

Abstract
Neisseria meningitidis autoaggregation is an important step during attachment to human cells. Aggregation is mediated by type IV pili and can be modulated by accessory pilus proteins, such as PilX, and posttranslational modifications of the major pilus subunit PilE. The mechanisms underlying the regulation of aggregation remain poorly characterized. Polynucleotide phosphorylase (PNPase) is a 3′–5′ exonuclease that is involved in RNA turnover and the regulation of small RNAs. In this study, we biochemically confirm that NMC0710 is the N. meningitidis PNPase, and we characterize its role in N. meningitidis pathogenesis. We show that deletion of the gene encoding PNPase leads to hyperaggregation and increased adhesion to epithelial cells. The aggregation induced was found to be dependent on pili and to be mediated by excessive pilus bundling. PNPase expression was induced following bacterial attachment to human cells. Deletion of PNPase led to global transcriptional changes and the differential regulation of 469 genes. We also demonstrate that PNPase is required for full virulence in an in vivo model of N. meningitidis infection. The present study shows that PNPase negatively affects aggregation, adhesion, and virulence in N. meningitidis.
INTRODUCTION
Neisseria meningitidis is a Gram-negative, encapsulated diplococcus with the potential to cause life-threatening epidemic disease. The human nasopharynx, where the bacterium typically resides during asymptomatic periods, is the only natural reservoir of N. meningitidis. However, the bacteria occasionally invade the mucosa, gain access to the bloodstream, and cross the blood-brain barrier to reach the cerebrospinal fluid. This pathological process clinically presents as severe sepsis and/or meningitis (1). To facilitate its colonization of the nasopharynx and its survival in the circulation, N. meningitidis has evolved intricate mechanisms for evading innate immune defenses (2). Specifically, the bacteria express or modify multiple surface structures, such as the polysaccharide capsule, lipopolysaccharides (LPSs), Neisseria hia homolog A (NhhA), and proteins that bind to human complement regulators to protect themselves from complement attack complexes (3–9).
A key virulence feature of N. meningitidis is type IV pili, which are thin membrane-spanning filaments that are involved in adhesion to human cells, bacterial aggregation, twitching motility, and competence (10–13). Bacterial aggregation is driven primarily by type IV pilus-mediated contact between bacteria, but these interactions can be modified by multiple factors, including the minor pilin PilX and posttranslational modifications of the major pilus subunit PilE (14–16). The formation of organized aggregates greatly increases initial adhesion to cells and protects the bacteria from shear stress (17). After the initial adhesion, the aggregates disperse, allowing a more-intimate adhesion that is characterized by the downregulation of pili and the capsule (18). The regulation of aggregation and the regulation of dispersion remain poorly characterized processes.
Polynucleotide phosphorylase (PNPase) is a 3′–5′ exonuclease that is involved in RNA degradation (19). It is expressed in both prokaryotic and eukaryotic cells, with the exception of fungi (20). In proteobacteria, PNPase is part of the RNA degradosome, as are RNase E, enolase, and an RNA helicase (21). In Escherichia coli, RNA degradation is initiated by endonucleic cleavage by RNase E. This exposes a free 3′ RNA end, at which PNPase and other 3′ RNases proceed to degrade the RNA (22). In addition to degradation of RNAs such as mRNA, PNPase has also been shown to be important for the stability of small RNAs (sRNA) (23). Because of its role in the regulation of mRNA and sRNA half-lives, PNPase is important for the regulation of many genes. PNPase is required for bacterial adaptation to cold shock (e.g., in E. coli, Salmonella enterica subsp. enterica serovar Typhimurium, Yersinia enterocolitica, and Campylobacter jejuni) (24–27). Importantly, PNPase is also involved in the regulation of virulence-associated gene expression. In S. Typhimurium, PNPase suppresses the expression of genes encoded by the Salmonella pathogenicity islands and by the SpvR virulence plasmid (27, 28). In Yersinia spp., PNPase positively regulates the activity of the type III secretion system and thereby enhances both resistance to macrophage-mediated killing and virulence in a mouse model (29, 30). In addition, PNPase negatively regulates bacterial biofilm formation in E. coli and S. Typhimurium, twitching motility and virulence in Dichelobacter nodosus, and swimming motility in C. jejuni (31–34). In N. meningitidis, the PNPase homolog has been shown to be upregulated during long-term colonization of human epithelial cells in vitro (35).
In this study, we show that N. meningitidis PNPase affects global gene regulation and negatively affects aggregation and adhesion to epithelial cells. The hyperaggregation observed in PNPase-deficient mutants depended on pili and involved increased bundling of pili. We also show that PNPase is required for full bacteremia in an in vivo model of N. meningitidis infection.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains and plasmids used in this paper are presented in Table 1. The encapsulated N. meningitidis serogroup C strain FAM20 (36) was used in experiments and is referred to as the “wild type.” N. meningitidis strains were grown on GC agar (Acumedia) plates or in GC broth with 1% Kellogg's supplement (37) in a humidified environment at 37°C under 5% CO2. Bacteria were lifted and placed on GC plates 2 days prior to the experiments and were restreaked once 16 to 18 h prior to the experiments. The E. coli strain DH5α was used as the host for cloning and plasmid propagation, and it was grown in lysogeny broth (LB) or on LB agar plates (Acumedia). To select for plasmids in E. coli, ampicillin (Amp) or kanamycin (Kan) was added at 100 μg/ml or 50 μg/ml, respectively. To select N. meningitidis transformants, Kan, chloramphenicol (Cap), or tetracycline (Tet) (Sigma-Aldrich) was added to the culture medium at a concentration of 50 μg/ml, 5 μg/ml, or 1 μg/ml, respectively. Where specified, isopropyl β-d-1-thiogalactopyranoside (IPTG) was added at a concentration of 0.5 mM.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype | Resistance marker(s) | Source or reference |
|---|---|---|---|
| Strains | |||
| N. meningitidis | |||
| Fam20 (wild type) | 36 | ||
| Δpnp | pnp::Kan | Kanr | This study |
| Δpnp/pnp | pnp::Kan; complemented with pnp under the control of the native promoter | Kanr Capr | This study |
| ΔsiaD | siaD::tet | Tetr | This study |
| ΔpilE | pilE::tet | Tetr | This study |
| PNPind | IPTG-inducible pnp | Capr | This study |
| Δpnp/PNPind | pnp::Kan; IPTG-inducible pnp | Kanr Capr Tetr | This study |
| Δpnp/PNPind ΔsiaD | pnp::Kan siaD::tet; IPTG-inducible pnp | Kanr Capr Tetr | This study |
| Δpnp/PNPind ΔpilE | pnp::Kan pilE::tet; IPTG-inducible pnp | Kanr Capr Tetr | This study |
| E. coli | |||
| DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK− mK+) λ− | ||
| BL21 | F− dcm ompT hsdS(rB− mB−) gal [malB+]K-12(λs) | Stratagene | |
| Plasmids | |||
| pTrcHis | Ampr | Invitrogen | |
| pTrcHis-PNP | Ampr | This study | |
| pBAD-lacI | Ampr | DNA2.0 | |
| pDEST-Δpnp | Ampr Kanr | This study | |
| pDEST-natPNP | Ampr Capr | This study | |
| pDEST-indPNP | Ampr Capr | This study | |
| pDEST-ΔpilE | Ampr Tetr | This study | |
| pTOPO-ΔsiaD | Kanr Tetr | This study |
Cell lines and culture conditions.
The FaDu human pharyngeal epithelial cell line (ATCC HTB-43) was maintained in Dulbecco's modified Eagle's medium (DMEM) with GlutaMAX and pyruvate (Invitrogen), supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich), in a humidified environment at 37°C under 5% CO2. For these experiments, FaDu cells were grown in 48-well plates in DMEM–10% FBS until 80% confluence was achieved.
Purification of PNPase.
PNPase was amplified from FAM20 genomic DNA using the PNPpurfw and PNPpurrev primers (Table 2). A PCR product was cloned into the pTrcHis vector (Invitrogen) according to the manufacturer's instructions, yielding a PNPase protein that was fused to an N-terminal His tag. The resulting plasmid was transformed into DH5α cells. The correct sequence was confirmed by sequencing, and the plasmid was then moved into the expression strain BL21. To induce the expression of PNPase, the bacteria were grown to log phase in LB with ampicillin, and 1 mM IPTG was added. The bacteria were harvested after 5 h of induction. The bacteria were resuspended in resuspension buffer (100 mM morpholinepropanesulfonic acid [MOPS], 500 mM NaCl [pH 7.4]). The bacteria were lysed using sonication and lysozyme treatment. The lysed bacteria were centrifuged at 100,000 × g for 1 h at 4°C to obtain the soluble fraction containing PNPase. The soluble fraction was purified twice in a Talon metal affinity resin (Clontech) and was finally dialyzed against 50 mM Tris-HCl (pH 7.4) at 4°C overnight. The purity of the samples was verified using Coomassie staining of proteins that were separated on an SDS-PAGE gel.
TABLE 2.
