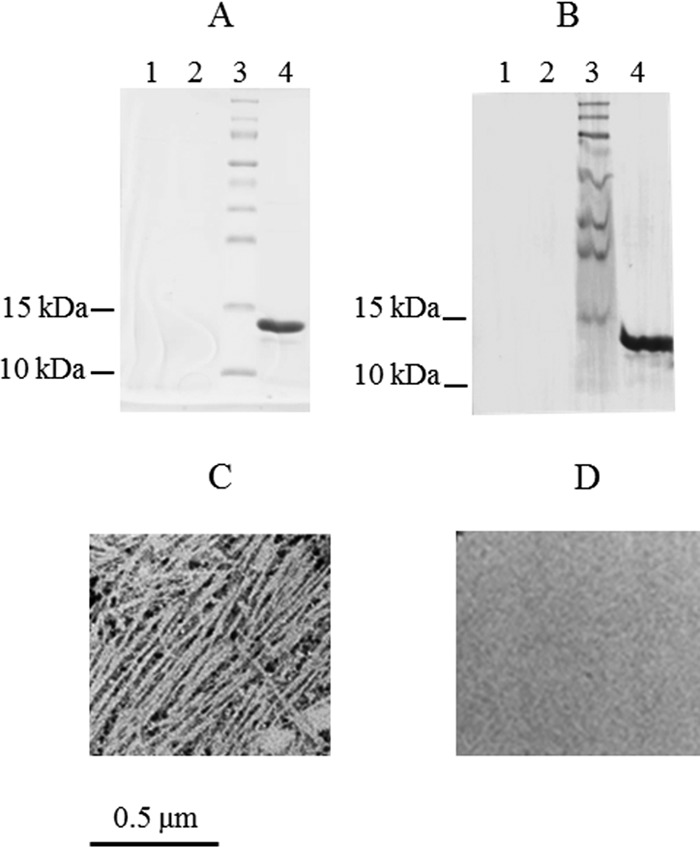
Analysis of pili purified from strains of E. coli expressing cfaABC and cfaABCE. Results of SDS-PAGE (A) and immunoblot (B) analyses of pilus preparations from negative-control strain MC4100 (lane 1), test strain MC4100/pGU1/pHSG576 (cfaABC) (lane 2), and positive-control strain MC4100/pGU1/pGU2 (cfaABCE) (lane 4) are shown. Lane 3 contains molecular size markers. (C, D) Transmission electron microscopy of negatively stained pilus preparations confirmed that although MC4100/pGU1/pGU2 produces CFA/I pili (C), test strain MC4100/pGU1/pHSG576 does not produce pili (D).
As expected, no pili were extracted from the negative-control strain, MC4100 (Fig. 1A and B, lanes 1). A protein of approximately 14 kDa that reacted with anti-CFA/I antiserum (Fig. 1A and B, lanes 4) was consistent with CfaB in the pilus preparation from MC4100/pGU1/GU2 (cfaABCE). Transmission electron microscopy showed the typical morphology expected of CFA/I pili (Fig. 1C). In contrast to MC4100/pGU1/GU2, pili could not be purified from strain MC4100/pGU1/pHSG576, as determined by SDS-PAGE, immunoblotting, and electron microscopy (Fig. 1A and B, lanes 2, and D). The ability of MC4100/pGU1/pHSG576 to produce pili was investigated further by electron microscopy of whole bacteria. As expected, the negative-control strain, MC4100, was nonpiliated (Fig. 2A), while the positive-control strain, MC4100/pGU1/GU2, was piliated (Fig. 2B). However, cells of MC4100/pGU1/pHSG576, which do not express CfaE, were nonpiliated (Fig. 2C). Examination of 500 individual cells by electron microscopy failed to reveal any piliated cells. It was therefore concluded that expression of the minor pilin is essential for the assembly of CFA/I pili.
FIG 2.
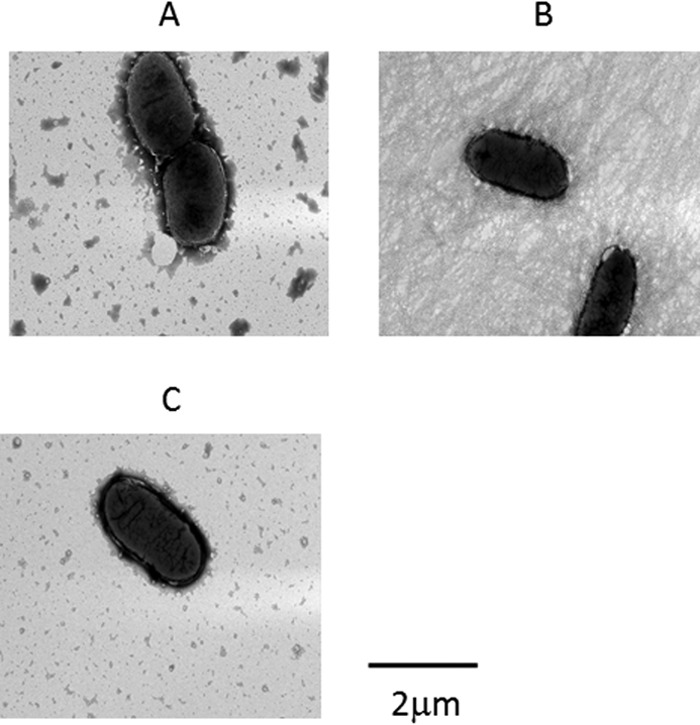
Expression of all four cfa genes is essential for CFA/I pilus production. (A) Negative-control strain MC4100, which lacks the cfa genes, does not express pili on the cell surface. (B) Positive-control strain MC4100/pGU1/pGU2 (cfaABCE) produces CFA/I pili. (C) Test strain MC4100/pGU1/pHSG576 (cfaABC) fails to produce pili.
