

Abstract
The carbohydrate recognition domains (CRDs) of lung collectin surfactant protein D (SP-D) recognize sugar patterns on the surface of lung pathogens and promote phagocytosis. Using Haemophilus influenzae Eagan strains expressing well-characterized lipopolysaccharide (LPS) surface structures of various levels of complexity, we show that bacterial recognition and binding by SP-D is inversely related to LPS chain extent and complexity. The crystal structure of a biologically active recombinant trimeric SP-D CRD complexed with a delipidated Eagan 4A LPS suggests that efficient LPS recognition by SP-D requires multiple binding interactions utilizing the three major ligand-binding determinants in the SP-D binding pocket, with Ca-dependent binding of inner-core heptose accompanied by interaction of anhydro-Kdo (4,7-anhydro-3-deoxy-d-manno-oct-2-ulosonic acid) with Arg343 and Asp325. Combined with enzyme-linked immunosorbent assays (ELISAs) and fluorescence-activated cell sorter (FACS) binding analyses, our results show that extended LPS structures previously thought to be targets for collectins are important in shielding the more vulnerable sites in the LPS core, revealing a mechanism by which pathogens with complex LPS extensions efficiently evade a first-line mucosal innate immune defense. The structure also reveals for the first time the dominant form of anhydro-Kdo.
INTRODUCTION
The innate immune proteins surfactant protein D (SP-D) and SP-A are pattern recognition receptors (PRRs) that belong to the superfamily of collagen-containing C-type lectins known as the collectins, which in humans also includes CL-L1, CL-P1, and mannan-binding lectin (1). Collectins can enhance proinflammatory molecules such as cytokines, and reactive oxygen species in phagocytes, by interacting with cell surface receptors or by the scavenging of bacterial molecules (2). As first-line host defense molecules in the lung, SP-D and SP-A target pathogens for neutralization and rapid clearance through interaction with surface carbohydrate structures and promotion of phagocytosis by alveolar macrophages. Bacterial surface lipopolysaccharide (LPS) is a target ligand for immune system defense molecules, and recognition by SP-D of the LPS of various Gram-negative bacterial species, including Escherichia coli (3), Haemophilus influenzae (4, 5), Salmonella enterica serovar Minnesota (3), Bordetella species (6), and Pseudomonas aeruginosa (5), has been previously reported. Many of those studies suggested that lung collectins bind to the LPS core and strongly implicated heptose (Hep) and glucose residues in this binding.
H. influenzae is a common cause of human disease worldwide. Serotype b capsular strains cause invasive bacteremic infections such as meningitis and septicemia (7, 8), while the more prevalent nonencapsulated or nontypeable H. influenzae strains are a common cause of otitis media (9), sinusitis, conjunctivitis, and acute lower respiratory tract infections. Surface-expressed LPS is involved in each stage of the pathogenesis of infections—colonization of the upper respiratory tract, systemic dissemination, and cytotoxic injury to target tissues (10). Host molecules that recognize the LPS of these organisms are important in limiting pathogenicity. In the newborn and in young infants, innate immune components play a key role in frontline defense against infection, while premature infants have reduced collectin levels and are more susceptible to infection (11).
The lipid A region of the LPS of H. influenzae bacteria is linked to a triheptosyl inner core by a single phosphorylated 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) moiety. While the LPS glycoform structures and the genetic blueprint for biosynthesis are known for many H. influenzae strains, the oligosaccharides of the exposed LPS outer core provide substantial heterogeneity within and between strains (12, 13), hindering detailed investigation of the interaction with the pulmonary collectins. The structure of the wild-type (WT) H. influenzae strain RM153 (Eagan) LPS (Fig. 1) indicates glycose extension from each inner-core heptose and a variety of phosphorylated substituents. The rfaF mutant Eagan 4A has a single heptose (HepI), and the orfH deletion mutant CA7 lacks the third heptose (HepIII), with the dominant glycoform having no hexose extension from HepII (14).
FIG 1.
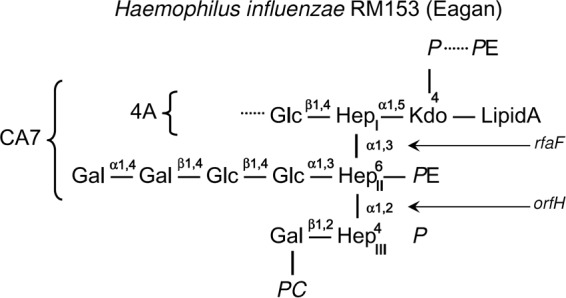
Schematic representation of LPS isolated from wild-type and mutant H. influenzae RM153 (Eagan) strains (adapted from reference 14). The relevant sugar links formed by the orfH and rfaF gene products are shown by arrows, with Eagan CA7 and Eagan 4A referring to the orfH and rfaF mutant strains, respectively. The dominant orfH mutant has no glycoside extension off of HepII. Glc, glucose; Gal, galactose; Hep, heptose; Kdo, 3-deoxy-d-manno-oct-2-ulosonic acid; PC, phosphocholine; P, phosphate; PE, phosphoethanolamine.