Primers used in this study
| Primer | Sequence |
|---|---|
| Cloning | |
| P1 | ATAGAAAAGTTGAGCGATTCAGCTTTATAC |
| P2 | TGTACAAACTTGAATACCGCACTGCTAAAAC |
| P3 | TGTACAAAGTGGCGCTTAGGGTGAAAGTGC |
| P4 | ATAATAAAGTTGGATACTCGGATTAATCGG |
| P5 | ATAGAAAAGTTGATGCCGTCTGAAAATTAAGTTAGAATTATCCC |
| P6 | GGGGACTGCTTTTTTGTACAAACTTGAAATCCAAAATCATACTG |
| P7 | GGGGACAAGTTTGTACAAAAAAGCAGGCTGACTTTGCTGACTCAGGATT |
| P8 | GGGGACCACTTTGTACAAGAAAGCTGGGTAAATACCGTCTGAAACCTG |
| K1 | AAAAAGCAGGCTGTATTAGTGACCTGTAGAATTCGAGC |
| K2 | AGAAAGCTGGGTTTAGAAAAACTCATCGAGCATCAAATG |
| Lac-pnpfw | TAACAATTTCACACAGGAAACAGCTTTGAAAGGAACAATAATGTTCAATAA |
| Pnp-down | CATTCATTCCCGATACCCGTAAAAA |
| Lac_up | TCCCCATCGGTGATGTCGTATAAGA |
| Lac-PNPrev | TTATTGAACATTATTGTTCCTTTCAAAGCTGTTTCCTGTGTGAAATTGTTA |
| PilEUHSfw | ATAGAAAAGTTGAGGCGGATGTGTAAGGTAGATG |
| PilEUHSrev | TGTACAAACTTGCGATCAGGGTGAAACCTT |
| PilEDHSfw | CTTGTACAAAGTGGACCTGCCGCACCAAATAA |
| PilEDHSrev | ATAATAAAGTTGGCAGGGAAATGGGTAGGGA |
| attB1_Tet_fw | AAAAAGCAGGCTACCTTTCCGAACAAGA |
| attB2_Tet_rev | CAAGAAAGCTGGGTTTCAGACGGCATTGGGT |
| TetRfw | TTTTTTTCAAACGAAGTCAGCCCCATACGATATAAG |
| TetRrev | TTCCATTCAGGTCGAGGTGGCC |
| NMC50fw | ATGTCAATCAATACGTTTGAAACCTTTATTTACG |
| NMC50rev | GGGCTGACTTCGTTTGAAAAAAAAGCGTG |
| SiaCfw | ATGCCGTCTGAATATGAACGATTTATCTGAAGC |
| SiaCrev | GATGAATAAGACTCCAGCTTAAATATATTTATTCAATATCAGTTTTTTTG |
| NMC49fw | ACCTGAATGGAATCAACTATTTCTTCTTCTAAACCATTTAG |
| NMC49rev | TTGACAATAGAGCGAAAAAGTACCGCG |
| SiaE-Tetfw | ATATATTTAAGCTGGAGTCTTATTCATCATGTCAATCAATACGTTTG |
| SiaE-Tetrev | AGAAATAGTTGATTCCATTCAGGTCGAGGTG |
| PNPpurfw | TTGAAAGGAACAATAATGTTCAATAAACAC |
| PNPpurrev | TTACTCGGCGGCATTTTC |
| qPCR | |
| PNP_qPCR_fw | TTGGGCGACGAAGACCACTT |
| PNP_qPCR_rev | TGTGCAGACGCGCTTCTTTG |
| S10_qPCR_fw | TTGGAAATCCGCACCCACTT |
| S10_qPCR_rev | TACATCAACACCGGCCGACAAA |
| PilN_qPCR_fw | GAGATGAACAAGCGCAAACA |
| PilN_qPCR_rev | AAGTGTGCGATGGAGGTTTC |
| PilO_qPCR_fw | CAGATGGAATCCCTTGAGGA |
| PilO_qPCR_rev | GAACCTGCCTGATGAAGCTC |
| PilW_qPCR_fw | AACTACGGCTGGTTCCTGTG |
| PilW_qPCR_rev | GTGCGCGCCAGTTCTTTAAA |
Measurement of PNPase enzymatic activity.
PNPase enzymatic activity was measured using a pyruvate kinase/lactate dehydrogenase-linked assay, as described previously for E. coli PNPase (38). Briefly, poly(A) RNA (Sigma) and phosphate were added as substrates for PNPase to yield ADP as a product. ADP was then used by pyruvate kinase with phosphoenolpyruvate (PEP) to obtain ATP and pyruvate. This pyruvate is itself used by lactate dehydrogenase to oxidize NADH to NAD+. The decrease in NADH was measured at 340 nm. Baseline activity was determined by running the reaction without phosphate. A 5 mM MgCl2 stock solution was used for all of the experiments. The experiments were performed at 28°C using 0.5, 1, 2, or 4 mM phosphate.
Generation of mutant strains.
Invitrogen's MultiSite Gateway three-fragment cloning system was used to construct the pDEST-Δpnp, pDEST-ΔpilE, pDEST-natPNP, and pDEST-indPNP plasmids. PCR amplification was performed using high-fidelity Phusion DNA polymerase (Thermo Scientific), and the primers used are presented in Table 2.
Briefly, the separated fragments were cloned into pDONR vectors. Three separate pDONR vectors containing the fragments of interest were cloned into the pDEST vector in the correct order using the LR Clonase Plus enzyme. The pnp deletion mutation sequences that corresponded to the sequences upstream and downstream of pnp were amplified from FAM20 chromosomal DNA using primer sets P1–P2 and P3–P4, respectively. A kanamycin resistance cassette was amplified from the pDONR P4-P1R vector (39) using primer set K1–K2. The three PCR products were first cloned into pDONR vectors and then fused into pDEST in order (first pnpUHS, then kanR, and finally pnpDHS) to create pDEST-Δpnp.
For the pilE deletion, mutation sequences corresponding to the sequences upstream and downstream of pilE were amplified from FAM20 chromosomal DNA using primer sets PilEUHSfw–PilEUHSrev and PilEDHSfw–PilEDHSrev, respectively. The tetracycline resistance cassette was amplified from pACYC184 using primers attB1_Tet_fw and attB2_Tet_rev. The three PCR products were then cloned into pDONR vectors and fused into pDEST in order (first pilEUHS and then TetR-pilEDHS) to create pDEST-ΔpilE.
To create plasmids for complementation of the Δpnp mutation, a downstream homologous sequence was amplified into the noncoding genomic region between NMC0075 and NMC0080 in FAM20 chromosomal DNA using primer set P5–P6 and was cloned into pDONR. The downstream fragment, together with a chloramphenicol resistance cassette, was obtained from the previously described plasmid pDONR P4-P1R (39). To obtain the pnp sequence under the control of the native promoter, the pnp gene, including a 243-bp upstream region containing its endogenous promoter, was amplified from FAM20 using primer set P7–P8 and was cloned into pDEST. To obtain an IPTG-inducible copy of pnp, the lac promoter was amplified from pBAD-lacI using primers Lac_up and Lac-PNPrev, and the PNPase coding sequence was sequenced from FAM20 using primers Lac-pnpfw and Pnp-down. The two fragments were joined using fusion PCR with primers attB1-lac and attB2-pnp and were cloned into pDEST. To obtain the final pDEST plasmids, the upstream and downstream pDONR plasmids were combined with either pDONR-natPNP or pDONR-indPNP to create pDEST-natPNP and pDEST-indPNP.
The ΔsiaD mutant was constructed using fusion PCR. Because siaD is part of the sacA-siaB-siaC-siaD-NMC0050 operon, we sought to construct a deletion mutant that allowed the native expression of NMC0050. To achieve this, fusion PCR was performed in two steps. In the first step, the tetracycline resistance cassettes in pACYC184 and NMC0050 were amplified using primer pairs TetRfw–TetRrev and NMC50fw–NMC50rev. Because the primers contain overlapping sequences, a fused NMC0050-Tet fragment was obtained from a PCR using the separate fragments as the template and NMC50fw and TetRrev as the primers. In the second fusion PCR, the downstream SiaC region was amplified using SiaCfw and SiaCrev, and the upstream NMC0049 region was amplified using NMC49fw and NMC49rev. The NMC0050-Tet fragment was amplified using the SiaE-Tetfw and SiaE-Tetrev primers, resulting in overlapping regions in the flanking fragments. In the final step, the SiaC, NMC0050-Tet, and NMC0049 fragments were joined and amplified using the SiaCfw and NMC49rev primers. The resulting siaC-NMC0050-tetR-NMC0049 fragment was cloned into pTOPO-ZeroII Blunt to yield pTOPO-ΔsiaD.
The constructs were integrated into the genome of N. meningitidis FAM20 using homologous allelic replacement following liquid or spot transformation with plasmid DNA (40). Mutants were selected by plating the bacteria on GC plates with the appropriate antibiotics. Because we were not able to transform the Δpnp mutant, the complemented strains were constructed by first integrating the complementation construct and then introducing the pnp mutation. For the double mutants, the ΔsiaD and ΔpilE mutations were introduced into the Δpnp/indPNP complemented strain in the presence of IPTG.
Generation of anti-PNPase antibodies.
Anti-PNPase antibodies were generated by EZBiolab. Three peptides were synthesized to match the predicted domains of the N. meningitidis PNPase: P1, C-KHLEADVVRSQILDGQPRIDGRDTRTVRP-NH2; P2, C-FFKREGKQSEKEILT-NH2; and P3, C-KALLDAPAREENAAE-COOH. Two rabbits were immunized per peptide, and the sera were then collected and pooled. Antisera (i.e., anti-P1, anti-P2, and anti-P3) were precipitated using 33% (NH4)2SO4, and the precipitates were dissolved in phosphate-buffered saline (PBS). The solution was dialyzed against PBS buffer and was lyophilized. For affinity purification, the antisera were purified using peptide affinity chromatography and were subsequently dialyzed against PBS buffer and lyophilized. Lyophilized antisera and affinity-purified antibodies were dissolved in PBS, and the aliquots were stored at −20°C. All three antibodies yielded PNPase-specific signals (data not shown).
Live-cell microscopy to analyze aggregation.
Bacteria were suspended in DMEM–1% FBS, filtered through 5-μm-pore-size filters to eliminate large aggregates, and diluted to 1 × 107 CFU/ml. They were then seeded into glass-bottom dishes (MatTek). Bacterial aggregation during growth was imaged using an inverted Axio Observer Z1 microscope (Carl Zeiss) at 37°C under 5% CO2. Images were captured after 30 to 240 min of incubation and were processed using AxioVision software, version 4.7 (Carl Zeiss). Live microscopy was performed more than twice for each setting.
Spectrophotometric quantification of bacterial aggregation.
Aggregation assays were performed as described previously (14). Briefly, the bacteria were suspended in DMEM–1% FBS, filtered through 5-μm-pore-size filters to eliminate large aggregates, and adjusted to an optical density at 600 nm (OD600) of 0.5. The bacteria were then grown to an OD600 of 1 at 37°C under 5% CO2 with agitation. Bacterial suspensions were subsequently transferred to room temperature and were left to sediment under static conditions. The OD600 of the supernatants was measured at 20-min intervals for 3 h. Aggregation assays were performed three times using duplicate samples.
Adherence assays.
Adherence assays were performed as described previously (3). Briefly, the bacteria were suspended in serum-free DMEM and were added to FaDu cells at a multiplicity of infection (MOI) of 100. Infected cells were incubated at 37°C under 5% CO2. After 2 h, the cells were gently washed five times with PBS to remove unbound bacteria. Eukaryotic cells were lysed using freshly prepared 1% saponin in PBS. The number of attached CFU was subsequently determined by serial dilution and by plating of the bacteria on GC plates. The bacteria were vortexed thoroughly before plating to break up aggregates. Adherence assays were performed three times in triplicate samples.
Preparation of cell lysates and Western blot analysis.