Purified CfaE subunits, but not CfaB subunits, bind to asialo-GM1.
As pili consisting of only the CfaB pilin could not be generated from strains expressing CfaA, -B, and -C, an alternative approach involving the isolation of soluble CfaB pilins was employed to study the interaction of CfaB with asialo-GM1. Stable, natively folded CfaB protein was purified independently of CfaE by generating a donor strand-complemented, hexahistidine-tagged form of the pilin as previously described (15, 17, 18, 31). Donor strand complementation refers to the noncovalent interaction between the N-terminal β-strand of one subunit in an assembled pilus with a C-terminal acceptor cleft in the adjacent subunit. The intersubunit donation of the N-terminal β-strand of CfaB simultaneously interlocks adjacent pilins and completes the immunoglobulin fold of adjacent pilins, whether they are CfaB or CfaE. When pilins are expressed in the absence of the pilus assembly machinery, they misfold and are subsequently degraded in the periplasm (17). However, in cis, donor strand complementation can be engineered into pilins to produce self-complementing proteins that are stable, soluble, and natively folded. These proteins are variants of the pilins that possess a C-terminal extension containing a flexible tetrapeptide linker, followed by a 19-amino-acid sequence corresponding to the complementing N-terminal β-strand of mature CfaB and a hexahistidine tag to facilitate purification by nickel affinity chromatography. The flexible linker allows the complementing β-strand to associate with the C-terminal cleft of the pilin to complete the Ig fold of the protein.
The donor strand-complemented fusion protein, termed dsc19CfaB(His)6, has an amino acid sequence that is identical to that of a CfaB fusion protein that was used to study donor strand complementation in CFA/I pili (17). Similarly, a donor strand-complemented CfaE fusion protein, dsc19CfaE(His)6, was constructed and purified as a negative control for asialo-GM1 binding experiments. The purified CfaB fusion protein reacted with an antihexahistidine antibody and had an apparent molecular mass of 19 to 20 kDa (Fig. 3A and B), as previously observed (17), which is consistent with its predicted molecular mass of 18.6 kDa. The purified CfaE fusion protein also reacted with the antihexahistidine antibody and had an apparent molecular mass of 41 kDa, consistent with its predicted amino acid sequence (Fig. 3C and D).
FIG 3.
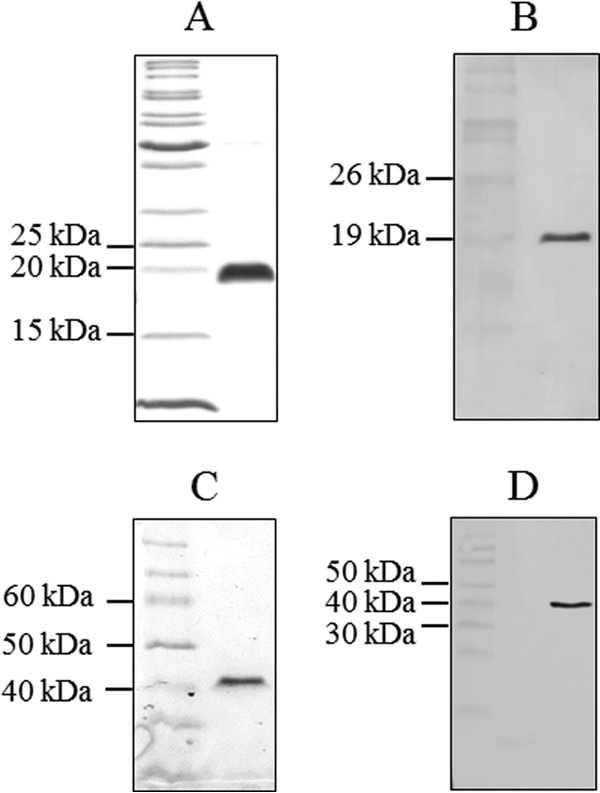
Purification of donor strand-complemented CfaB and CfaE subunits. The identity and purity of donor strand-complemented CfaB (A, B) and CfaE (C, D) were confirmed by SDS-PAGE (A, C) and immunoblotting with an antihexahistidine antibody (B, D). The electrophoretic migrations of CfaB and CfaE fusion proteins were consistent with their predicted molecular sizes of 18.6 and 41 kDa, respectively.