Structurally collectins are usually multimers of trimers, with the numbers of trimeric units differing within the collectin family (15). The C-terminal carbohydrate recognition domain (CRD) mediates calcium-dependent binding of microbial cell wall components, with the binding affinity increasing through multimerization (15). The separation of the carbohydrate binding sites favors recognition of the widely spaced repetitive sugars of microbial cell surfaces over host cell components (16). Structural studies of the interaction between human SP-D (hSP-D) and simple ligands have utilized a recombinant homotrimeric fragment (rfhSP-D), comprised of the neck region plus three CRDs. rfhSP-D retains significant biological activity in vitro and in vivo (17, 18), exhibiting therapeutic activity in murine models of pulmonary hypersensitivity and infection induced by an opportunistic fungus, Aspergillus fumigatus (19). Reported structures of ligand-bound complexes are restricted to simple but representative ligands (20–24). Recognition is achieved through CRD Ca-dependent binding of the terminal monosaccharide through a mannose-type equatorial hydroxyl pair or, as in the case of inositol-1-phosphate, galactose and l,d-heptose, a stereochemically equivalent pair.
On the basis of the reported SP-D recognition of the Gram-negative bacterial LPS core, the well-characterized H. influenzae strain Eagan LPS and the truncated inner core mutants, representing the extreme ends of the spectrum of oligosaccharide content, provide a model to establish the structural basis of H. influenzae recognition by the pulmonary collectins in vivo. Study of rfhSP-D, with accessible ligand-binding sites in an extensively characterized crystal form, allows determination of the role of each component of the inner and terminal LPS core in binding by hSP-D. We report here the use of enzyme-linked immunosorbent assays (ELISAs), fluorescence-activated cell sorter (FACS) analysis, and X-ray crystallography to study the interactions of hSP-D and rfhSP-D with wild-type and mutant Eagan bacteria and purified LPS and to demonstrate the functional importance of extended LPS structures in evading host collectin-mediated defenses.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and LPS purification.
The RM153 Eagan type b strain (7) is a disease isolate obtained from the Netherlands (12). Eagan 4A and CA7 are derived mutant strains containing kanamycin resistance cassettes that interrupt the rfaF and orfH genes, respectively, producing truncated LPS glycoforms (14). Before each experiment, bacteria from frozen stocks were grown overnight on chocolate agar plates, which were supplemented with 2 μg/ml kanamycin for the mutants. A single colony from the overnight plate was inoculated into 30 ml brain heart infusion broth (Oxoid) supplemented with 2 μg/ml NAD and 10 μg/ml hemin (Sigma) and with kanamycin (10 μg/ml) for the mutant strains. Bacteria were grown at 37°C with shaking until the desired culture density at 490 nm was reached (optical density at 490 nm [OD490] of 0.6 = 107 CFU/ml). LPS was extracted by the phenol-chloroform/light petroleum method (12) and then was purified by repeated ultracentrifugation (105,000 × g, 4°C, processed twice for 5 h each time). LPS purity was confirmed by SDS-PAGE and by staining with silver (25).
Purification of NhSP-D.
Native human SP-D (NhSP-D) was purified from bronchoalveolar fluid obtained from therapeutic saline solution lung washings of alveolar proteinosis patients from the Royal Brompton Hospital, London, using the method of Strong et al. (26). SP-D was passed through a polymyxin B column (Detoxi-Gel; Pierce, United Kingdom) to remove endotoxin as previously described (18). The final amount of endotoxin present in the SP-D preparations was <0.1 endotoxin U (EU)/μg protein assayed by the use of a QCL-1000 Limulus amoebocyte lysate system (BioWhittaker, Walkersville, MD) according to the manufacturer's instructions.
Generation and purification of a recombinant fragment of human SP-D.
The expression and purification of rfhSP-D, a recombinant trimeric neck/CRD fragment of SP-D, including a short region of the collagen stalk (8 Gly-X-Y) and representing residues 179 to 355, have been described previously (18, 19). The contaminating level of endotoxin present in the rhSP-D preparation was minimized (18) as described for NhSP-D (see above). The assay was linear over a range of 0.1 to 1.0 EU/ml (10 EU = 1 ng of endotoxin).
ELISA.
The three versions of H. influenzae LPS used were from the Eagan wild-type (3 heptoses), Eagan mutant CA7 (2 heptoses), and Eagan mutant 4A (1 heptose) strains. The LPS was resuspended in phosphate-buffered saline (PBS)–0.5 mM EDTA. MaxiSorp 96-well microtiter plates (Nunc) were coated with 1 μg LPS/100 μl coating buffer (0.15 M sodium carbonate, 0.35 M sodium bicarbonate) per well and incubated overnight at 4°C. Plates were washed three times with Tris-buffered saline (TBS; 20 mM Tris, 150 mM NaCl) and blocked with 150 μl/well blocking buffer (TBS with 2% bovine serum albumin [BSA] [wt/vol]) at room temperature for 2 h. The plates were washed three times before serial dilutions of surfactant proteins were made from 1 μg to 10 μg/100 μl/well in blocking buffer with 5 mM CaCl2 or 2 mM EDTA (control). The plates were incubated for 2 h at room temperature and then washed with blocking buffer with 5 mM CaCl2 or 2 mM EDTA. Rabbit anti-rfhSP-D was diluted 1:1,000 in blocking buffer with 5 mM CaCl2 or 2 mM EDTA and incubated for 2 h at 37°C and then washed four times with blocking buffer with 5 mM CaCl2 or 2 mM EDTA. Goat anti-rabbit-Alk phosphatase was diluted 1:10,000 in blocking buffer; 100 μl was added per well and incubated for 2 h at 37°C. Following four washes with blocking buffer with 5 mM CaCl2 or 2 mM EDTA, color was developed with 1 mg/ml pNPP (p-nitrophenyl phosphate) diluted in 25 mM Tris-HCl–5 mM CaCl2–5 mM MgCl2 (pH 7.4) and read using a plate reader. Sugar competition assays were performed by adding maltose at various concentrations to the microtiter plates.