To measure protein expression, the bacteria were suspended in DMEM supplemented with 1 or 10% FBS to obtain an OD600 of 0.1. They were then grown while being agitated at 37°C under 5% CO2 for 4 h, unless stated otherwise. Bacteria attached to the FaDu cells were washed four times in prewarmed DMEM to remove unbound bacteria. The cells were lysed in PBS containing 1% saponin and cOmplete protease inhibitor (Roche) on ice to release the bacteria. The samples were vortexed to break up aggregates and were then filtered (pore size, 5 μm) to remove FaDu cell debris. The bacteria were collected using centrifugation and were resuspended in PBS, and the samples were adjusted to an OD600 of 1.0. The samples were diluted in 4× sample buffer containing 1% β-mercaptoethanol, separated on 12% SDS-PAGE gels, and transferred to Immobilon-P transfer membranes (Millipore) according to the manufacturer's instructions. PNPase was detected using the rabbit polyclonal antibody against peptide P1 (described above). PilC, PilE, and PilT were detected using polyclonal rabbit antibodies. Opa proteins were detected using monoclonal mouse antibodies 4B12/C11, detecting all Opa proteins, and H.22.1, detecting Opa540 and Opa1800, as described previously (41). For the quantification of protein expression levels, a monoclonal antibody against EF-Tu (Hycult Biotech) was used as a normalization control. Primary antibodies were detected using a goat anti-mouse antibody conjugated with the infrared (IR)-reactive dye 680LT and/or a goat anti-rabbit antibody conjugated with the IR-reactive dye 800CW as a secondary antibody (Li-Cor) and were visualized using an Odyssey IR scanner (Li-Cor). Alternatively, primary antibodies were detected using peroxidase-conjugated anti-rabbit antibodies (Bio-Rad) and a SuperSignal chemiluminescence detection kit (Pierce Biotechnology). A Precision Plus All Blue prestained protein standard (Bio-Rad) or a PageRuler Plus prestained protein ladder (Fermentas) was used as a molecular mass marker. Analyses of raw image data files were performed using ImageJ analysis software (version 1.43). Protein expression was quantified from 3 to 6 biological replicates.
Analysis of pnp expression by quantitative real-time PCR (qPCR).
For the measurement of PNPase mRNA levels, bacteria were grown in DMEM–1% FBS to an OD600 of 0.3 with agitation at 37°C under 5% CO2. The log-phase bacteria were added to FaDu cells at an MOI of 100 and were incubated under static conditions. After 2 h, the cells were gently washed four times with serum-free DMEM to remove unbound bacteria. As a control, log-phase bacteria were incubated under static conditions in cell culture plates without FaDu cells. To obtain bacteria before and after dispersion, bacterial infection was performed under an inverted Axio Observer Z1 microscope (Carl Zeiss) at 37°C under 5% CO2. Before and after dispersion, unattached cells were washed twice in prewarmed DMEM. To stabilize RNA, RNAprotect (Qiagen) was added to the cultures. RNAprotect also lysed the FaDu cells, allowing the collection of the attached bacteria. The bacteria were collected by centrifugation at 5,000 × g for 10 min. The bacterial pellet was treated with 15 mg/ml lysozyme (Sigma) and protease K (Qiagen) for 10 min to lyse the bacteria. RNA was isolated with the RNeasy Plus minikit (Qiagen). Two hundred nanograms of RNA was reverse transcribed using the SuperScript Vilo master mix (Thermo Fisher). PCR amplification was performed using a Roche LightCycler 480 machine with the LightCycler 480 SYBR green I master mix (Roche Diagnostics) and primer pairs specific for pnp, pilN, pilO, pilW, and S10 (NMC0129). The primer sequences are presented in Table 2. The PCR program was adapted from Roche's LightCycler 480 SYBR green I master mix instructions, with 40 cycles of amplification and an annealing temperature of 60°C. The threshold cycle (CT) values were determined using the second derivative maximum method. Relative expression was analyzed with LightCycler 480 software, version 1.5, using S10 as an internal standard, and was normalized to that of log-phase bacteria. The specificity of the primers was verified by melting curve analysis. mRNA expression was analyzed in three biological replicates with triplicate samples.
Microarray analysis.
Log-phase cultures grown in GC medium were harvested, and RNA was immediately stabilized using a 2% phenol–38% ethanol solution for 30 min at 4°C. The bacteria were then collected by centrifugation and were lysed with 50 mg/ml of lysozyme in Tris-EDTA buffer for 5 min at room temperature. RNA was isolated using an SV Total RNA purification kit (Promega), including an on-column DNase I digest, and was eluted in 100 μl of H2O. The concentration and purity of RNA were analyzed using a NanoDrop spectrophotometer, and RNA integrity was confirmed using denaturing agarose gel electrophoresis. cDNA synthesis and microarray analyses were performed by Roche's NimbleGen gene expression services using a custom-made whole-genome open reading frame (ORF) expression array for N. meningitidis FAM18 (GenBank accession no. NC_008767), with a 2.1M 12-plex array format that contained 10 probes per gene (18,780 probes) with 7 replicates (131,460 probes in total on the final array). Gene expression data were analyzed using DNAStar ArrayStar software. Normalized expression values were generated using quantile normalization and the Robust Multichip Average (RMA) algorithm (42–44) with a ≥2.0-fold change as the cutoff. Microarray analyses were performed using six replicates for each sample. One wild-type sample was excluded from the analysis. The genes were clustered into COG (Clusters of Orthologous Groups) categories according to the COG classification for FAM18 on the NeMeSys website (45).
Capsule ELISA.
Capsular polysaccharides were quantified using whole-cell enzyme-linked immunosorbent assays (ELISA) with a monoclonal anti-serogroup C capsule antibody (code 95/678; National Institute for Biological Standards and Control), as described previously (46). Purified serogroup C polysaccharides (code 07/318; National Institute for Biological Standards and Control) were used as the positive control. The ELISA plates were coated with 100 μl of bacteria in PBS and were incubated overnight at 4°C. The plates were subsequently blocked in 5% bovine serum albumin (BSA) for 2 h and were incubated with a 1:400 dilution of the anti-capsule monoclonal antibody overnight at 4°C. For detection, the plates were incubated with a 1:5,000 dilution of horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G for 1 h. Plate-bound peroxidase was detected using 3,3′,5,5′-tetramethylbenzidine (TMB) (Invitrogen), and absorbance was measured at 490 nm. Capsule expression was assayed once using triplicate samples.
MATS.
A MATS (microbial adhesion to solvents) assay was performed using the modified procedures described by Ly et al. (47). N. meningitidis was harvested following overnight growth on GC agar plates, resuspended in PBS, and filtered through a 5-μm-pore-size filter to remove clumps. The OD600 was measured, and the cell suspensions were equalized to an OD600 of 0.4. The bacteria were pelleted using centrifugation at 3,000 × g and were resuspended in PBS. Bacterial suspensions were mixed at a 4:1 ratio with hexadecane in microcentrifuge tubes, vortexed for 30 s to mix the two phases thoroughly, and left to stand at room temperature for 15 min to allow the phases to separate. An aliquot of the aqueous phase was measured as the OD600. The percentage of hydrophobicity was calculated using the formula [1 − (Abs2/Abs1)] × 100%, where Abs1 is absorbance before mixing with hexadecane and Abs2 is absorbance after mixing with hexadecane. Hydrophobicity was assayed three times using triplicate samples.
Biofilm assay.
Bacteria were grown on GC agar overnight. Bacteria were resuspended in GC liquid with 1% Kellogg's supplement to an OD of 0.05 in 96-well microtiter plates and were allowed to grow for 24 h at 37°C under 5% CO2. Unbound bacteria were removed by washing the samples twice with PBS, and bacteria were then stained for 2 min with 0.3% crystal violet. Unbound dye was removed by washing the samples twice in PBS. The stained biofilms were finally resuspended in 33% acetic acid, and absorbance was measured at 630 nm. Biofilm formation was assayed three times using triplicate samples.
LOS detection.
Lipooligosaccharide (LOS) was isolated, separated on a Tricine SDS-PAGE gel, and visualized by silver staining as described previously (48).
Electron microscopy.
To stain the bacteria, we used a protocol described previously (14). Briefly, the bacteria were gently suspended in PBS and were then added to Formvar-coated grids for 5 min. They were then fixed in 1% glutaraldehyde for 5 min. The grids were washed twice with water, incubated with 1% ammonium molybdate for 20 s, air dried, and analyzed using a TECNAI G2 Spirit BioTWIN electron microscope (FEI). Pilus bundling was quantified manually using 75 frames (34 for wild-type and 41 for Δpnp bacteria) using ImageJ (NIH, Bethesda, MD, USA). The quantifications were performed in a blinded manner.
Mouse model of infection.
The hCD46Ge transgenic mouse line (CD46+/+) harbors the complete human CD46 gene and expresses CD46 in a human-like pattern (41, 49, 50). To study the clearance of bacteria from the blood, transgenic mice (n = 7) were challenged intraperitoneally with 5 × 107 CFU of bacteria suspended in 100 μl PBS. Blood samples were obtained from the tail vein at different time points after infection (2 h, 6 h, and 24 h). Bacterial counts were determined by plating serial dilutions of the samples on GC plates. The bacteria were vortexed thoroughly before plating to break up aggregates. The mouse experiments described in the present study were conducted at the animal facility of Stockholm University. Animal care and experiments were performed according to institutional guidelines. All protocols were approved by the Swedish Ethical Committee on Animal Experiments.
Motility.
Motility was analyzed as described previously (51). Briefly, bacteria were suspended in 3 ml of prewarmed GC broth containing 10% Kellogg's supplement in 35-mm poly-d-lysine-coated glass-bottom dishes (MatTek) and were incubated for 1 h prior to microscopy. The cell culture dishes were transferred to a humidified incubation chamber (37°C, 5% CO2) connected to an inverted fluorescence microscope (Axio Observer Z1; Carl Zeiss). The images captured during the time lapse experiments were further processed using AxioVision software (Carl Zeiss). Bacteria were tracked using an automatic tracking module in the AxioVision software suite, version 4.7, and each individual track was manually inspected for automatic tracking errors. The velocity distributions of 37 individual 60-s tracks were analyzed.
Statistical analysis.
Differences between two groups were analyzed using Student's t test. Differences between multiple groups were analyzed using ANOVA (analysis of variance) followed by the Bonferroni post hoc test. Significant differences between ratios or percentages were analyzed after log transformation of the data. P values below 0.05 were considered statistically significant. Statistical analyses were performed using Graph Pad Prism software, version 5.
RESULTS
NMC0710 is a PNPase homolog.