The binding of dsc19CfaB(His)6 to asialo-GM1 was tested in an ELISA-based assay (Fig. 4). The dsc19CfaE(His)6 protein and heat-inactivated preparations of both dsc19CfaB(His)6 and dsc19CfaE(His)6 served as negative controls for asialo-GM1 binding. Surprisingly, the negative-control protein, dsc19CfaE(His)6, bound strongly to asialo-GM1-coated ELISA plates, while the test protein, dsc19CfaB(His)6, bound poorly. The level of dsc19CfaB(His)6 binding was identical to that of heat-inactivated dsc19CfaB(His)6 and dsc19CfaE(His)6, indicating that this low level of binding was nonspecific. The ability of CfaE to bind asialo-GM1 has not been reported previously. However, the inability of dsc19CfaB(His)6 to bind asialo-GM1 is inconsistent with a previous report that pili consisting solely of CfaB bind to asialo-GM1 (25). One potential explanation for these apparently conflicting data is that both CfaE and CfaB bind asialo-GM1 but that dsc19CfaB(His)6 does not fold into an active form with the normal activities of CfaB. To ensure that CfaB retained its native conformation for tests of its ability to bind asialo-GM1, we took the approach of producing pili in E. coli strains expressing CfaA, -B, and -C and variant forms of CfaE that were unable to bind asialo-GM1. Pili consisting of mutant CfaE and wild-type CfaB could then be tested for any residual asialo-GM1 binding activity that might be associated with CfaB.
FIG 4.
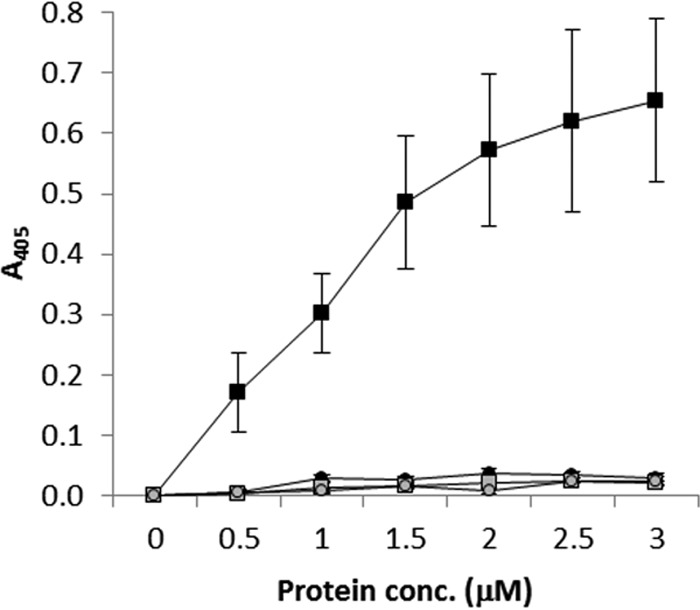
Binding of donor strand-complemented CfaB and CfaE subunits to asialo-GM1. The asialo-GM1 binding of donor strand-complemented subunits and that of negative-control, heat-inactivated subunits were compared in an ELISA-based assay. Purified CfaE subunits (black squares) bound to asialo-GM1, while binding of CfaB (black circles) was no greater than that of negative-control heat-inactivated CfaE and CfaB subunits (gray squares and circles, respectively). Data points are the mean values of three biological repeats, each consisting of three technical repeats, ± the standard error of the mean.
Binding of CFA/I pili to asialo-GM1 is mediated by CfaE.
It appeared likely that amino acid residues within a pocket of CfaE that are required for binding to erythrocyte receptors might also be required for binding to asialo-GM1. It has been demonstrated that alanine substitution of the arginine residues at positions 67 and 181 in CfaE abolish the hemagglutination activity of CfaE-coated polystyrene beads (18). To test whether these residues are also involved in asialo-GM1 binding and whether pili containing a nonbinding variant of CfaE could be generated, site-directed mutations were introduced into the cfaE gene in pGU2 (Table 1). The plasmids generated included pGU3, which encodes CfaE1 (R181A substitution); pGU4, which encodes CfaE2 (R67A substitution); and pGU5, which encodes CfaE3 (R181A and R67A substitutions). E. coli strain MC4100/pGU1 (cfaA, -B, -C) was complemented with pGU2, pGU3, or pGU4 to produce pili containing either wild-type or mutant pili. Pili were extracted from each of these strains, purified, and examined by SDS-PAGE, immunoblotting, and electron microscopy. Pilus extracts from all of the strains contained a 14-kDa protein, consistent with the CfaB pilin (Fig. 5A), that reacted with anti-CFA/I antiserum as expected (Fig. 5B). Pilus extracts examined by electron microscopy showed morphology typical of CFA/I pili.
FIG 5.