Interactions between collectins and whole bacteria.
Cultures of H. influenzae (30 ml) were grown to 107 CFU/ml as described earlier. The bacteria were pelleted and washed three times with TBS (20 mM Tris, 150 mM NaCl, pH 7.4). The final pellet was resuspended in TBS/C (TBS with 2 mM CaCl2), containing 10 μg/ml of either biotinylated collectins or biotinylated BSA (control), and incubated for 30 min on ice. After pelleting and washing were performed three times, bacteria were incubated with streptavidin-phycoerythrin for 30 min on ice. The samples were washed and fixed in 4% (vol/vol) paraformaldehyde before being analyzed by flow cytometry.
FITC labeling of bacteria.
Fluorescein isothiocyanate (FITC; Sigma) (1 mg) was dissolved in 1 ml of pH 9.6 buffer (1 M NaHCO3 [pH 8.2] adjusted to pH 9.6 with 1 M Na2CO3 [pH 11.6]) to make a stock solution which was diluted 10-fold for labeling the bacteria. Bacteria grown overnight were diluted into fresh media the following day until the desired OD was reached. The bacteria were washed twice in PBS and then resuspended with 1 ml FITC (100 μg/ml) solution and incubated for 1 h at room temperature. Unbound FITC was removed by washing twice with PBS. The pellet of FITC-labeled bacteria was resuspended in storage buffer and stored at 20°C. Viability was verified through plating.
Statistical analysis.
Means and standard deviations were calculated by the use of GraphPad Prism (Version 6) software, and P values for the differences between means were determined by the Student t test (unless otherwise stated) with equal variances. Results were considered significant if the P value was <0.05.
Preparation of Eagan 4A core oligosaccharide for crystallographic study.
Mild acid hydrolysis was carried out on both Eagan wild-type LPS and Eagan 4A LPS based on the procedure described by Masoud et al. (12). The starting LPS volumes were 53 mg for strain 4A and 50 mg for the wild type, giving yields of 18 mg LPS core and ∼80% for strain 4A and of 30 mg LPS core and ∼90% for the wild type.
Crystallization and data collection.
Native crystals of rfhSP-D were grown and cryoprotected as described previously (23, 24). Buffers for cryocooling and ligand soaking were prepared by addition of LPS oligosaccharide to cryobuffers prepared using MPD (2,4-methylpentane diol) in precipitant buffer to provide a final concentration of ligand within the cryobuffer of 30 mM. Data were collected at 100°K on an ADSC Quantum 4R charge-coupled-device (CCD) detector on Daresbury SRS station 14.1 (λ = 1.488 Å). Integrated intensities were calculated with the program MOSFLM (27).
Structure solution and refinement.
Isomorphism was sufficient to allow the coordinates of the 1.6-Å native rfhSP-D structure (PDB code 1PW9 [23]) to be used as a starting model for the rfhSP-D–Eagan 4A oligosaccharide complex. Electron density maps were calculated using Crystallography & NMR System (CNS) software (28) and the CCP4 program suite (29), including density modification (solvent flattening and histogram matching but not NCS averaging). Model building was carried out using maximum likelihood refinement with CNS (28) in alternation with rounds of manual model building with the program O (30). Ligand topology and parameter files were obtained from the HIC-Up server (31). Final refinement details are given in Table 1, and the quality of the final structure was verified by MolProbity (32) and PROCHECK (33). All main and side-chain stereochemical parameters either fall inside the expected limits or are better than those expected (PROCHECK), with 98% of residues in favored regions of the Ramachandran plot and with no outliers. Molecular figures were generated using MOLSCRIPT (34) and the PyMOL Molecular Graphics System Version 1.4 (Schrödinger, LLC, 2011).
TABLE 1.
Data collection and processinga
| Parameter | Result(s) |
|---|---|
| Data collection | |
| Synchrotron station | SRS 14.1 |
| Wavelength (Å) | 1.488 |
| Space group | P21 |
| Cell dimensions | a = 55.35 Å, b = 107.99 Å, c = 55.65Å, α = 90°, β = 92.14°, γ = 90° |
| Maximal resolution (Å) | 1.70 |
| Highest-resolution bin (Å) | 1.79–1.70 |
| No. of observations | 217,593 (31,047) |
| No. of unique reflections | 71,591 (10,454) |
| Completeness (%) | 99.9 (99.9) |
| Rmergeb | 0.075 (0.295) |
| I/σ(I) | 5.1 (2.4) |
| Refinement | |
| No. of protein atomsc | 3,483 |
| No. of residues, chains A, B, and C | 204–355 |
| No. of water molecules | 531 |
| No. of calcium ions | 9 |
| No. of ligand atoms | 44 |
| Rworkd (%) | 19.2 |
| Rfreee (%) | 21.0 |
| RMSD bond length (Å) | 0.007 |
| RMSD bond angle (°) | 1.2 |
| Average B-values (Å2) | |
| Protein main chain | 21.6 |
| Water | 33.3 |
| Other heteroatoms | 29.4 |
| Ramachandran plot valuesf (%) | |
| Favored | 98.0 |
| Outliers | 0.0 |
Numbers in parentheses refer to the highest-resolution bin. RMSD, root mean square deviation.
