

Abstract
Coxiella burnetii is an intracellular bacterial pathogen that causes human Q fever, an acute debilitating flu-like illness that can also present as chronic endocarditis. Disease typically occurs following inhalation of contaminated aerosols, resulting in an initial pulmonary infection. In human cells, C. burnetii generates a replication niche termed the parasitophorous vacuole (PV) by directing fusion with autophagosomes and lysosomes. C. burnetii requires this lysosomal environment for replication and uses a Dot/Icm type IV secretion system to generate the large PV. However, we do not understand how C. burnetii evades the intracellular immune surveillance that triggers an inflammatory response. We recently characterized human alveolar macrophage (hAM) infection in vitro and found that avirulent C. burnetii triggers sustained interleukin-1β (IL-1β) production. Here, we evaluated infection of ex vivo human lung tissue, defining a valuable approach for characterizing C. burnetii interactions with a human host. Within whole lung tissue, C. burnetii preferentially replicated in hAMs. Additionally, IL-1β production correlated with formation of an apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC)-dependent inflammasome in response to infection. We also assessed potential activation of a human-specific noncanonical inflammasome and found that caspase-4 and caspase-5 are processed during infection. Interestingly, although inflammasome activation is closely linked to pyroptosis, lytic cell death did not occur following C. burnetii-triggered inflammasome activation, indicating an atypical response after intracellular detection. Together, these studies provide a novel platform for studying the human innate immune response to C. burnetii.
INTRODUCTION
Coxiella burnetii is a Gram-negative, obligate intracellular pathogen that causes Q fever in humans (1). Acute Q fever presents with flu-like symptoms; however, infection can persist and cause life-threatening endocarditis. C. burnetii is a category B select agent due to an aerosol mode of transmission, low infectious dose, and environmental stability (2). The pathogen has a unique intracellular lifestyle that requires replication within an acidic lysosome-like parasitophorous vacuole (PV) in macrophages (3). Using a Dot/Icm type IV secretion system (T4SS) to secrete bacterial proteins into the host cytoplasm (4, 5), C. burnetii hijacks the host cell to control signaling cascades and heterotypic fusion with endosomes, autophagosomes, and lysosomes to establish the PV (6). Although C. burnetii intracellular trafficking has been well characterized, understanding of the initial interactions between the pathogen and its human host is lacking. Although small animal models provide beneficial information about the host response to C. burnetii, they do not accurately mimic the human lung response to infection, presenting the need for improved systems. For example, mouse alveolar macrophages degrade C. burnetii (7), while the pathogen replicates efficiently in primary human alveolar macrophages (hAMs) (8).
Macrophages are critical for Q fever development and dictate disease outcome by responding to invading pathogens. Macrophages detect bacteria using extracellular Toll-like receptors (TLRs) and intracellular nucleotide-binding oligomerization domain proteins (NODs). TLR signaling ultimately results in nuclear factor-κB (NF-κB) activation and expression of genes encoding proinflammatory cytokines such as interleukin-1β (IL-1β). TLRs recognize bacterial lipopolysaccharide (LPS), composed of lipid A and O antigen. The lipid A moiety of LPS is a conserved invariant portion present in all Gram-negative bacteria and is recognized by TLRs, while the O antigen varies among species and is not typically recognized (9). The LPS structure differs between virulent and avirulent C. burnetii strains and is a major determinant of Q fever (10). Virulent organisms, referred to as being in phase I, produce full-length LPS and cause significant disease in guinea pigs and humans. However, continued passage in culture results in truncation of LPS, resulting in attenuation of the pathogen and its characterization as phase II. One clonal phase II isolate of C. burnetii (clone 4, RSA439) is exempt from select agent regulations and has been used extensively to study the cell biology of infection. However, we recently demonstrated differing primary hAM cytokine responses to C. burnetii pathotypes (8), indicating the need for comparison of virulent and avirulent bacteria.
In addition to TLRs, cytosolic inflammasomes form in response to microbes and result in caspase activation (11, 12). Active caspases process the proinflammatory cytokines IL-1β and IL-18 to mature secreted forms and promote pyroptotic cell death (13, 14). This response typically requires an initial signal that triggers expression of pro-IL-1β and pro-IL-18 and a second signal that initiates complex formation (15). A canonical inflammasome contains a NOD-like receptor or PYHIN (pyrin domain and hematopoietic expression, interferon-inducible nature and nuclear localization [HIN] domain-containing), the adapter molecule apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), and caspase-1 (16). Although the inflammasome-mediated response has been described for many intracellular pathogen systems, there is a lack of information regarding intracellular sensing of C. burnetii.
In this study, we defined a novel approach to study C. burnetii infection using ex vivo human lung tissue and primary hAMs. Using these infection platforms, we demonstrated that avirulent C. burnetii specifically replicates in hAMs within the lung environment and triggers caspase-dependent IL-1β production. We also discovered activation of a potential noncanonical caspase-5-dependent hAM inflammasome that may direct secretion of IL-1β in response to C. burnetii. These findings advance understanding of the innate immune response to C. burnetii in the human lung.