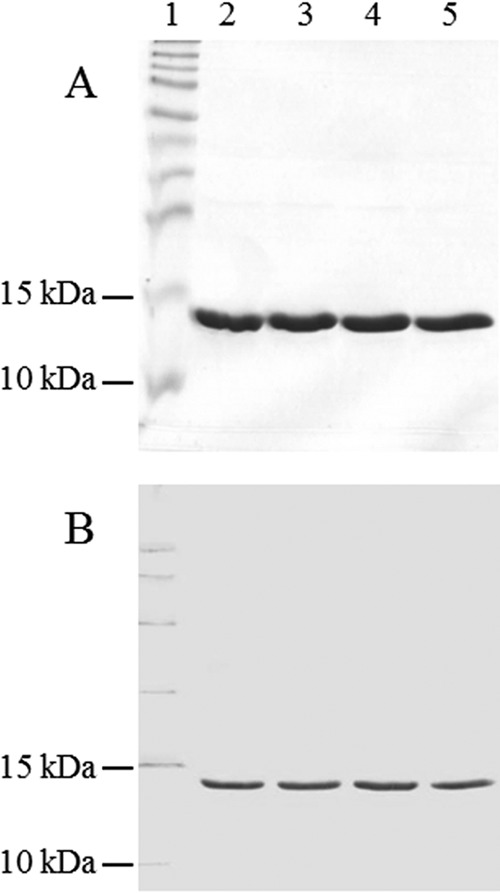
Purification of wild-type and mutant CFA/I pili. Purified pili were analyzed by SDS-PAGE (A) and immunoblotting with anti-CFA/I antiserum (B). Lanes: 1, protein molecular size markers; 2, wild-type pili; 3, pili containing CfaE1 (R181A substitution); 4, pili containing CfaE2 (R67A substitution); 5, pili containing CfaE3 (R67A and R181A substitutions).
Wild-type and mutant pili were then compared for their asialo-GM1 binding activity in an ELISA-based assay (Fig. 6A). Heat-inactivated pili containing wild-type CfaE or CfaE3 (R181A and R67A substitutions) served as negative controls for asialo-GM1 binding activity. In addition, the specificity of pilus binding was tested with the glycosphingolipid GM1, which does not bind to CFA/I (25). Pili containing wild-type CfaE bound to asialo-GM1 (Fig. 6A) but not to GM1 (Fig. 6B), as expected. Binding of CFA/I to asialo-GM1 was significantly reduced in pili containing CfaE1 or CfaE2 (P < 0.05 at all pilus concentrations). These data confirmed that CFA/I binding is specific for the glycosphingolipid asialo-GM1 and that CfaE residues from the same binding pocket interact with both erythrocyte receptors and asialo-GM1. Pili containing CfaE3 showed a greater reduction of asialo-GM1 binding than pili containing CfaE1 or CfaE2. However, the residual binding of pili containing CfaE3 was no greater than that of heat-inactivated wild-type pili (P ≥ 0.37 at all pilus concentrations) or heat-inactivated mutant pili containing CfaE3 (P > 0.31 at 10 to 80 μg/ml; P = 0.10 at 100 μg/ml), indicating that it was nonspecific. Collectively, these data show that CfaE confers CFA/I pilus binding activity toward asialo-GM1 and that this occurs via residues that were previously reported to be required for binding to erythrocyte receptors (18, 20). Since pilus binding to asialo-GM1 was abolished by mutations in CfaE, it was concluded that CfaB does not bind asialo-GM1 independently of CfaE.
FIG 6.
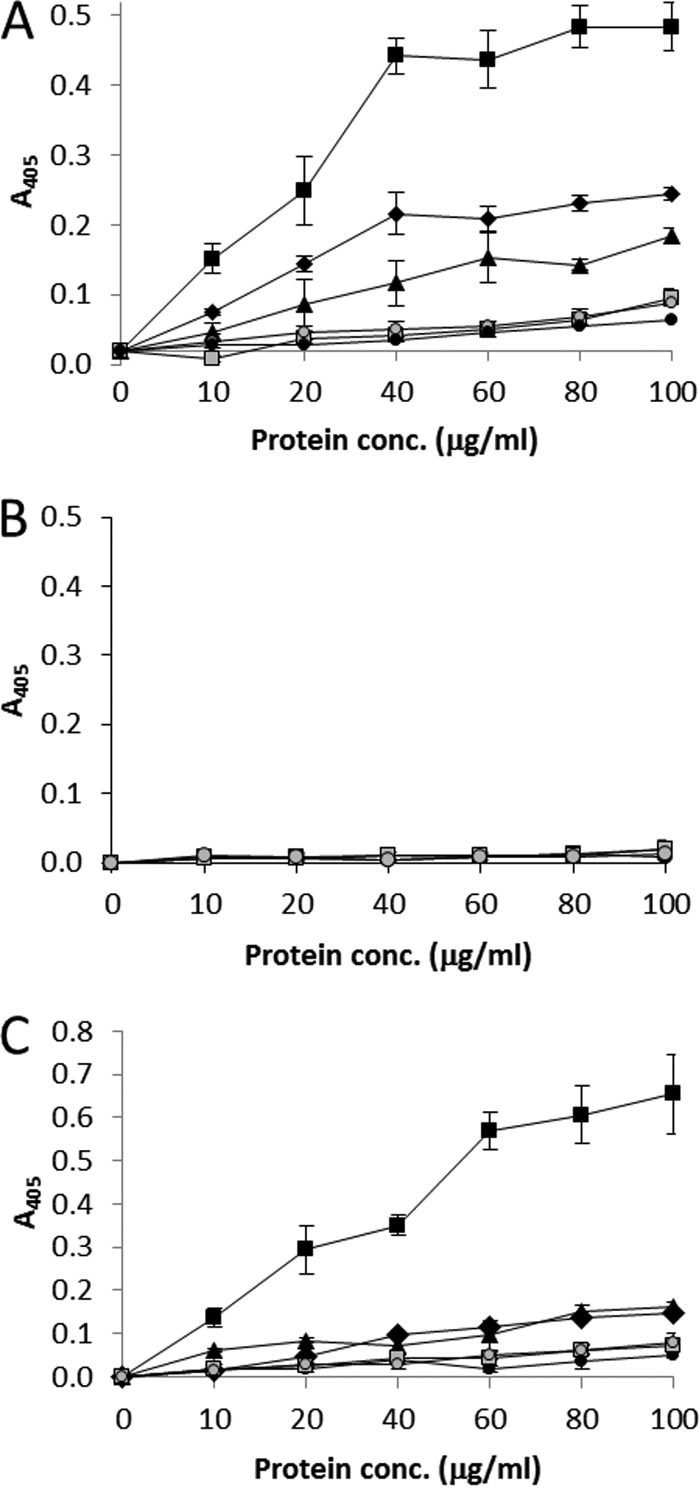
Binding of mutant and wild-type CFA/I pili to glycosphingolipids and Caco-2 intestinal cells. ELISA measurements of pilus binding to asialo-GM1 (A), GM1 (B), and Caco-2 cells (C) are shown. Wild-type CFA/I pili (black squares) bound strongly to asialo-GM1 and Caco-2 cells, while pili containing CfaE1 (R181A substitution, black diamonds) and CfaE2 (R67A, black triangles) showed significantly reduced binding (P < 0.05 at all pilus