Rmerge = ΣhΣj |Ih,j − Ih|/ΣhΣj |Ih,j|, where Ih,j is the jth observation of reflection h and Ih is the mean of the j measurements of reflection h.
Excluding alternative side chain conformations.
Rwork = Σh ||Foh| − |Fch||/Σh |Foh|, where Foh and Fch are the observed and calculated structure factor amplitudes, respectively, for reflection h.
Rfree is equivalent to Rwork for a randomly selected subset (5%) of reflections not used in the refinement.
Defined according to Molprobity.
Protein structure accession number.
Coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4E52.
RESULTS
Binding of hSP-D to H. influenzae Eagan LPS.
The interactions of native hSP-D (NhSP-D) and rfhSP-D with LPS from H. influenzae were investigated by ELISA. NhSP-D binding to Eagan 4A was concentration dependent (<1 μg/ml, P < 0.05; >1 μg/ml, P < 0.001) (Fig. 2a) and calcium dependent, with substitution of the 2 mM calcium buffer with 5 mM EDTA buffer resulting in no detectable binding (data not shown). NhSP-D bound more efficiently to LPS from mutant strains of bacteria expressing fewer heptoses in their core region (CA7, P < 0.01; 4A, P < 0.001) than to the LPS from Eagan wild-type bacteria (Fig. 2a). The relative binding affinities of NhSP-D for LPS were 4A (1 heptose) ≫ CA7 (2 heptoses) > WT (3 heptoses). Addition of 100 mM maltose inhibited the binding of NhSP-D to the wild-type LPS completely (Fig. 2a) but inhibited binding to Eagan 4A LPS only partially (∼40% reduction at >5 μg/ml, P < 0.01) (Fig. 2c).
FIG 2.

SP-D interactions with H. influenzae Eagan wild-type and mutant strains of isolated LPS and whole bacteria. Error bars represent standard deviations (SD). Student's t test P values: *, P < 0.05; **, P < 0.01; ***, P < 0.001. (a and b) interactions of (a) native hSP-D and (b) recombinant SP-D with H. influenzae LPS. (c and d) Sugar competition assays with Eagan 4A LPS (c) native hSP-D and (d) recombinant SP-D. (e and f) FACS analysis of binding of whole bacteria by (e) native hSP-D (NhSP-D) and (f) recombinant SP-D.
Similarly, rfhSP-D bound much more strongly to Eagan 4A LPS than to either Eagan CA7 LPS or Eagan wild-type LPS (P < 0.001 [Fig. 2b]). This interaction with Eagan 4A was completely inhibited by EDTA (data not shown) and was rfhSP-D concentration dependent (>1 μg/ml, P < 0.001). Binding of both wild-type LPS and CA7 LPS by rfhSP-D was seen at a concentration of 5 μg/ml (P < 0.01 [compared to 0 μg/ml]). The binding was also shown to be inhibited by maltose in a dose-dependent manner, with serial dilution showing that 25 mM maltose was sufficient for significant inhibition (P < 0.05 [Fig. 2d]) and that 100 mM maltose was sufficient for complete inhibition (Fig. 2b and d).
Binding of hSP-D to whole bacteria.
We used FACS analysis to investigate whether the binding of NhSP-D and rfhSP-D to isolated LPS was reflected in their interactions with different strains of whole bacteria. Consistent with the ELISA findings, NhSP-D bound more efficiently to both Eagan 4A and Eagan CA7 mutant bacteria than to wild-type strain Eagan (Fig. 2e). However, the binding of rfhSP-D to Eagan 4A was markedly more efficient than that to the WT and CA7 strains (Fig. 2f). Binding was inhibited by maltose and abrogated in the presence of EDTA (data not shown).
Crystal structure.
Data collection and processing statistics are given in Table 1. Since data from all rfhSP-D crystals soaked with the wild-type LPS oligosaccharide failed to reveal bound sugar in the ligand-binding site, only results for the 4A ligand are presented here. The crystal structure of rfhSP-D complexed with the Eagan 4A core saccharide isolated from Eagan 4A LPS reveals Ca-dependent binding of the ligand via the Kdo-linked inner-core heptose (HepI) utilizing the O6′ and O7′ hydroxyls of HepI (Fig. 3a). The electron density map reveals bound saccharide in only two of the three CRDs, with ligand absent in chain A.
FIG 3.

Bound Eagan 4A in subunit B of the rfhSP-D–ligand complex. (a) The coordination of the calcium ion Ca1 and HepI. (b) Stereo electron density map for the bound Glc-Hep and the putative anhydro-Kdo (4,7-dehydro-3-deoxy-d-manno-oct-2-ulosonic acid) ligand. The original Kdo numbering has been retained. The portion of the electron density map labeled A corresponds to the putative position of O2′ (original Kdo numbering) in either or both of the enantiomeric substituents of anhydro-Kdo C4. The glucose is not visible in the electron density map.