A Blast search was performed using E. coli PNPase as a query. It yielded a potential PNPase homolog at the meningococcal locus NMC0710. The proteins share 62.55% identity and are similar in size (Fig. 1A). To confirm that NMC0710 has the same enzymatic function as PNPase, a His-tagged version of NMC0710 was overexpressed in E. coli. Following metal affinity purification, a single band with a size similar to that of the expected protein was visible on a Coomassie blue-stained gel (Fig. 1B). Like the E. coli protein, N. meningitidis NMC0710 was predicted to have no transmembrane segments and no translocation signals. During the purification of N. meningitidis, His6-NMC0710 was found in the soluble fraction, suggesting that this protein is not membrane associated. PNPase can degrade RNA polymers by phosphorolysis to release nucleotide diphosphates. This activity was determined for NMC0710 by use of an enzymatic assay in which the formation of ADP from poly(A) was linked to NADH reduction through lactate dehydrogenase and pyruvate kinase, as described previously (38). The reactions are shown in Fig. 1C. Km and Vmax were estimated to be 0.50 mM and 5.55 U/mg, respectively, by varying the phosphate concentration and plotting the inverse concentrations and activities on a Hill plot (Fig. 1D). While Km was similar to the 0.7 mM value reported for E. coli, Vmax was substantially higher than the 0.7-U/mg value reported previously (52). In summary, these data show that NM0710 has PNPase activity. Consequently, NMC0710 is referred to below as the PNPase of N. meningitidis.
FIG 1.

NMC0710 is an N. meningitidis PNPase. (A) Alignment of the protein sequences for PNPase in Escherichia coli and Neisseria meningitidis FAM20. The alignment was constructed using Clustal Omega and was visualized using Jalview. The alignment is color-coded according to Blosum62 amino acid conservation scores (deep blue indicates identical residues; white indicates low conservation). The peptides used to produce the antibodies are boxed in red. (See Materials and Methods for details.) (B) Coomassie blue-stained SDS-PAGE gel showing the His-tagged N. meningitidis PNPase that was purified from E. coli. (C) Diagram of the reactions used to assay the phosphorolytic activity of PNPase. In this assay, the production of ADP was coupled to the oxidation of NADH via pyruvate kinase and lactate dehydrogenase, and the loss of NADH was measured at 340 nm. Phosphoenolpyruvate (PEP) and NADH were added in excess to ensure that the PNPase reaction was rate-limiting. (D) Hill plot showing the relation between the concentration of phosphate and activity.
PNPase is required for optimal growth.
With the PNPase enzymatic activity of NMC0710 established, a PNPase deletion mutant (Δpnp) was created in the N. meningitidis FAM20 background in order to investigate the role of PNPase in meningococcal physiology and pathogenesis. A complemented strain (Δpnp/pnp), which contained a copy of the pnp gene under the control of its native promoter at a separate chromosomal location, was also constructed. For the determination of PNPase levels, antibodies were raised against three peptides in the protein sequence (for peptide regions, see Materials and Methods; also Fig. 1). These antibodies confirmed that the Δpnp mutant lacked the PNPase protein and that the Δpnp/pnp strain had PNPase levels similar to those of the wild-type strain (see Fig. S1A in the supplemental material). During the cloning experiments, it was observed that the Δpnp mutant formed smaller colonies on the plates than the wild-type strain. To analyze these data in more detail, growth curve analysis was performed. The growth curve confirmed that the Δpnp mutant had a lower growth rate than the wild type (Fig. 2). The doubling time was increased from 50 to 85 min for the Δpnp mutant. This was similar to the situation in which a PNPase deletion mutation in E. coli increased the doubling time from 30 to 60 min (53). The growth rate of the Δpnp/pnp strain was similar to that of the wild type. Because the Δpnp mutant showed altered growth properties, we speculated that PNPase expression might be differentially regulated during different growth phases. However, the expression of PNPase was shown to be stable throughout the growth phases (see Fig. S1B). Taking the data together, PNPase-deficient meningococci displayed a decreased growth rate, but wild-type bacteria expressed stable amounts of PNPase during different growth phases in liquid medium.
FIG 2.
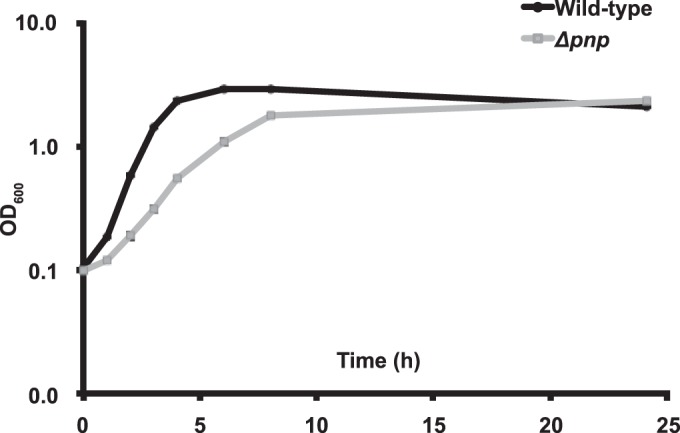
PNPase deficiency leads to a reduced growth rate. Wild-type FAM20 and the Δpnp mutant were inoculated to an OD600 of 0.1 and were grown in GC medium supplemented with 1% Kellogg's solution at 37°C under 5% CO2 with shaking. Growth was monitored for 24 h by measuring the OD at 600 nm.
PNPase negatively regulates bacterial aggregation.
During the growth studies, it was observed by visual inspection that the Δpnp mutant cultures had a tendency to aggregate. To confirm this observation, aggregation was analyzed using live-cell microscopy and culture sedimentation. Live-cell microscopy of growing bacteria revealed that the Δpnp mutant formed larger aggregates than the wild-type and Δpnp/pnp strains at the same time points (Fig. 3A). For the analysis of sedimentation, bacteria were grown to an OD600 of 1 with agitation. The cultures were then transferred to static conditions, and the OD of the surface layer was measured over time. As shown in Fig. 3B, the Δpnp mutant sedimented faster than the wild-type and Δpnp/pnp strains, confirming the observations made under microscopy. To determine whether the phenotypes observed were caused by the inactivation of PNPase or by a secondary mutation that could potentially have arisen in a slow-growing mutant, the Δpnp mutant was complemented with a copy of pnp under the control of an IPTG-inducible promoter. When the Δpnp/indPNP strain was grown in the absence of IPTG, its growth and aggregation were similar to those of the Δpnp mutant. When IPTG was added, Δpnp/indPNP bacteria again grew normally (see Fig. S2 in the supplemental material), and no excessive aggregation was observed (data not shown). These data show that PNPase deficiency leads to increased aggregation of N. meningitidis.
FIG 3.
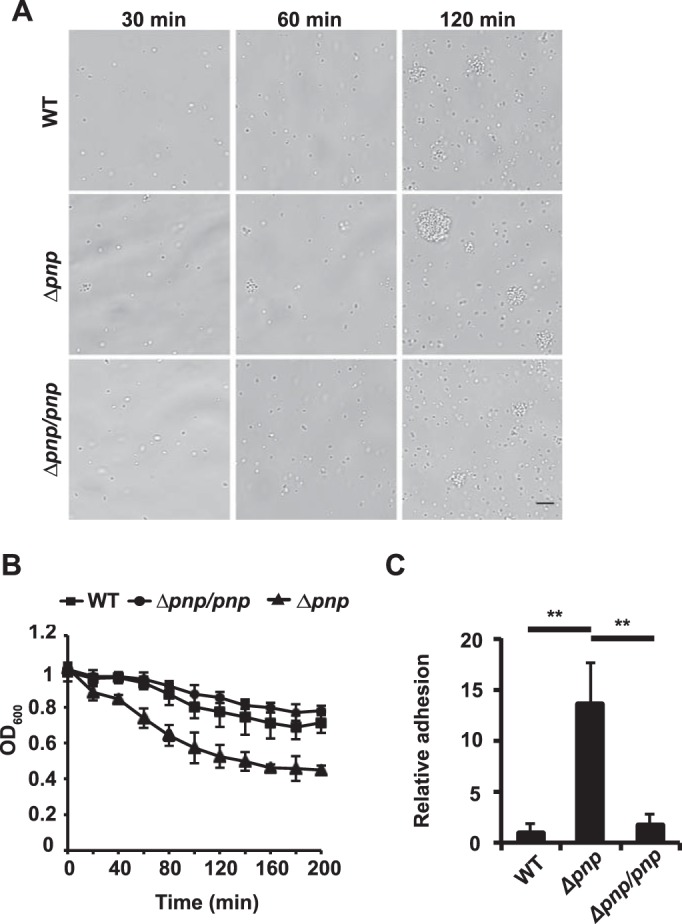
Aggregation and adhesion are enhanced in the Δpnp mutant. (A) Live-cell microscopy of wild-type, Δpnp, and Δpnp/pnp bacteria. The bacteria were imaged after 30, 60, and 120 min. Representative images are shown. Bar, 10 μm. (B) Sedimentation of wild-type, Δpnp, and Δpnp/pnp bacteria in liquid cultures. The bacteria were grown with shaking to an OD600 of 1 (0 h) and were subsequently moved to static conditions. The OD600 of the upper layer of the culture was measured every 20 min. (C) Adhesion of wild-type, Δpnp, and Δpnp/pnp bacteria to FaDu epithelial cells. FaDu cells were grown to 80% confluence and were then infected with log-phase bacteria to an MOI of 100 for 2 h. Unbound bacteria were washed away, and bound bacteria were released by treating the cells with saponin. The bound bacteria were quantified by viable counting on GC plates. Statistical significance (determined by ANOVA followed by a Bonferroni post hoc test) is indicated by asterisks (**, P < 0.01).
Lack of PNPase leads to increased adhesion.
The formation of aggregates is believed to be important during the initial stage of infection, when the bacteria attempt to adhere to human cells (14). Therefore, the increased aggregation of the Δpnp mutant may also influence its ability to adhere. To investigate this, adhesion assays with human epithelial FaDu cells were performed. The cells were infected with exponentially growing bacteria at an MOI of 100 for 2 h, and unbound bacteria were washed away. The bound bacteria were plated in order to obtain viable counts. The results shown in Fig. 3C demonstrate that the Δpnp mutant adheres to the host cells significantly better than the wild-type and Δpnp/pnp strains. Because PNPase negatively affects aggregation and adhesion, we wanted to investigate the expression of PNPase before and after attachment to cells. When planktonic bacteria and bacteria attached to epithelial cells were compared, we found that PNPase expression was higher in the attached bacteria than in the planktonic bacteria at both the mRNA level (Fig. 4A) and the protein level (Fig. 4B). Attached bacteria also expressed increased amounts of PNPase mRNA after aggregate dispersion (see Fig. S3 in the supplemental material).