concentrations). Binding of pili containing CfaE3 (R67A R181A; black circles) was no greater than that of heat-inactivated wild-type pili (gray squares) or heat-inactivated CfaE3-containing pili (gray circles). Active and heat-inactivated wild-type pili and pili containing CfaE3 failed to bind to GM1 (B), which served as a negative control, confirming the specificity of glycosphingolipid binding by CFA/I.
To confirm the significance of R67 and R181 in the binding of CFA/I pili to both asialo-GM1 and erythrocytes, E. coli strains MC4100/pGU1/pGU2 (wild-type CFA/I pili), MC4100/pGU1/pGU3 (CFA/I pili containing CfaE1), MC4100/pGU1/pGU4 (CFA/I pili containing CfaE2), and MC4100/pGU1/pGU5 (CFA/I pili containing CfaE3) were tested for the ability to agglutinate human type A erythrocytes. As expected, E. coli expressing wild-type pili was positive for hemagglutination, while the E. coli strains expressing the mutant pili were negative for hemagglutination (data not shown). Therefore, residues R67 and R181 of CfaE are involved in CFA/I binding to both asialo-GM1 and erythrocyte receptors.
CfaE mutations that abolish asialo-GM1 binding also abolish adherence to Caco-2 cells.
To test the significance of CfaE residues required for asialo-GM1 binding in a model that has greater biological relevance, the binding of wild-type and mutant pili to cultured Caco-2 intestinal cells was determined (Fig. 6B). In comparison to wild-type pili, mutant pili containing CfaE1 or CfaE2 had greatly reduced binding activity (P < 0.05 at all pilus concentrations). CfaE3 mutant pili did not exhibit any greater binding activity than heat-inactivated wild-type or CfaE3 pili at any pilus concentration, indicating that low-level binding to the cell culture was nonspecific. Therefore, the reduced binding of mutant pili to Caco-2 cells was consistent with the phenotypes observed in asialo-GM1 binding assays.
To determine whether the observations of pilus binding to Caco-2 cells also applied to whole piliated bacteria, the adherence abilities of E. coli MC4100/pGU1/pGU2 (wild-type CFA/I pili) and MC4100/pGU1/pGU5 (pili containing CfaE3) and nonpiliated negative-control strain MC4100 were compared (Fig. 7). The adherence of MC4100/pGU1/pGU2 was >9-fold greater than that of both MC4100/pGU1/pGU5 (P < 0.05) and MC4100 (P < 0.05). However, the abilities of MC4100/pGU1/pGU5 and MC4100 to adhere to Caco-2 cells were not significantly different (P = 0.37).
FIG 7.