The electron density map shows no density for the β1–4 glucose attached to HepI. While there is clear density for the α1–5 Hep-Kdo glycosidic bond, the density of the attached sugar suggests a closed 5-membered ring rather than the expected Kdo pyranose ring. The best fit to this density (Fig. 3b) is a modified ribose ring with side groups placed according to the density, agreeing with the proposed enantiomeric furanoid derivative (Fig. 4) resulting from mild acid hydrolysis of O5-substituted Kdo (35). The anhydro-Kdo C4 enantiomeric substitution is not visible in the electron density map, although there is evidence of either or both enantiomeric positions of O2′ (original Kdo numbering) in a position which suggests interaction with Arg343 (Fig. 3b). The modified Kdo is positioned toward Asp325 and Arg343, with hydrogen bonds between Asp325 and one of the hydroxyl groups (Fig. 5). On one side of the binding pocket, residues Asn323 and Asp325 are positioned similarly to those in the mannobiose and inositol phosphate structures; on the opposite side, Arg343 is tilted toward the Hep as seen in the galactose-bound structure where hydrogen bonds between the galactose and Arg343 are formed (24).
FIG 4.

Formation of the 4,7 closure furanoid derivative (anhydro-Kdo) following mild acid hydrolysis and β-elimination of the phosphate from O4 of Kdo. The original Kdo numbering has been retained. (Adapted from reference 35.)
FIG 5.

The Hep-Kdo(anhydro) disaccharide in the ligand-binding site. Kdo O6′ is positioned to interact with Asp325, while the enantiomeric C4 substituent is not identifiable in the electron density except for an indication of O2′ (original Kdo numbering), directed toward Arg343, which may be similarly positioned in the two enantiomers. The original Kdo numbering has been retained.
DISCUSSION
The interactions between lung surfactant protein D and H. influenzae strain Eagan have been investigated in vitro by a variety of techniques using whole bacteria and isolated LPS. In vitro studies of binding, complemented by resolution of the three-dimensional structure of the rfhSP-D complex with Eagan 4A core oligosaccharide, combine to give a consistent and compelling picture of recognition by SP-D being inversely related to LPS complexity. Recognition of the Eagan 4A LPS core by the SP-D CRD is mediated through calcium-dependent binding of HepI enhanced by additional interactions between the HepI-Kdo core and the ligand-binding determinants Asp325 and Arg343 which flank the ligand-binding pocket of the protein.
Both native hSP-D (NhSP-D) and the biologically active (17–19) recombinant trimeric fragment rfhSP-D bind weakly to LPS from WT Eagan and the CA7 mutant strain but bind more avidly to Eagan 4A LPS, which has a more truncated core with fewer heptoses (Fig. 2a and b). In all cases, binding is calcium dependent and completely inhibited by EDTA. While the levels of maltose inhibition of LPS binding appear to differ significantly between NhSP-D and rfhSP-D (Fig. 2a, c, and d), this is not the case, as the effect of 100 mM maltose for NhSP-D (Fig. 2c) is equivalent to that of ∼15 mM maltose for rfhSP-D (Fig. 2d) in terms of the molar excess of maltose over CRD. The binding of both forms of SP-D to whole bacterial cells (Fig. 2e and f) shows the same pattern as that seen with isolated LPS, with efficient binding to Eagan 4A bacteria but less-efficient binding, particularly for rfhSP-D, to the WT and CA7 strains. Binding of NhSP-D, but not rfhSP-D, to Eagan WT and CA7 relative to 4A appears to be markedly more efficient for whole bacteria than for isolated LPS. This may reflect the LPS pattern on the bacterial surface rather than the pattern present under conditions of fixation on ELISA plates, allied to the broader repertoire of pattern recognition of the oligomerized NhSP-D, allowing efficient multiple binding of LPS targets on the bacterial surface.
The results of the NhSP-D and rfhSP-D binding studies performed with both whole bacteria and isolated LPS suggest that the preferred interaction of SP-D with Haemophilus Eagan strains is calcium-mediated CRD binding to the LPS proximal core region rather than to the distal core terminal sugars (3), with the more extended LPS structures of the WT and CA7 bacteria interfering with this recognition. The crystal structure (see below) confirms that hSP-D binds this preferred, inner-core target when it is exposed as in the case of Eagan 4A. Both NhSP-D and rfhSP-D may, however, target the terminal core sugars when the inner core is shielded as in the case in the WT and CA7 strains, although the results of the binding studies suggest that only NhSP-D is able to accomplish the multimeric binding required for significant attachment.