FIG 4.
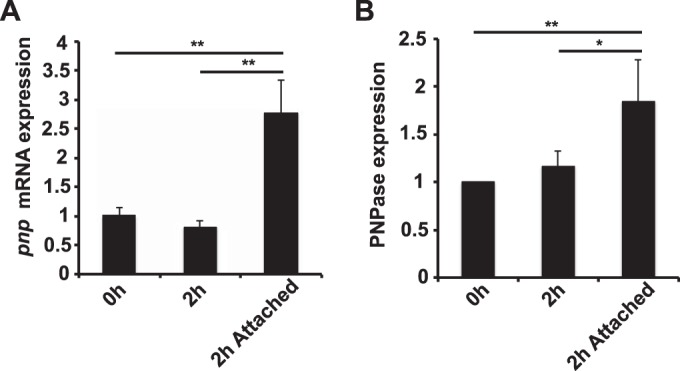
PNPase expression is induced by host attachment. The expression of PNPase in wild-type bacteria was measured at the mRNA level using qPCR (A) and at the protein level using quantitative immunoblotting (B). S10 expression was used as an internal control for the qPCR, and EF-Tu expression was used as an internal control for the immunoblotting. The expression levels are normalized to that for the 0-h sample. The bacteria were grown in DMEM–1% FBS either to log phase with shaking, to log phase followed by 2 h under static conditions, or to log phase followed by 2 h under static conditions while the bacteria were attached to FaDu cells. Statistical significance (determined by ANOVA followed by a Bonferroni post hoc test) is indicated as follows: *, P < 0.05; **, P < 0.01.
These data suggest that PNPase-deficient meningococci adhered in larger numbers than wild-type bacteria to epithelial host cells and that PNPase was upregulated in bacteria attached to host cells.
PNPase affects global gene expression in N. meningitidis.
In E. coli, PNPase regulates the expression levels of many genes through its roles in mRNA degradation (53) and sRNA regulation (23). To investigate whether PNPase also plays a major role in gene regulation in N. meningitidis, microarray analysis of wild-type and Δpnp mutant bacteria was performed using log-phase cultures grown in GC liquid. The microarray revealed 469 differentially expressed genes (>2-fold change in expression), 263 of which were upregulated and 206 of which were downregulated. A summary of the expression changes based on the COG (Clusters of Orthologous Groups) classification of all of the genes is presented in Table 3. The full list of all the differentially expressed genes is presented in Table S1 in the supplemental material. The transcriptomic data revealed dramatic changes in the expression of genes involved in central metabolism. In the “Energy production and conversion” COG category, 25 of the 120 genes were downregulated, especially those affecting the Krebs cycle. Conversely, the increased expression of genes involved in the glycolytic Entner-Doudoroff pathway suggested that the Δpnp mutants utilize this alternative ATP-generating pathway to compensate for the low Krebs cycle activity and increased glycolytic activity. Additionally, there were many downregulated genes in the “Amino acid transport and metabolism” COG group. Among the upregulated genes, those in the “Translation, ribosomal structure and biogenesis” COG class stood out, with nearly one-third (49 of 159) of the genes upregulated.
TABLE 3.
Genes differentially regulated in the pnp mutanta
| COG category | Total no. of genes | No. of genes differentially regulated |
|
|---|---|---|---|
| Up | Down | ||
| RNA processing and modification | 1 | 0 | 0 |
| Chromatin structure and dynamics | 1 | 0 | 0 |
| Energy production and conversion | 120 | 8 | 25 |
| Cell cycle control, cell division, chromosome partitioning | 35 | 5 | 2 |
| Amino acid transport and metabolism | 165 | 9 | 34 |
| Nucleotide transport and metabolism | 49 | 4 | 4 |
| Carbohydrate transport and metabolism | 72 | 7 | 11 |
| Coenzyme transport and metabolism | 88 | 8 | 13 |
| Lipid transport and metabolism | 47 | 7 | 4 |
| Translation, ribosomal structure and biogenesis | 159 | 49 | 8 |
| Transcription | 80 | 9 | 1 |
| Replication, recombination, and repair | 183 | 23 | 8 |
| Cell wall/membrane/envelope biogenesis | 154 | 17 | 16 |
| Cell motility | 31 | 3 | 1 |
| Posttranslational modification, protein turnover, chaperones | 79 | 2 | 10 |
| Inorganic ion transport and metabolism | 105 | 4 | 12 |
| Secondary metabolites biosynthesis, transport, and catabolism | 28 | 6 | 2 |
| General function prediction only | 209 | 21 | 32 |
| Function unknown | 160 | 20 | 20 |
| Signal transduction mechanisms | 45 | 3 | 7 |
| Intracellular trafficking, secretion, and vesicular transport | 65 | 7 | 6 |
| Defense mechanisms | 21 | 3 | 2 |
| Extracellular structures | 2 | 0 | 0 |
| CDSb without COG classification | 416 | 67 | 1 1 |
Differentially regulated genes were analyzed on the basis of their COG classifications. The genes were clustered into COG categories according to the COG classification for FAM18 at the NeMeSys web site. The total number of genes in each COG class is listed for comparison. The full list of genes can be found in Table S1 in the supplemental material.
CDS, coding sequence.
Bacterial surface properties are unchanged in Δpnp mutants.
The microarray revealed that several genes involved in capsule synthesis, including siaD (6.7-fold), sacA (2.4-fold), ctrA (2.05-fold), and ctrD (2.29-fold), were upregulated in the Δpnp mutant. Because the capsule is an important factor during adhesion to both biotic and abiotic surfaces (54), we sought to investigate the status of the capsule further. To determine whether these changes in expression affected the capsule in the Δpnp mutant, the amount of capsule was measured using whole-cell ELISA, as described previously (46). The ELISA revealed that there was no significant change in the amount of capsule present on the bacterial surface (Fig. 5A). Because the capsule is much more hydrophilic than the underlying bacterial cell surface, the degree of capsulation can also be determined by measuring cellular hydrophobicity (54). We measured hydrophobicity using MATS (microbial adhesion to solvents) assays (47). In these assays, the bacteria were suspended in a buffered aqueous (polar) solution and were mixed with a nonpolar n-alkene, hexadecane. The percentage of bacteria present in the polar phase was measured after phase separation. The results showed that there was no significant difference between the Δpnp mutant and the wild-type or Δpnp/pnp strain. The capsule-negative ΔsiaD mutant was significantly more hydrophobic than the other strains as a result of the loss of its capsule (Fig. 5B).
FIG 5.

Bacterial surface properties remain unchanged in the Δpnp mutant. (A) Capsular polysaccharides were quantified using ELISA with anti-serogroup C capsule antibodies. Abs, absorbance. (B) The surface hydrophobicities of wild-type, Δpnp, and Δpnp/pnp bacteria were assayed using the MATS assay. The ΔsiaD mutant was included as a capsule-negative control. Statistical significance or nonsignificance (determined by ANOVA followed by a Bonferroni post hoc test) is indicated as follows: ns, P > 0.05; *, P < 0.05. (C) Formation of static biofilms by wild-type, Δpnp, and Δpnp/pnp bacteria. Bacteria were incubated for 24 h in GC liquid plus 1% Kellogg's solution at 37°C under 5% CO2 under static conditions. The ΔsiaD mutant was included as a capsule-negative control. (D) The expression of Opa protein in wild-type, Δpnp, and Δpnp/pnp bacteria was measured using quantitative immunoblotting. EF-Tu expression was used as an internal control.
Because the Δpnp mutants showed an increased ability to adhere to human cells, we also wanted to investigate whether this ability extended to abiotic surfaces and to examine the ability of the bacteria to form biofilms, as the E. coli PNPase mutant does (34). However, there was no change in the ability of the Δpnp mutant to form stationary biofilms (Fig. 5C). This correlates well with previous studies that have shown that changes in aggregation did not increase binding to plastic surfaces by N. meningitidis (17).
To directly analyze the role of the capsule in the aggregation of the Δpnp strain, a Δpnp/indPNP ΔsiaD double mutant strain was analyzed using time lapse microscopy in the absence of IPTG (see Fig. S4 in the supplemental material). The aggregation of the double mutant was similar to that observed for the Δpnp mutant, providing direct evidence that the capsule was not involved in the aggregation observed.
The levels of Opa proteins were also analyzed (Fig. 5D), because they play roles in adhesion and because they are prone to antigenic shifts (55). The microarray showed that the opacity protein OpaB (NMC1551) was upregulated 3.3-fold in the Δpnp mutant (for details, see Table S1 in the supplemental material). The total level of Opa proteins, determined using immunoblotting, was not significantly affected; neither were the levels of Opa1800 and Opa540 (see Fig. S5 in the supplemental material). This indicates that changes in Opa expression did not increase adhesion to host cells. The LOS pattern of the Δpnp mutant was also investigated and found to be similar to that of the wild type (see Fig. S5 in the supplemental material). Taken together, these results show that the Δpnp mutants retained capsule levels, hydrophobicity, biofilm formation, Opa expression, and LOS patterns similar to those of the wild type.
The level of pilus bundling is increased in the Δpnp mutant.
The process of aggregation and adhesion in N. meningitidis is dependent on type IV pili (56–58). To investigate whether the aggregation of Δpnp bacteria was dependent on pilus formation, a Δpnp/indPNP ΔpilE double mutant strain was monitored using time lapse microscopy in the absence of IPTG. The double mutant presented a dramatic reduction in aggregate formation from that for the Δpnp/indPNP strain, as expected (Fig. 6). At later time points, some aggregates were observed, but these might have resulted from improper cell division or segregation. This possibility is supported by the fact that many bacteria showed aberrant cell shapes and were present as small clusters that did not resemble normal aggregates. Growth curve analysis of the Δpnp/indPNP ΔpilE double mutant also revealed that it grew more slowly than the Δpnp mutant (see Fig. S6 in the supplemental material), showing that the growth defect of the Δpnp mutant is not related to hyperaggregation. Overall, these data show that pili are important, if not essential, for aggregate formation by the Δpnp mutant. To exclude the possibility that the phenotypes observed were caused by phase variation of pilE, the pilE gene of the Δpnp mutant was sequenced and was found to be identical to that of the wild type.
FIG 6.

Pili are required for Δpnp mutant-induced aggregation. Live-cell microscopy of wild-type, Δpnp/PNPind, Δpnp/PNPind ΔpilE, and ΔpilE bacteria was performed. The bacteria were imaged after 30, 60, 120, and 240 min. Representative images are shown. Bar, 10 μm.