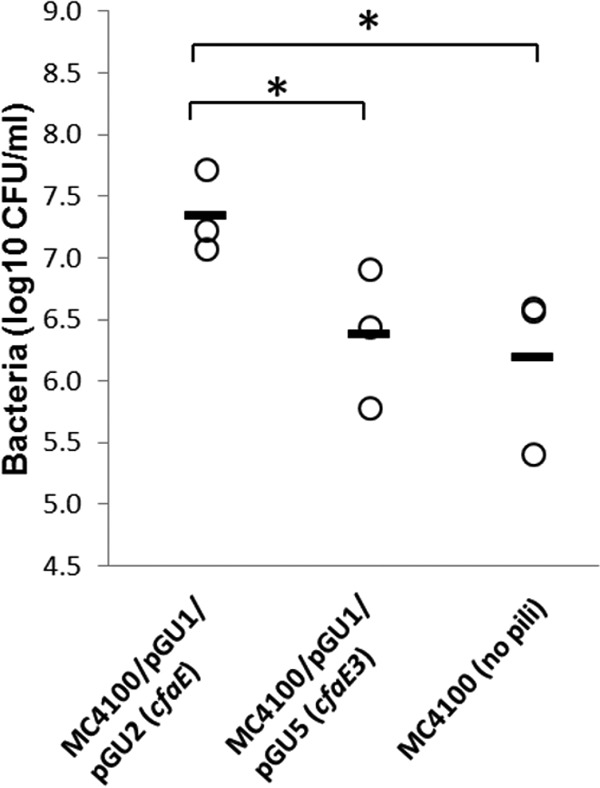
Effect of cfaE3 mutations on the adherence of CFA/I-piliated E. coli to Caco-2 intestinal cells. MC4100 strains expressing CFA/I pili containing CfaE3 (R67A and R181A substitutions) adhere significantly less to Caco-2 cells than do strains expressing pili with the wild-type CfaE subunit. Significant differences in adherence (P < 0.05) are indicated by asterisks.
DISCUSSION
The initial aim of this study was to elucidate the mechanism by which CfaB binds to asialo-GM1, which would serve as a model for other glycosphingolipids that are bound by CFA/I pili. The first approach used to develop a binding assay involved the generation of CFA/I pili consisting of only the major pilin CfaB. This approach involved the expression of three of the four cfa genes, cfaA, -B, and -C, on the basis of a previous report that expression of these genes is sufficient to assemble pili devoid of CfaE (25). However, in this study, although CFA/I pili could be detected in extracts of an E. coli strain expressing all four cfa genes, cfaA, -B, -C, and -E, they could not be detected in extracts of a strain expressing only cfaA, -B, and -C by SDS-PAGE, immunoblotting, or electron microscopy. To examine the possibility that pilus assembly occurs at a very low frequency in the absence of CfaE, 500 independent CfaE− bacteria were examined for piliation. However, none of the cells were piliated. Therefore, it was concluded that the expression of CfaE is an absolute requirement for CFA/I pilus assembly. The reason for the discrepancy between our results and those of Jansson et al. (25) is not clear. However, the results presented here are consistent with our understanding of the assembly of the closely related CS1 pilus, in which the minor tip-associated pilin CooD controls the initiation of, and is essential for, pilus assembly (19, 32). Our findings are also consistent with a prior report that insertional mutation of cfaE abolishes CFA/I assembly in ETEC strain H10407 (24).
As it was not possible to generate pili consisting solely of CfaB, an alternative approach involving the isolation of CfaB pilin for assays of asialo-GM1 binding was taken. This approach took advantage of prior work demonstrating that natively folded CFA/I pilins can be purified by generating in cis donor strand-complemented pilins (17). Binding assays with donor strand-complemented pilins showed that dsc19CfaE(His)6, which was included as a negative control, bound strongly to asialo-GM1, whereas dsc19CfaB(His)6 did not bind at all. Although the asialo-GM1 binding activity of CfaE had not previously been tested, our finding on CfaB contradicts the results reported by Jansson et al. (25). Although dsc19CfaB(His)6 was expected to be natively folded and therefore to retain activity, it was possible that our specific expression and purification procedure resulted in an inactive protein. Therefore, an alternative strategy for testing the binding activity of CfaB was devised.
To ensure the production of natively folded CfaB, we sought to produce CFA/I pili containing a mutant CfaE protein that is unable to bind asialo-GM1. By abolishing the asialo-GM1 binding activity of CfaE, the residual asialo-GM1 binding activity of CfaB could be measured. It seemed possible that amino acid residues in CfaE that are involved in erythrocyte binding may also be required for asialo-GM1 binding. Therefore, alanine substitutions were introduced into CfaE to test the roles of R181 and R67 in asialo-GM1 binding. Substitution of either residue reduced the binding of whole pili to asialo-GM1, while substitution of both residues completely abolished pilus binding. This shows that CfaB does not bind asialo-GM1 independently of CfaE and is consistent with the result of the binding experiments performed with donor strand-complemented CfaB and CfaE. In addition, we found that although CFA/I-piliated E. coli bound to cultured intestinal cells, a piliated strain carrying the R181A and R67A substitutions in CfaE did not show significantly greater adherence than nonpiliated E. coli. Therefore, we conclude that, contrary to the existing model describing the adhesive properties of CFA/I pili, asialo-GM1 binding is not a distinct activity that resides in CfaB independently of CfaE.
How can the discrepancy between the conclusions drawn from this study and a those from a previous study by Jansson et al. be explained? It was concluded that E. coli expressing cfaA, -B, and -C assemble small amounts of CFA/I pili that are devoid of CfaE (25). This result could not be replicated in our study and is inconsistent with a previous study indicating that CfaE is required for pilus assembly (24). One potential explanation of how pili could be recovered from such a strain relies on the possibility that cfaE was indeed expressed in the strain generated by Jansson et al. Although the strain was described as a cfaE mutant, there is no description of how the cfaE mutation was made and there was no independent confirmation of the absence of CfaE in pili by immunoblot analysis of the pili. Regardless of the explanation, in this report, we present two independent lines of evidence that CFA/I binding to asialo-GM1 is not a distinct binding activity that resides in CfaB. Instead, the activities of CFA/I pilus binding to asialo-GM1, erythrocytes, and intestinal cells are inseparable and depend on the same amino acid residues within CfaE.