MATERIALS AND METHODS
Bacteria and eukaryotic cell culture.
C. burnetii NMII (RSA439, avirulent clone 4) was cultured in acidified citrate cysteine medium (ACCM) for 7 days at 37°C with 5% CO2 and 2.5% O2. Cultures were washed three times and resuspended in sucrose phosphate (SP) buffer prior to infection experiments. All experiments using virulent C. burnetii were performed in the Centers for Disease Control and Prevention-approved biosafety level 3 facility at the University of Arkansas for Medical Sciences. hAMs were infected with a multiplicity of infection of 10, and precision-cut lung slices (PCLS) were infected with 1.5 × 107 bacteria by addition of organisms to the medium (0 h postinfection [hpi]). At 24 hpi, cells were washed to remove extracellular bacteria.
Primary hAMs were obtained by bronchoalveolar lavage (BAL) from human lungs acquired from the National Disease Research Interchange postmortem. BAL fluid cells were collected by centrifugation and red blood cells lysed by incubation with 0.86% ammonium chloride for 10 min. Ammonium chloride was neutralized by addition of culture medium and removed by centrifugation. Cells were resuspended in Dulbecco's modified Eagle's medium-F12 (DMEM-F12) containing 10% fetal bovine serum (FBS), penicillin (50 U/ml), streptomycin sulfate (50 μg/ml), gentamicin sulfate (50 μg/ml), and amphotericin B (0.25 μg/ml) (Invitrogen). Cells were allowed to adhere to tissue culture dishes for 2 h, and then nonadherent cells were removed by washing, leaving a >95% population of hAMs as confirmed by immunofluorescence microscopy (data not shown).
After BAL, lungs were filled with low-melting-point agarose and allowed to solidify at 4°C. Lung tissue was then processed into two-inch sections. Sections were then cored with an 8-mm-diameter tool, and tissue was sectioned into 750-μm precision-cut lung slices (PCLS) using a vibrating microtome (Compresstome; Precisionary Instruments). PCLS were incubated with shaking in DMEM-F12 containing antibiotics and antimycotics at 37°C and 5% CO2. PCLS and hAMs were cultured in antibiotic- and antimycotic-containing medium for 3 days, and then antibiotic- and antimycotic-free medium was added and samples incubated for an additional 24 h before infection to ensure absence of contamination.
Confocal fluorescence microscopy.
hAMs or PCLS were fixed in 4% paraformaldehyde (PFA) and blocked in solution containing 0.5% bovine serum albumin (BSA) and 0.3% Triton X-100 for 1 h at room temperature. Samples were then incubated with primary antibodies in blocking solution for 1 h at room temperature, followed by Alexa Fluor-conjugated secondary antibodies (Alexa Fluor-488, -594, or -647) for 1 h. Caspase-1 antibody (Cell Signaling) was used at a concentration of 1:100. ASC (Santa Cruz Biotechnologies) and IL-1β (R&D Systems) antibodies were used at a concentration of 1:50. Cells were imaged using a Nikon Ti-Eclipse confocal microscope and images analyzed with Nikon Elements software.
Immunoblot analysis.
Following infection, hAMs were lysed in buffer containing 1% SDS by passage through a 26-gauge needle. Total protein was quantified using a DC protein assay (Bio-Rad) and 10 μg of total protein separated by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a 0.2-μm polyvinylidene fluoride membrane and blocked overnight in Tris-buffered saline (150 mM NaCl, 100 mM Tris-HCl, pH 7.6) containing 0.1% Tween 20 and 5% nonfat milk. After blocking, total protein levels were confirmed by probing with mouse β-tubulin antibody (Sigma). Lysates were probed with caspase-4 or caspase-5 antibodies (Cell Signaling) or an IL-1β antibody (R&D Systems). Appropriate secondary antibodies conjugated to horseradish peroxidase were used with ECL-Plus chemiluminescence reagent to visualize proteins. Positive-control cells were treated with Escherichia coli LPS (100 ng/ml; Sigma) for 6 h followed by ATP (5 mM; Sigma) for 30 min.
Cytokine analysis.
IL-1β enzyme-linked immunosorbent assays (ELISAs) (BD Biosciences) were performed using supernatants harvested from infected cells or PCLS. Precoated wells were incubated with samples for 2 h at room temperature and then washed, and detection antibody was added and left for 1 h. After removing excess antibody, samples were fixed in 4% paraformaldehyde (PFA) for 30 min to inactivate any remaining virulent bacteria. PFA was removed, and enzyme working reagent was added and left for 30 min, followed by washing and treatment with tetramethylbenzidine (TMB) one-step substrate for 30 min. The reaction was then stopped with stop solution and absorbance measured at 450 nm using a BioTek H2 Synergy plate reader. A standard curve was established using duplicate standards ranging from 0 to 125 pg/ml, and unknown sample concentrations were calculated.