To find out whether the Δpnp mutant showed altered pilation, the pili were visualized using transmission electron microscopy. The electron micrographs shown in Fig. 7A demonstrate that the Δpnp mutant is pilated and that it displays an increased number of pilus bundles. Quantification of pilus bundling (Fig. 7B) showed that the majority of the pili were present in bundles in the Δpnp mutant, whereas in wild-type cells, the majority were present as single pili (nonbundled). Previous data showing that increased pilus bundling is correlated with increased aggregation and adhesion might explain the changes observed in these properties (39). The Δpnp mutant bacteria were also shown to retain their twitching motility (see Fig. S7 in the supplemental material), which is also dependent on pili (59). However, the speed of twitching motility was slightly reduced in the mutant. To assess whether the bundling of pili in the Δpnp mutant could be explained by changes in pilus proteins, the major pilin subunit PilE, the tip protein PilC, and the retraction ATPase PilT were investigated using Western blot analysis. A slight increase in the level of PilE was indeed observed in the Δpnp mutant, and this might have contributed to the phenotype observed (Fig. 8A). No significant differences were observed in the levels of PilC and PilT (Fig. 8B and C). The transcription data (see Table S1 in the supplemental material) showed that the genes encoding the three pilus assembly proteins were upregulated in the Δpnp mutant (pilN, 2.45-fold; pilO, 2.07-fold; pilW, 2.48-fold). While PilN and PilO are involved in early pilus assembly, PilW functions to stabilize the pilus and pilus bundles (12). In the absence of pilW, the pili are unable to form bundles, preventing aggregation (60). However, the expression of pilN, pilO, and pilW was found to be upregulated during attachment to FaDu cells, at the same time as pnp upregulation, suggesting that PNPase does not directly inhibit the expression of the pilus assembly genes (see Fig. S3 in the supplemental material). Taken together, these results show that the excessive aggregation observed in the Δpnp mutants is dependent on pili and is correlated with increased pilus bundling and elevated PilE expression levels.
FIG 7.
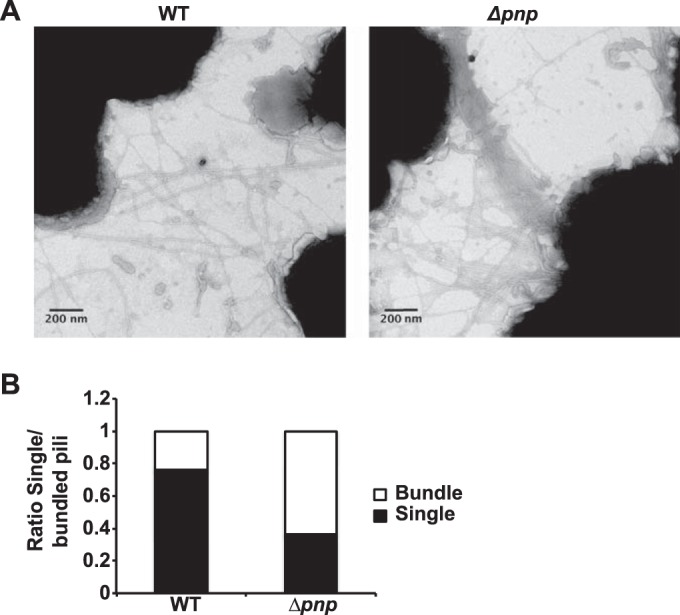
Pilus bundling is increased in the Δpnp mutant. (A) Electron micrographs of wild-type and Δpnp bacteria at ×125,000 magnification. The micrographs shown are representative examples. (B) Pilus bundling was quantified and was analyzed by determining the number of pili in bundles compared to the number of unbundled pili. A total of 74 frames were analyzed (34 with wild-type and 40 with Δpnp bacteria).
FIG 8.
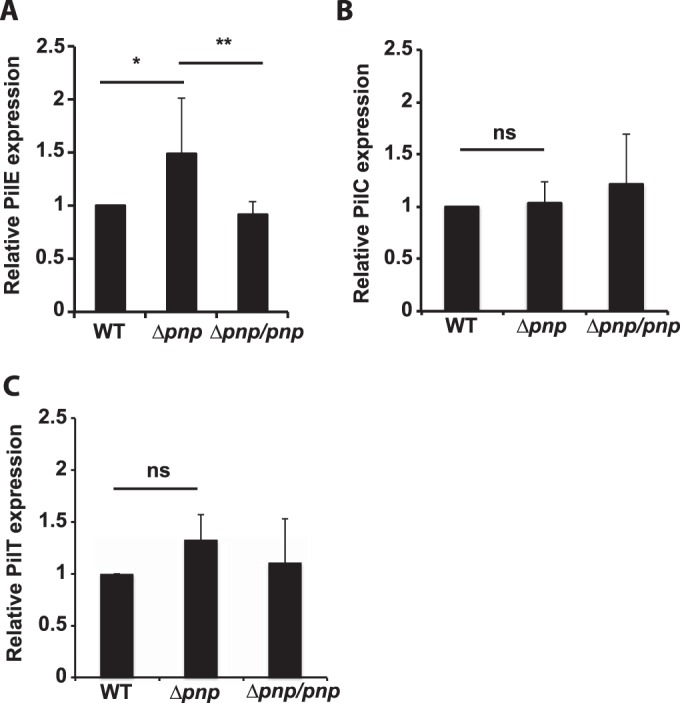
Quantification of the expression of pilus proteins. Quantitative immunoblots were analyzed using antibodies against PilE (A), PilC (B), and PilT (C). The expression of EF-Tu was used as an internal control. Statistical significance or nonsignificance (determined by ANOVA followed by a Bonferroni post hoc test) is indicated as follows: ns, P > 0.05; *, P < 0.05; **, P < 0.01.
The virulence of the Δpnp strain is attenuated in vivo.
To evaluate the importance of PNPase in vivo, we analyzed the survival of the Δpnp mutants in a mouse model of meningococcal infection. Mice were infected intraperitoneally with 5 × 107 CFU of either wild-type or Δpnp bacteria. Venous blood samples were collected after 2, 6, and 24 h for determination of the level of bacteremia The Δpnp mutants presented significantly lower bacterial counts in the blood than the wild-type strain at 6 h postinfection (Fig. 9). A nonsignificant decrease was also observed at 24 h postinfection. At 2 h postinfection, however, there was no difference in bacterial numbers between the strains. This shows that the Δpnp mutant had a reduced ability to sustain bacteremia in vivo.
FIG 9.
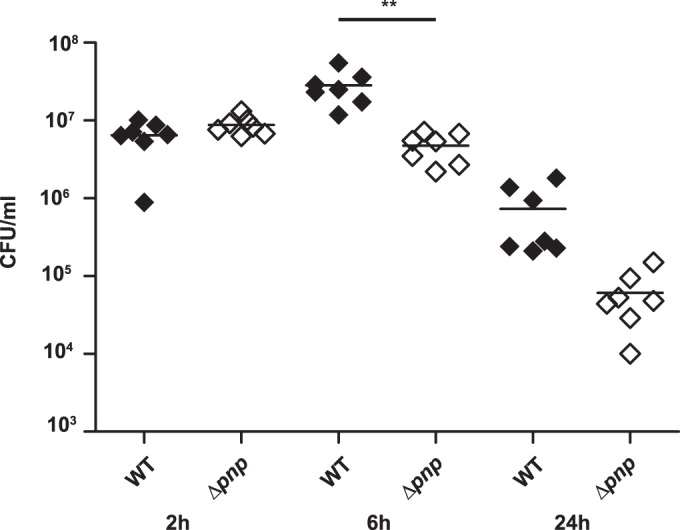
Reduced bacteremia with the Δpnp mutant in a CD46 in vivo mouse model of bacterial infection. hCD46Ge transgenic mice (n, 7 per group) were challenged intraperitoneally with 5 × 107 CFU of either wild-type or Δpnp bacteria. Blood samples were obtained from the tail vein at 2, 6, and 24 h postinfection, and bacteria were quantified by determining the number of viable bacteria on GC plates. Statistical significance (determined by ANOVA followed by a Bonferroni post hoc test) is indicated by asterisks (**, P < 0.01).
DISCUSSION
PNPase plays a central role in RNA turnover and regulation in most bacteria. PNPase has been shown previously to regulate virulence properties in several species of bacteria (24–34). The results presented in this paper show that PNPase plays an important role in regulating aggregation, adhesion, and virulence in N. meningitidis. The phenotypes observed in the mutants in this study were different from those reported for PNPase mutants previously. This is not surprising given the wide range of phenotypes associated with these mutants. These results demonstrate that although the enzymatic function of PNPase is conserved, its downstream targets differ considerably for different bacteria.
In this study, we purified the protein encoded by the NMC0710 locus of FAM20 and confirmed PNPase enzyme activity. In E. coli, PNPase controls the mRNA levels of a large number of genes by regulating their half-lives and is required for an optimal growth rate (53). In similar findings, a PNPase-deficient strain of N. meningitidis showed an impaired growth rate, and microarray analysis revealed 469 differentially expressed genes in the PNPase mutant. Large transcriptional changes were found in genes associated with central metabolism, which is interesting, because these processes are also affected in PNPase mutants of E. coli. Indeed, PNPase enzymatic activity is, in turn, regulated by both citrate and ATP in E. coli, providing feedback from metabolic pathways (52, 61). A recent study described a set of 98 putative sRNA molecules that were transcribed in N. meningitidis, some of which might be involved in the regulatory changes observed in the pnp mutant (62). Because the microarray used in this study included only predicted protein-coding genes and not sRNAs, analysis of the contributions of these genes was not within the scope of this study.
Deletion of PNPase in meningococci resulted in increased bacterial aggregation and pilus bundling. N. meningitidis aggregation and dispersion are regulated processes that are thought to be important for host colonization. In the literature, there are many examples of mutations that abolish aggregation, and these alterations notably involve markers related to pilus biogenesis, such as PilE, PilW, and PilX (14, 60). There are also examples of mutations that increase aggregation. Most of these are also involved in pilus function and modification. Notable examples include the pilus retraction ATPase PilT (60) and the gene encoding the pilus phosphoglycerol ligase, pptB (15). In these mutants, increased aggregation was associated with increased pilus bundling. It has also been shown that glycosylation plays a role in pilus bundling, because purified pili from a PilE S63A mutant that lacks glycosylation at S63 form more insoluble aggregates than the wild type (16). Mutations in genes encoding proteins that have no known interactions with the pilus, such as NafA, have also been shown to result in excessive aggregation (39). Our results show an upregulation of PilE protein levels in the Δpnp mutant that might have either contributed to or resulted from increased pilus bundling. The microarray data showed that the pilus assembly genes pilN, pilO, and pilW were upregulated, which might also have contributed to the observed increase in bundling.