ACKNOWLEDGMENTS
We acknowledge Saeed Hashimi for valuable discussions and the support provided by the Central Analytical Research Facility at the Queensland University of Technology.
REFERENCES
- 1.Steffen R, Castelli F, Nothdurft HD, Rombo L, Zuckerman JN. 2005. Vaccination against enterotoxigenic Escherichia coli, a cause of travelers' diarrhea. J Travel Med 12:102–107. [DOI] [PubMed] [Google Scholar]
- 2.Wennerås C, Erling V. 2004. Prevalence of enterotoxigenic Escherichia coli-associated diarrhoea and carrier state in the developing world. J Health Popul Nutr 22:370–382. [PubMed] [Google Scholar]
- 3.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 4.Northey G, Evans MR, Sarvotham TS, Thomas DR, Howard TJ. 2007. Sentinel surveillance for travellers' diarrhoea in primary care. BMC Infect Dis 7:126. doi: 10.1186/1471-2334-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nada RA, Armstrong A, Shaheen HI, Nakhla I, Sanders JW, Riddle MS, Young S, Sebeny P. 2013. Phenotypic and genotypic characterization of enterotoxigenic Escherichia coli isolated from U.S. military personnel participating in Operation Bright Star, Egypt, from 2005 to 2009. Diagn Microbiol Infect Dis 76:272–277. doi: 10.1016/j.diagmicrobio.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 6.Monteville MR, Riddle MS, Baht U, Putnam SD, Frenck RW, Brooks K, Moustafa M, Bland J, Sanders JW. 2006. Incidence, etiology, and impact of diarrhea among deployed US military personnel in support of Operation Iraqi Freedom and Operation Enduring Freedom. Am J Trop Med Hyg 75:762–767. [PubMed] [Google Scholar]
- 7.Svennerholm AM, Lundgren A. 2012. Recent progress toward an enterotoxigenic Escherichia coli vaccine. Expert Rev Vaccines 11:495–507. doi: 10.1586/erv.12.12. [DOI] [PubMed] [Google Scholar]
- 8.Madhavan TP, Sakellaris H. 2015. Colonization factors of enterotoxigenic Escherichia coli. Adv Appl Microbiol 90:155–197. doi: 10.1016/bs.aambs.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 9.Evans DG, Evans DJ Jr, Tjoa W. 1977. Hemagglutination of human group A erythrocytes by enterotoxigenic Escherichia coli isolated from adults with diarrhea: correlation with colonization factor. Infect Immun 18:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anantha RP, McVeigh AL, Lee LH, Agnew MK, Cassels FJ, Scott DA, Whittam TS, Savarino SJ. 2004. Evolutionary and functional relationships of colonization factor antigen I and other class 5 adhesive fimbriae of enterotoxigenic Escherichia coli. Infect Immun 72:7190–7201. doi: 10.1128/IAI.72.12.7190-7201.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bao R, Fordyce A, Chen YX, McVeigh A, Savarino SJ, Xia D. 2014. Structure of CfaA suggests a new family of chaperones essential for assembly of class 5 fimbriae. PLoS Pathog 10:e1004316. doi: 10.1371/journal.ppat.1004316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Esser L, Interlandi G, Kisiela DI, Tchesnokova V, Thomas WE, Sokurenko E, Xia D, Savarino SJ. 2013. Tight conformational coupling between the domains of the enterotoxigenic Escherichia coli fimbrial adhesin CfaE regulates binding state transition. J Biol Chem 288:9993–10001. doi: 10.1074/jbc.M112.413534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattopadhyay S, Tchesnokova V, McVeigh A, Kisiela DI, Dori K, Navarro A, Sokurenko EV, Savarino SJ. 2012. Adaptive evolution of class 5 fimbrial genes in enterotoxigenic Escherichia coli and its functional consequences. J Biol Chem 287:6150–6158. doi: 10.1074/jbc.M111.303735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tchesnokova V, McVeigh AL, Kidd B, Yakovenko O, Thomas WE, Sokurenko EV, Savarino SJ. 2010. Shear-enhanced binding of intestinal colonization factor antigen I of enterotoxigenic Escherichia coli. Mol Microbiol 76:489–502. doi: 10.1111/j.1365-2958.2010.07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li YF, Poole S, Nishio K, Jang K, Rasulova F, McVeigh A, Savarino SJ, Xia D, Bullitt E. 2009. Structure of CFA/I fimbriae from enterotoxigenic Escherichia coli. Proc Natl Acad Sci U S A 106:10793–10798. doi: 10.1073/pnas.0812843106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mu XQ, Savarino SJ, Bullitt E. 2008. The three-dimensional structure of CFA/I adhesion pili: traveler's diarrhea bacteria hang on by a spring. J Mol Biol 376:614–620. doi: 10.1016/j.jmb.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole ST, McVeigh AL, Anantha RP, Lee LH, Akay YM, Pontzer EA, Scott DA, Bullitt E, Savarino SJ. 2007. Donor strand complementation governs intersubunit interaction of fimbriae of the alternate chaperone pathway. Mol Microbiol 63:1372–1384. doi: 10.1111/j.1365-2958.2007.05612.x. [DOI] [PubMed] [Google Scholar]