The crystal structure of rfhSP-D complexed with Eagan 4A oligosaccharide reveals Ca-dependent ligand binding via the HepI O6′ and O7′ hydroxyls of the Kdo-linked inner-core heptose HepI, despite the availability of mannose-type O3′ and O4′ pairs (24) on HepI and the HepI terminal Glc. HepI shows a significant rotation away from Asp325 (Fig. 5) compared to the bound Hep in the heptose disaccharide-bound rfhSP-D structure (22). The β1–4 Glc attached to HepI O4 is not visible in the electron density map, presumably due to conformational freedom about the glycosidic linkage and a lack of constraining protein or crystal contacts. There is clear electron density for the α1–5 Hep-Kdo glycosidic bond, but the Kdo electron density does not resemble a pyranose ring. Previous analyses of hydrolyzed H. influenzae LPS consistently indicate uncharacterized anhydro-Kdo forms (12, 25, 36). The best fit to the electron density map (Fig. 3b) is the proposed (35) 5-membered anhydro-Kdo ring arising from β-elimination of phosphate or pyrophosphate from KdoO4, accompanied by pyranose ring opening (35, 37) and subsequent 4,7 ring closure to form a furanoid derivative with a chiral center at C4 carrying an extended substitution (Fig. 4). Density for the C4 substituents is absent except in a position (Fig. 3b) which may correspond to either or both of the enantiomeric positions of O2′ (original Kdo numbering; Fig. 4). This demonstrates for the first time the putative structure of the dominant anhydro-Kdo formed by the hydrolysis procedures used in the characterization of a variety of bacterial LPSs.
Asp325 is hydrogen bonded to the anhydro-Kdo C6 OH group, which would be similarly positioned in a normal Kdo oriented similarly with respect to HepI (Fig. 5). Crucially, C4 in the anhydro-Kdo is positioned such that normal Kdo would present the C4 phosphate to Arg343. Alternatively, rotation of the normal Kdo about the glycosidic α1–5 HepI-Kdo bond would direct the Kdo C7 and C8 OH groups toward Asp325 and the acidic group off of Kdo C2 toward Arg343. This position of the normal Kdo acidic group aligns with the isolated density seen here off of the anhydro-Kdo C4 (Fig. 3b). Thus, recognition of the Eagan 4A inner core involves not only the primary calcium ion but also both binding site flanking residues (20–24). This concerted binding suggests high affinity for the HepI-Kdo disaccharide, consistent with the binding studies where Eagan 4A competes strongly with the high-affinity SP-D ligand maltose. This optimal mode of SP-D binding via the inner core is consistent with studies of Bordetella mutants (6) and S. Minnesota strains with incomplete cores (3), while mannose-binding protein binds to Hep and GlcNAc in the rough core oligosaccharide of the E. coli K-12 cell wall and to Hep in the incomplete core of E. coli B (38).
The reduced affinity of SP-D for Eagan CA7 (both bacterial LPS and purified LPS) compared to that for Eagan 4A is at first sight puzzling since the dominant CA7 glycoform lacks any hexose extension off of HepII (14). The additional substituted sugar off of HepI (HepII at O3′) for CA7, in addition to Kdo and Glc at O4′, introduces steric constraints which may affect the ability of HepI, and hence the ability of Kdo, to achieve the preferred high-affinity, multiple protein and LPS interactions demonstrated by the structure presented here. The weak affinity of rfhSP-D for the Eagan CA7 and WT LPS may then arise from alternative recognition such as of the terminal glucose or the O3′ and O4′ pair on HepII.
A picture thus emerges of enhanced bacterial resistance to SP-D through expression of extended LPS glycoforms, with the crystal structure revealing that optimal binding is achieved through multiple interactions of the inner-core HepI-Kdo pair with all three major ligand-binding determinants in the SP-D binding pocket. If this optimal binding is precluded by the spatial and conformational constraints of additional sugars, recognition may still be achieved through other hydroxyl pairs, although there is a clear inverse relationship between binding and LPS complexity.
The enhanced binding of the mutant with exposed core targets for the SP-D CRD demonstrates the efficiency of recognition in the absence of more-complex LPS structures, with considerable selective pressure on the bacterium to develop such structures to shield these vulnerable sites and to subvert SP-D-mediated host defenses, as is the case for the more resistant WT Eagan strains. Strategies designed to disrupt LPS synthesis in Haemophilus and other Gram-negative organisms could thus be effective in rendering bacteria susceptible to host innate immune defenses mediated by collectins. Whether rfhSP-D can act as an opsonin for strains with truncated LPS cores is not clear from the results presented here, but the potential of high doses of the recombinant fragment to enhance bacterial clearance is worthy of further investigation, since Eagan 4A bacteria and isolated LPS are recognized and bound by SP-D even in the absence of the collagen tail and N-terminal region, and the multimeric binding potential, of the native oligomer.
The results of the current studies demonstrate a potential general mechanism by which Gram-negative bacteria have evolved extended LPS structures to escape innate immune surveillance by collectins. Increasing LPS complexity may have provided selective pressure for the recombination of host genes encoding nonfibrillary collagens and CRDs (39), enabling multimerization and increased avidity for complex LPS structures on the surface of pathogens. This may represent a model demonstrating how microbial and host genes have coevolved.
ACKNOWLEDGMENTS
Help from the beamline scientists at the Daresbury SRS and the Diamond Light Source is gratefully acknowledged.
This work was supported by the Medical Research Council (A.K.S., T.J.G., K.B.M.R., H.W.C., D.W.H., M.E.D.), CLRC Daresbury Laboratory (T.J.G., A.K.S.), STFC (T.J.G., A.K.S.), Diamond Light Source (T.J.G., A.K.S.), the Sir Halley Stewart Trust (H.W.C.), and the Beit Memorial Fellowships (H.W.C.).