Despite the existence of studies of different aggregation mutants, the underlying regulation of pilus bundling is largely unknown. In this paper, we show that PNPase, which is unlikely to be directly involved in pilus-related functions, acts as an antiaggregation factor by suppressing excessive pilus bundling.
The Δpnp mutant attached to epithelial host cells more efficiently than the wild-type strain. N. meningitidis adheres to human cells either in microcolonies or as single diplococci that subsequently join together or grow to form microcolonies. In this study, we show that PNPase mutant bacteria attach in greater numbers than the wild type. Such attachment could occur either directly, through increased bacterium-cell binding, or indirectly, through increased bacterium-bacterium interactions, leading to the attachment of larger aggregates. After the initial adhesion, the microcolonies break up, allowing for more-intimate adhesion (63). For wild-type meningococci, PNPase expression was induced in bacteria adhering to epithelial cells more than in planktonic bacteria. This PNPase upregulation might destabilize the aggregates, paving the way toward more-intimate adhesions and subsequent bacterial invasion. Indeed, we show that PNPase expression is higher in attached bacteria after aggregate dispersion. It is also interesting that PNPase acts as a negative regulator of several genes known to be involved in capsule synthesis (see Table S1 in the supplemental material), a process that has been shown to be downregulated upon intimate adhesion (18).
In this study, we show that infection of mice with the Δpnp mutant led to less bacteremia than infection with the wild-type strain. While this shows that PNPase is required for full virulence in this mouse model, the mechanism for decreased bacterial survival is unclear. Previous studies of bacteremia that used the hyperaggregative nafA- and pilT-deficient N. meningitidis mutants showed that these mutants also displayed decreased survival (39, 64). We cannot exclude the possibility that the decreased bacterial counts in blood were due to an increase in bacterial binding to host cells as a result of excessive aggregation. Indeed, it has been shown that the hyperaggregative pptB mutant releases significantly fewer cells into the media after attachment than the wild type (15). An important factor to consider is that the growth rate of the Δpnp mutant is lower than that of wild-type bacteria, placing it at a disadvantage with regard to the host in an in vivo setting. The fact that the numbers of bacteria in the blood did not differ at 2 h postinfection suggests that the mutant was not hindered from entering the bloodstream.
In conclusion, we have shown that PNPase negatively affects aggregation and adhesion in N. meningitidis. Aggregation was shown to be dependent on pili and likely to be mediated by increased pilus bundling. PNPase was also shown to be required for full survival of the bacteria in an in vivo setting, in a mouse model of infection.
Supplementary Material
Funding Statement
This work was funded by the Swedish Research Council for Medicine and Health (2013-2434), Torsten Söderbergs Foundation (MT13/12), the Swedish Cancer Society (CAN2014/533), and Ragnar Söderbergs Foundation (MF8/10).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01463-15.
REFERENCES
- 1.Stephens DS. 2009. Biology and pathogenesis of the evolutionarily successful, obligate human bacterium Neisseria meningitidis. Vaccine 27(Suppl 2):B71–B77. doi: 10.1016/j.vaccine.2009.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lo H, Tang CM, Exley RM. 2009. Mechanisms of avoidance of host immunity by Neisseria meningitidis and its effect on vaccine development. Lancet Infect Dis 9:418–427. doi: 10.1016/S1473-3099(09)70132-X. [DOI] [PubMed] [Google Scholar]
- 3.Sjölinder H, Eriksson J, Maudsdotter L, Aro H, Jonsson AB. 2008. Meningococcal outer membrane protein NhhA is essential for colonization and disease by preventing phagocytosis and complement attack. Infect Immun 76:5412–5420. doi: 10.1128/IAI.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis LA, Ngampasutadol J, Wallace R, Reid JE, Vogel U, Ram S. 2010. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog 6:e1001027. doi: 10.1371/journal.ppat.1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seib KL, Serruto D, Oriente F, Delany I, Adu-Bobie J, Veggi D, Arico B, Rappuoli R, Pizza M. 2009. Factor H-binding protein is important for meningococcal survival in human whole blood and serum and in the presence of the antimicrobial peptide LL-37. Infect Immun 77:292–299. doi: 10.1128/IAI.01071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis LA, Shafer WM, Dutta Ray T, Ram S, Rice PA. 2013. Phosphoethanolamine residues on the lipid A moiety of Neisseria gonorrhoeae lipooligosaccharide modulate binding of complement inhibitors and resistance to complement killing. Infect Immun 81:33–42. doi: 10.1128/IAI.00751-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarva H, Ram S, Vogel U, Blom AM, Meri S. 2005. Binding of the complement inhibitor C4bp to serogroup B Neisseria meningitidis. J Immunol 174:6299–6307. doi: 10.4049/jimmunol.174.10.6299. [DOI] [PubMed] [Google Scholar]
- 8.Vogel U, Weinberger A, Frank R, Muller A, Kohl J, Atkinson JP, Frosch M. 1997. Complement factor C3 deposition and serum resistance in isogenic capsule and lipooligosaccharide sialic acid mutants of serogroup B Neisseria meningitidis. Infect Immun 65:4022–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahler CM, Martin LE, Shih GC, Rahman MM, Carlson RW, Stephens DS. 1998. The (α2→8)-linked polysialic acid capsule and lipooligosaccharide structure both contribute to the ability of serogroup B Neisseria meningitidis to resist the bactericidal activity of normal human serum. Infect Immun 66:5939–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Virji M, Kayhty H, Ferguson DJ, Alexandrescu C, Heckels JE, Moxon ER. 1991. The role of pili in the interactions of pathogenic Neisseria with cultured human endothelial cells. Mol Microbiol 5:1831–1841. doi: 10.1111/j.1365-2958.1991.tb00807.x. [DOI] [PubMed] [Google Scholar]
- 11.Wall D, Kaiser D. 1999. Type IV pili and cell motility. Mol Microbiol 32:1–10. doi: 10.1046/j.1365-2958.1999.01339.x. [DOI] [PubMed] [Google Scholar]
- 12.Carbonnelle E, Hélaine S, Nassif X, Pelicic V. 2006. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol 61:1510–1522. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- 13.Rudel T, Facius D, Barten R, Scheuerpflug I, Nonnenmacher E, Meyer TF. 1995. Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc Natl Acad Sci U S A 92:7986–7990. doi: 10.1073/pnas.92.17.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hélaine S, Carbonnelle E, Prouvensier L, Beretti J-L, Nassif X, Pelicic V. 2005. PilX, a pilus-associated protein essential for bacterial aggregation, is a key to pilus-facilitated attachment of Neisseria meningitidis to human cells. Mol Microbiol 55:65–77. doi: 10.1111/j.1365-2958.2004.04372.x. [DOI] [PubMed] [Google Scholar]
- 15.Chamot-Rooke J, Mikaty G, Malosse C, Soyer M, Dumont A, Gault J, Imhaus AF, Martin P, Trellet M, Clary G, Chafey P, Camoin L, Nilges M, Nassif X, Dumenil G. 2011. Posttranslational modification of pili upon cell contact triggers N. meningitidis dissemination. Science 331:778–782. doi: 10.1126/science.1200729. [DOI] [PubMed] [Google Scholar]
- 16.Marceau M, Forest K, Béretti J-L, Tainer J, Nassif X. 1998. Consequences of the loss of O-linked glycosylation of meningococcal type IV pilin on piliation and pilus-mediated adhesion. Mol Microbiol 27:705–715. doi: 10.1046/j.1365-2958.1998.00706.x. [DOI] [PubMed] [Google Scholar]
- 17.Mikaty G, Soyer M, Mairey E, Henry N, Dyer D, Forest KT, Morand P, Guadagnini S, Prévost MC, Nassif X, Duménil G. 2009. Extracellular bacterial pathogen induces host cell surface reorganization to resist shear stress. PLoS Pathog 5:e1000314. doi: 10.1371/journal.ppat.1000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deghmane A-E, Giorgini D, Larribe M, Alonso J-M, Taha M-K. 2002. Down-regulation of pili and capsule of Neisseria meningitidis upon contact with epithelial cells is mediated by CrgA regulatory protein. Mol Microbiol 43:1555–1564. doi: 10.1046/j.1365-2958.2002.02838.x. [DOI] [PubMed] [Google Scholar]
- 19.Donovan WP, Kushner SR. 1986. Polynucleotide phosphorylase and ribonuclease II are required for cell viability and mRNA turnover in Escherichia coli K-12. Proc Natl Acad Sci U S A 83:120–124. doi: 10.1073/pnas.83.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sarkar D, Fisher PB. 2006. Polynucleotide phosphorylase: an evolutionary conserved gene with an expanding repertoire of functions. Pharmacol Ther 112:243–263. doi: 10.1016/j.pharmthera.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Bandyra KJ, Bouvier M, Carpousis AJ, Luisi BF. 2013. The social fabric of the RNA degradosome. Biochim Biophys Acta 1829:514–522. doi: 10.1016/j.bbagrm.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stead MB, Marshburn S, Mohanty BK, Mitra J, Castillo LP, Ray D, van Bakel H, Hughes TR, Kushner SR. 2011. Analysis of Escherichia coli RNase E and RNase III activity in vivo using tiling microarrays. Nucleic Acids Res 39:3188–3203. doi: 10.1093/nar/gkq1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Lay N, Gottesman S. 2011. Role of polynucleotide phosphorylase in sRNA function in Escherichia coli. RNA 17:1172–1189. doi: 10.1261/rna.2531211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamanaka K, Inouye M. 2001. Selective mRNA degradation by polynucleotide phosphorylase in cold shock adaptation in Escherichia coli. J Bacteriol 183:2808–2816. doi: 10.1128/JB.183.9.2808-2816.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haddad N, Burns CM, Bolla JM, Prévost H, Fédérighi M, Drider D, Cappelier JM. 2009. Long-term survival of Campylobacter jejuni at low temperatures is dependent on polynucleotide phosphorylase activity. Appl Environ Microbiol 75:7310–7318. doi: 10.1128/AEM.01366-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henry A, Shanks J, van Hoof A, Rosenzweig JA. 2012. The Yersinia pseudotuberculosis degradosome is required for oxidative stress, while its PNPase subunit plays a degradosome-independent role in cold growth. FEMS Microbiol Lett 336:139–147. doi: 10.1111/j.1574-6968.12000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clements MO, Eriksson S, Thompson A, Lucchini S, Hinton JC, Normark S, Rhen M. 2002. Polynucleotide phosphorylase is a global regulator of virulence and persistency in Salmonella enterica. Proc Natl Acad Sci U S A 99:8784–8789. doi: 10.1073/pnas.132047099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ygberg SE, Clements MO, Rytkonen A, Thompson A, Holden DW, Hinton JC, Rhen M. 2006. Polynucleotide phosphorylase negatively controls spv virulence gene expression in Salmonella enterica. Infect Immun 74:1243–1254. doi: 10.1128/IAI.74.2.1243-1254.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenzweig JA, Weltman G, Plano GV, Schesser K. 2005. Modulation of Yersinia type three secretion system by the S1 domain of polynucleotide phosphorylase. J Biol Chem 280:156–163. doi: 10.1074/jbc.M405662200. [DOI] [PubMed] [Google Scholar]