- 18.Li YF, Poole S, Rasulova F, McVeigh AL, Savarino SJ, Xia D. 2007. A receptor-binding site as revealed by the crystal structure of CfaE, the colonization factor antigen I fimbrial adhesin of enterotoxigenic Escherichia coli. J Biol Chem 282:23970–23980. doi: 10.1074/jbc.M700921200. [DOI] [PubMed] [Google Scholar]
- 19.Sakellaris H, Penumalli VR, Scott JR. 1999. The level of expression of the minor pilin subunit, CooD, determines the number of CS1 pili assembled on the cell surface of Escherichia coli. J Bacteriol 181:1694–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakellaris H, Munson GP, Scott JR. 1999. A conserved residue in the tip proteins of CS1 and CFA/I pili of enterotoxigenic Escherichia coli that is essential for adherence. Proc Natl Acad Sci U S A 96:12828–12832. doi: 10.1073/pnas.96.22.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voegele K, Sakellaris H, Scott JR. 1997. CooB plays a chaperone-like role for the proteins involved in formation of CS1 pili of enterotoxigenic Escherichia coli. Proc Natl Acad Sci U S A 94:13257–13261. doi: 10.1073/pnas.94.24.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakellaris H, Balding DP, Scott JR. 1996. Assembly proteins of CS1 pili of enterotoxigenic Escherichia coli. Mol Microbiol 21:529–541. doi: 10.1111/j.1365-2958.1996.tb02562.x. [DOI] [PubMed] [Google Scholar]
- 23.Froehlich BJ, Karakashian A, Sakellaris H, Scott JR. 1995. Genes for CS2 pili of enterotoxigenic Escherichia coli and their interchangeability with those for CS1 pili. Infect Immun 63:4849–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker KK, Levine MM, Morison J, Phillips A, Barry EM. 2009. CfaE tip mutations in enterotoxigenic Escherichia coli CFA/I fimbriae define critical human intestinal binding sites. Cell Microbiol 11:742–754. doi: 10.1111/j.1462-5822.2009.01287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jansson L, Tobias J, Lebens M, Svennerholm AM, Teneberg S. 2006. The major subunit, CfaB, of colonization factor antigen I from enterotoxigenic Escherichia coli is a glycosphingolipid binding protein. Infect Immun 74:3488–3497. doi: 10.1128/IAI.02006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orø HS, Kolsto AB, Wennerås C, Svennerholm AM. 1990. Identification of asialo GM1 as a binding structure for Escherichia coli colonization factor antigens. FEMS Microbiol Lett 60:289–292. [DOI] [PubMed] [Google Scholar]
- 27.Scott JR. 1974. A turbid plaque-forming mutant of phage P1 that cannot lysogenize Escherichia coli. Virology 62:344–349. doi: 10.1016/0042-6822(74)90397-3. [DOI] [PubMed] [Google Scholar]
- 28.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 29.Hanahan D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166:557–580. doi: 10.1016/S0022-2836(83)80284-8. [DOI] [PubMed] [Google Scholar]
- 30.Casadaban MJ. 1976. Regulation of the regulatory gene for the arabinose pathway, araC. J Mol Biol 104:557–566. doi: 10.1016/0022-2836(76)90120-0. [DOI] [PubMed] [Google Scholar]
- 31.Li YF, Poole S, Rasulova F, McVeigh AL, Savarino SJ, Xia D. 2009. Crystallization and preliminary X-ray diffraction analyses of several forms of the CfaB major subunit of enterotoxigenic Escherichia coli CFA/I fimbriae. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:242–247. doi: 10.1107/S1744309109001584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Froehlich BJ, Karakashian A, Melsen LR, Wakefield JC, Scott JR. 1994. CooC and CooD are required for assembly of CS1 pili. Mol Microbiol 12:387–401. doi: 10.1111/j.1365-2958.1994.tb01028.x. [DOI] [PubMed] [Google Scholar]
- 33.Takeshita S, Sato M, Toba M, Masahashi W, Hashimoto-Gotoh T. 1987. High-copy-number and low-copy-number plasmid vectors for lacZ alpha complementation and chloramphenicol- or kanamycin-resistance selection. Gene 61:63–74. doi: 10.1016/0378-1119(87)90365-9. [DOI] [PubMed] [Google Scholar]
- 34.Girón JA, Xu JG, Gonzalez CR, Hone D, Kaper JB, Levine MM. 1995. Simultaneous expression of CFA/I and CS3 colonization factor antigens of enterotoxigenic Escherichia coli by delta aroC, delta aroD Salmonella typhi vaccine strain CVD 908. Vaccine 13:939–946. doi: 10.1016/0264-410X(95)00003-J. [DOI] [PubMed] [Google Scholar]