REFERENCES
- 1.Holmskov UL. 2000. Collectins and collectin receptors in innate immunity. APMIS Suppl 100:1–59. [PubMed] [Google Scholar]
- 2.Sano H, Kuroki Y. 2005. The lung collectins, SP-A and SP-D, modulate pulmonary innate immunity. Mol Immunol 42:279–287. doi: 10.1016/j.molimm.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 3.Kuan SF, Rust K, Crouch E. 1992. Interactions of surfactant protein-D with bacterial lipopolysaccharides—surfactant protein-D is an Escherichia coli-binding protein in bronchoalveolar lavage. J Clin Invest 90:97–106. doi: 10.1172/JCI115861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, Korfhagen TR. 2000. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol 165:3934–3940. doi: 10.4049/jimmunol.165.7.3934. [DOI] [PubMed] [Google Scholar]
- 5.Restrepo CI, Dong Q, Savov J, Mariencheck WI, Wright JR. 1999. Surfactant protein D stimulates phagocytosis of Pseudomonas aeruginosa by alveolar macrophages. Am J Respir Cell Mol Biol 21:576–585. doi: 10.1165/ajrcmb.21.5.3334. [DOI] [PubMed] [Google Scholar]
- 6.Schaeffer LM, McCormack FX, Wu H, Weiss AA. 2004. Interactions of pulmonary collectins with Bordetella bronchiseptica and Bordetella pertussis lipopolysaccharides elucidate the structural basis of their antimicrobial activities. Infect Immun 72:7124–7130. doi: 10.1128/IAI.72.12.7124-7130.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moxon RE, Vaughn KA. 1981. The type b capsular polysaccharide as a virulence determinant of Haemophilus influenzae: studies using clinical isolates and laboratory transformants. J Infect Dis 143:517–524. doi: 10.1093/infdis/143.4.517. [DOI] [PubMed] [Google Scholar]
- 8.Moxon ER, Maskell D. 1992. Haemophilus influenzae lipopolysaccharide: the biochemistry and biology of a virulence factor, p 75–96. In Harmaeche CE, Penn CW, Smyth CJ (ed), Molecular biology of bacterial infection: current status and future perspectives. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 9.Murphy TF, Apicella MA. 1987. Nontypable Haemophilus influenzae: a review of clinical aspects, surface antigens, and the human immune response to infection. Rev Infect Dis 9:1–15. doi: 10.1093/clinids/9.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Moxon ER. 1985. The molecular basis of Haemophilus influenzae virulence. J R Coll Physicians Lond 19:174–178. [PMC free article] [PubMed] [Google Scholar]
- 11.Malhotra R, Willis AC, Lopez Bernal A, Thiel S, Sim RB. 1994. Mannan-binding protein levels in human amniotic fluid during gestation and its interaction with collectin receptor from amnion cells. Immunology 82:439–444. [PMC free article] [PubMed] [Google Scholar]
- 12.Masoud H, Moxon ER, Martin A, Krajcarski D, Richards JC. 1997. Structure of the variable and conserved lipopolysaccharide oligosaccharide epitopes expressed by Haemophilus influenzae serotype b strain Eagan. Biochemistry 36:2091–2103. doi: 10.1021/bi961989y. [DOI] [PubMed] [Google Scholar]
- 13.Hood DW, Richards JC, Moxon ER. 1999. Haemophilus influenzae lipopolysaccharide. Biochem Soc Trans 27:493–498. doi: 10.1042/bst0270493. [DOI] [PubMed] [Google Scholar]
- 14.Hood DW, Deadman ME, Allen T, Masoud H, Martin A, Brisson JR, Fleischmann R, Venter JC, Richards JC, Moxon ER. 1996. Use of the complete genome sequence information of Haemophilus influenzae strain Rd to investigate lipopolysaccharide biosynthesis. Mol Microbiol 22:951–965. doi: 10.1046/j.1365-2958.1996.01545.x. [DOI] [PubMed] [Google Scholar]
- 15.Crouch EC. 1998. Structure, biologic properties, and expression of surfactant protein D (SP-D). Biochim Biophys Acta 1408:278–289. doi: 10.1016/S0925-4439(98)00073-8. [DOI] [PubMed] [Google Scholar]
- 16.Weiss WI, Drickamer K. 1996. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem 65:441–473. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 17.Clark HW, Reid KBM. 2002. Structural requirements for SP-D function in vitro and in vivo: therapeutic potential of recombinant SP-D. Immunobiology 205:619–631. doi: 10.1078/0171-2985-00159. [DOI] [PubMed] [Google Scholar]
- 18.Singh M, Madan T, Waters P, Parida SK, Sarma PU, Kishore U. 2003. Protective effects of a recombinant fragment of human surfactant protein D in a murine model of pulmonary hypersensitivity induced by dust mite allergens. Immunol Lett 86:299–307. doi: 10.1016/S0165-2478(03)00033-6. [DOI] [PubMed] [Google Scholar]