- 30.Rosenzweig JA, Chromy B, Echeverry A, Yang J, Adkins B, Plano GV, McCutchen-Maloney S, Schesser K. 2007. Polynucleotide phosphorylase independently controls virulence factor expression levels and export in Yersinia spp. FEMS Microbiol Lett 270:255–264. doi: 10.1111/j.1574-6968.2007.00689.x. [DOI] [PubMed] [Google Scholar]
- 31.Haddad N, Tresse O, Rivoal K, Chevret D, Nonglaton Q, Burns CM, Prévost H, Cappelier JM. 2012. Polynucleotide phosphorylase has an impact on cell biology of Campylobacter jejuni. Front Cell Infect Microbiol 2:30. doi: 10.3389/fcimb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palanisamy SK, Fletcher C, Tanjung L, Katz ME, Cheetham BF. 2010. Deletion of the C-terminus of polynucleotide phosphorylase increases twitching motility, a virulence characteristic of the anaerobic bacterial pathogen Dichelobacter nodosus. FEMS Microbiol Lett 302:39–45. doi: 10.1111/j.1574-6968.2009.01831.x. [DOI] [PubMed] [Google Scholar]
- 33.Rouf SF, Ahmad I, Anwar N, Vodnala SK, Kader A, Römling U, Rhen M. 2011. Opposing contributions of polynucleotide phosphorylase and the membrane protein NlpI to biofilm formation by Salmonella enterica serovar Typhimurium. J Bacteriol 193:580–582. doi: 10.1128/JB.00905-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carzaniga T, Antoniani D, Dehò G, Briani F, Landini P. 2012. The RNA processing enzyme polynucleotide phosphorylase negatively controls biofilm formation by repressing poly-N-acetylglucosamine (PNAG) production in Escherichia coli C. BMC Microbiol 12:270. doi: 10.1186/1471-2180-12-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hey A, Li M-S, Hudson MJ, Langford PR, Kroll JS. 2013. Transcriptional profiling of Neisseria meningitidis interacting with human epithelial cells in a long-term in vitro colonization model. Infect Immun 81:4149–4159. doi: 10.1128/IAI.00397-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahman M, Källström H, Normark S, Jonsson AB. 1997. PilC of pathogenic Neisseria is associated with the bacterial cell surface. Mol Microbiol 25:11–25. doi: 10.1046/j.1365-2958.1997.4601823.x. [DOI] [PubMed] [Google Scholar]
- 37.Kellogg DS Jr, Cohen IR, Norins LC, Schroeter AL, Reising G. 1968. Neisseria gonorrhoeae. II. Colonial variation and pathogenicity during 35 months in vitro. J Bacteriol 96:596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fontanella L, Pozzuolo S, Costanzo A, Favaro R, Dehò G, Tortora P. 1999. Photometric assay for polynucleotide phosphorylase. Anal Biochem 269:353–358. doi: 10.1006/abio.1999.4042. [DOI] [PubMed] [Google Scholar]
- 39.Kuwae A, Sjölinder H, Eriksson J, Eriksson S, Chen Y, Jonsson AB. 2011. NafA negatively controls Neisseria meningitidis piliation. PLoS One 6:e21749. doi: 10.1371/journal.pone.0021749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elkins C, Thomas CE, Seifert HS, Sparling PF. 1991. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J Bacteriol 173:3911–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sjölinder H, Jonsson AB. 2007. Imaging of disease dynamics during meningococcal sepsis. PLoS One 2:e241. doi: 10.1371/journal.pone.0000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolstad BM, Irizarry RA, Åstrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 43.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 44.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rusniok C, Vallenet D, Floquet S, Ewles H, Mouze-Soulama C, Brown D, Lajus A, Buchrieser C, Medigue C, Glaser P, Pelicic V. 2009. NeMeSys: a biological resource for narrowing the gap between sequence and function in the human pathogen Neisseria meningitidis. Genome Biol 10:R110. doi: 10.1186/gb-2009-10-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jones A, Geörg M, Maudsdotter L, Jonsson AB. 2009. Endotoxin, capsule, and bacterial attachment contribute to Neisseria meningitidis resistance to the human antimicrobial peptide LL-37. J Bacteriol 191:3861–3868. doi: 10.1128/JB.01313-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ly MH, Vo NH, Le TM, Belin JM, Waché Y. 2006. Diversity of the surface properties of lactococci and consequences on adhesion to food components. Colloids Surf B Biointerfaces 52:149–153. doi: 10.1016/j.colsurfb.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 48.Albiger B, Johansson L, Jonsson A-B. 2003. Lipooligosaccharide-deficient Neisseria meningitidis shows altered pilus-associated characteristics. Infect Immun 71:155–162. doi: 10.1128/IAI.71.1.155-162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johansson L, Rytkonen A, Bergman P, Albiger B, Kallstrom H, Hokfelt T, Agerberth B, Cattaneo R, Jonsson AB. 2003. CD46 in meningococcal disease. Science 301:373–375. doi: 10.1126/science.1086476. [DOI] [PubMed] [Google Scholar]
- 50.Johansson L, Rytkonen A, Wan H, Bergman P, Plant L, Agerberth B, Hokfelt T, Jonsson AB. 2005. Human-like immune responses in CD46 transgenic mice. J Immunol 175:433–440. doi: 10.4049/jimmunol.175.1.433. [DOI] [PubMed] [Google Scholar]
- 51.Eriksson J, Eriksson OS, Maudsdotter L, Palm O, Engman J, Sarkissian T, Aro H, Wallin M, Jonsson A-B. 2015. Characterization of motility and piliation in pathogenic Neisseria. BMC Microbiol 15:92. doi: 10.1186/s12866-015-0424-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Del Favero M, Mazzantini E, Briani F, Zangrossi S, Tortora P, Dehò G. 2008. Regulation of Escherichia coli polynucleotide phosphorylase by ATP. J Biol Chem 283:27355–27359. doi: 10.1074/jbc.C800113200. [DOI] [PubMed] [Google Scholar]
- 53.Mohanty BK, Kushner SR. 2003. Genomic analysis in Escherichia coli demonstrates differential roles for polynucleotide phosphorylase and RNase II in mRNA abundance and decay. Mol Microbiol 50:645–658. doi: 10.1046/j.1365-2958.2003.03724.x. [DOI] [PubMed] [Google Scholar]
- 54.Bartley SN, Tzeng Y-L, Heel K, Lee CW, Mowlaboccus S, Seemann T, Lu W, Lin Y-H, Ryan CS, Peacock C, Stephens DS, Davies JK, Kahler CM. 2013. Attachment and invasion of Neisseria meningitidis to host cells is related to surface hydrophobicity, bacterial cell size and capsule. PLoS One 8:e55798. doi: 10.1371/journal.pone.0055798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Virji M, Makepeace K, Ferguson DJP, Achtman M, Moxon ER. 1993. Meningococcal Opa and Opc proteins: their role in colonization and invasion of human epithelial and endothelial cells. Mol Microbiol 10:499–510. doi: 10.1111/j.1365-2958.1993.tb00922.x. [DOI] [PubMed] [Google Scholar]
- 56.Virji M, Makepeace K, Peak IRA, Ferguson DJP, Jennings MP, Moxon ER. 1995. Opc- and pilus-dependent interactions of meningococci with human endothelial cells: molecular mechanisms and modulation by surface polysaccharides. Mol Microbiol 18:741–754. doi: 10.1111/j.1365-2958.1995.mmi_18040741.x. [DOI] [PubMed] [Google Scholar]
- 57.Imhaus A-F, Duménil G. 2014. The number of Neisseria meningitidis type IV pili determines host cell interaction. EMBO J 33:1767–1783. doi: 10.15252/embj.201488031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taktikos J, Lin YT, Stark H, Biais N, Zaburdaev V. 2015. Pili-induced clustering of N. gonorrhoeae bacteria. PLoS One 10:e0137661. doi: 10.1371/journal.pone.0137661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merz AJ, So M, Sheetz MP. 2000. Pilus retraction powers bacterial twitching motility. Nature 407:98–102. doi: 10.1038/35024105. [DOI] [PubMed] [Google Scholar]
- 60.Carbonnelle E, Hélaine S, Prouvensier L, Nassif X, Pelicic V. 2005. Type IV pilus biogenesis in Neisseria meningitidis: PilW is involved in a step occurring after pilus assembly, essential for fibre stability and function. Mol Microbiol 55:54–64. doi: 10.1111/j.1365-2958.2004.04364.x. [DOI] [PubMed] [Google Scholar]
- 61.Nurmohamed S, Vincent HA, Titman CM, Chandran V, Pears MR, Du D, Griffin JL, Callaghan AJ, Luisi BF. 2011. Polynucleotide phosphorylase activity may be modulated by metabolites in Escherichia coli. J Biol Chem 286:14315–14323. doi: 10.1074/jbc.M110.200741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fagnocchi L, Bottini S, Golfieri G, Fantappiè L, Ferlicca F, Antunes A, Guadagnuolo S, Del Tordello E, Siena E, Serruto D, Scarlato V, Muzzi A, Delany I. 2015. Global transcriptome analysis reveals small RNAs affecting Neisseria meningitidis bacteremia. PLoS One 10:e0126325. doi: 10.1371/journal.pone.0126325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pujol C, Eugène E, Marceau M, Nassif X. 1999. The meningococcal PilT protein is required for induction of intimate attachment to epithelial cells following pilus-mediated adhesion. Proc Natl Acad Sci U S A 96:4017–4022. doi: 10.1073/pnas.96.7.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eriksson J, Eriksson OS, Jonsson A-B. 2012. Loss of meningococcal PilU delays microcolony formation and attenuates virulence in vivo. Infect Immun 80:2538–2547. doi: 10.1128/IAI.06354-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.