- 19.Madan T, Kishore U, Singh M, Strong P, Clark HW, Hussain EM, Reid KBM, Sarma PU. 2001. Surfactant proteins A and D protect mice against pulmonary hypersensitivity induced by Aspergillus fumigatus antigens and allergens. J Clin Invest 107:467–475. doi: 10.1172/JCI10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crouch E, Hartshorn K, Horlacher T, McDonald B, Smith K, Carafella T, Seaton B, Seeberger PH, Head J. 2009. Recognition of mannosylated ligands and influenza A virus by human surfactant protein D: contributions of an extended site and residue 343. Biochemistry 48:3335–3345. doi: 10.1021/bi8022703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crouch E, McDonald B, Smith K, Roberts M, Mealy T, Seaton B, Head J. 2007. Critical role of Arg/Lys343 in the species-dependent recognition of phosphatidylinositol by pulmonary surfactant protein D. Biochemistry 46:5160–5169. doi: 10.1021/bi700037x. [DOI] [PubMed] [Google Scholar]
- 22.Wang H, Head J, Kosma P, Brade H, Muller-Loennies S, Sheikh S, McDonald B, Smith K, Cafarella T, Seaton B, Crouch E. 2008. Recognition of heptoses and the inner core of bacterial lipopolysaccharides by surfactant protein D. Biochemistry 47:710–720. doi: 10.1021/bi7020553. [DOI] [PubMed] [Google Scholar]
- 23.Shrive AK, Tharia HA, Strong P, Kishore U, Burns I, Rizkallah PJ, Reid KBM, Greenhough TJ. 2003. High resolution structural insights into ligand binding and immune cell recognition by human lung surfactant protein D. J Mol Biol 331:509–523. doi: 10.1016/S0022-2836(03)00761-7. [DOI] [PubMed] [Google Scholar]
- 24.Shrive AK, Martin C, Burns I, Paterson JM, Martin JD, Townsend JP, Waters P, Clark HW, Kishore U, Reid KBM, Greenhough TJ. 2009. Structural characterisation of ligand-binding determinants in human lung surfactant protein D: influence of Asp325. J Mol Biol 394:776–788. doi: 10.1016/j.jmb.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deadman ME, Hermant P, Engskog M, Makepeace K, Moxon ER, Schweda EKH, Hood DW. 2009. Lex2B, a phase-variable glycosyltransferase, adds either a glucose or a galactose to Haemophilus influenzae lipopolysaccharide. Infect Immun 77:2376–2384. doi: 10.1128/IAI.01446-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strong P, Kishore U, Morgan C, Lopez Bernal A, Singh M, Reid KBM. 1998. A novel method of purifying lung surfactant proteins A and D from the lung lavage of alveolar proteinosis patients and from pooled amniotic fluid. J Immunol Methods 220:139–149. doi: 10.1016/S0022-1759(98)00160-4. [DOI] [PubMed] [Google Scholar]
- 27.Leslie AGW. 1992. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EACMB newsletter on protein crystallography, issue 26. Daresbury Laboratory, Warrington, United Kingdom. [Google Scholar]
- 28.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54:905–921. doi: 10.1107/S0907444998003254. [DOI] [PubMed] [Google Scholar]
- 29.Collaborative Computational Project Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 30.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47:110–119. doi: 10.1107/S0108767390010224. [DOI] [PubMed] [Google Scholar]
- 31.Kleywegt GJ, Henrick K, Dodson EJ, van Aalten DMF. 2003. Pound-wise but penny-foolish—how well do micromolecules fare in macromolecular refinement? Structure 11:1051–1059. doi: 10.1016/S0969-2126(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB III, Headd JB, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laskowski R, MacArthur MW, Moss DS, Thornton JM. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:283–291. doi: 10.1107/S0021889892009944. [DOI] [Google Scholar]
- 34.Kraulis PJ. 1991. MOLSCRIPT: a program package to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24:946–950. doi: 10.1107/S0021889891004399. [DOI] [Google Scholar]
- 35.Auzanneau F-I, Charon D, Szabó L. 1991. Phosphorylated sugars. Part 27. Synthesis and reactions, in acid medium, of 5-O-substituted methyl 3-deoxy-α-d-manno-oct-2-ulopyranosidonic acid 4-phosphates. J Chem Soc Perkin Trans 1 3:509–517. doi: 10.1039/P19910000509. [DOI] [Google Scholar]
- 36.Yildirim HH, Hood DW, Moxon ER, Schweda EKH. 2003. Structural analysis of lipopolysaccharides from Haemophilus influenzae serotype f: structural diversity observed in three strains. Eur J Biochem 270:3153–3167. doi: 10.1046/j.1432-1033.2003.03693.x. [DOI] [PubMed] [Google Scholar]
- 37.Danan A, Mondange M, Sarfati SR, Szabo P. 1982. Synthesis and behaviour under acidic conditions of 2-deoxy-d-arabino-hexopyranose and 3-deoxy-2-ketoaldonic acids bearing O-phosphono or O-glucosyl substituents at position β to the carbonyl function. J Chem Soc Perkin Trans 1 3:1275–1282. doi: 10.1039/P19820001275. [DOI] [Google Scholar]
- 38.Kawasaki N, Kawasaki T, Yamashina I. 1989. A serum lectin (mannan-binding protein) has complement-dependent bactericidal activity. J Biochem 106:483–489. [DOI] [PubMed] [Google Scholar]
- 39.Bezouska K, Crichlow GV, Rose JM, Taylor ME, Drickamer K. 1991. Evolutionary conservation of intron position in a subfamily of genes encoding carbohydrate-recognition domains. J Biol Chem 266:11604–11609. [PubMed] [Google Scholar]