FLICA activity assay.
hAMs (1.0 ×105 cells/well) were cultured in black-wall 96-well glass-bottom plates and either left uninfected and untreated or infected for the indicated times. Uninfected hAMs were treated with E. coli LPS and ATP as described above (positive control). Cells were then incubated with 6-carboxyfluorescein (FAM)–caspase-1 fluorescein (FLICA) (Immunochemistry Technologies) for 60 min at 37°C. Unbound probe was removed by adding medium to wells and incubating for 60 min to allow unbound probe to diffuse from cells. Medium was then aspirated, cells placed in phosphate-buffered saline (PBS), and fluorescence measured using a BioTek H2 Synergy plate reader (excitation, 490 nm; emission, 525 nm).
LDH assay.
hAMs (4 ×105/well) were plated in 24-well plates. hAMs were then either left uninfected and untreated or infected for the indicated times. Positive-control hAMs were treated with E. coli LPS and ATP as described above. Supernatants were then harvested and lactate dehydrogenase (LDH) measured using a CytoTox 96 nonradioactive cytotoxicity assay (Promega) according to the manufacturer's protocol. The positive control was set to 100%, and values for experimental samples were calculated as percentages of the positive-control value.
RNA isolation and quantitative reverse transcription-PCR (qRT-PCR).
Uninfected or infected hAMs were harvested in TRIzol (Invitrogen) by repeated pipetting. Phase separation was performed by addition of 0.2 ml chloroform/ml of TRIzol. Tubes were shaken, and then samples were centrifuged at 12,000 × g for 15 min at 4°C. The aqueous phase was collected and RNA precipitated with 1 volume of 70% ethanol. RNA was then isolated using an RNeasy kit according to the manufacturer's instructions (Qiagen), performing two on-column DNase digestions (Qiagen). Total RNA (500 ng) was converted to cDNA using a Superscript III first-strand synthesis kit (Life Technologies) and random hexamers. TaqMan-based PCR was performed for each sample using primers and probe (Life Technologies) specific for each gene of interest and TaqMan Fast Advanced master mix (Life Technologies). Samples were normalized to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene and compared to uninfected samples.
Statistical analysis.
All experiments were performed with material from at least three separate donors. Statistical analyses were performed using Prism 6 software and two-way analysis of variance (ANOVA).
RESULTS
C. burnetii preferentially replicates within hAMs in ex vivo human lung tissue.
We recently developed a primary hAM approach to study C. burnetii host-pathogen interactions in primary human cells (8). This study showed that virulent and avirulent C. burnetii strains replicate to high numbers in hAMs and trigger pathotype-specific innate inflammatory responses. However, no previous study has proven that alveolar macrophages are the pathogen's target cell in vivo. To determine if hAMs are the true C. burnetii target cell, we extended our studies to intact ex vivo human lung tissue to study initial interactions between C. burnetii and its human host. Lung tissue was sectioned into 750-μm-thick slices termed precision-cut lung slices (PCLS) (17) and incubated in tissue culture plates. We visualized PCLS by bright-field microscopy (Fig. 1A and B) and found cells morphologically indicative of macrophages in alveolar spaces (Fig. 1C, arrowhead). PCLS were then infected with C. burnetii for 72 h and visualized by bright-field microscopy, which indicated large bacterium-containing vacuoles in cells on the walls of alveolar spaces (Fig. 1B). It is important to note that the tissue structure was maintained during infection, suggesting that C. burnetii does not trigger disruption of the lung structure during intracellular growth, unlike the damage resulting from other bacterial respiratory infections (18, 19).
FIG 1.

Cells morphologically similar to alveolar macrophages are present in PCLS. PCLS were incubated with avirulent C. burnetii for 72 h and then processed for histology and observed by bright-field microscopy. Bars, 50 μm. (A) Four-micrometer-sectioned PCLS showing alveolar spaces (AS) and an airway (AW). (B) AS containing an expanded C. burnetii PV (arrowhead). (C) AS containing dense cells morphologically similar to macrophages (arrowhead).
To determine if cells that support C. burnetii replication are macrophages, PCLS were infected for 72 h and then processed for confocal fluorescence microscopy. As shown in Fig. 2A, a subset of alveolar cells contained an expanded CD63-positive PV that supported robust C. burnetii replication. Accumulation of bacteria in these PVs was not the result of a large inoculum, as bacterial numbers continually increased throughout the infection time course and type IV secretion system-defective C. burnetii did not accumulate in macrophages (data not shown). Although individual C. burnetii cells were present in numerous alveolar cells, only macrophages supported bacterial replication, as indicated by the presence of expanded PVs only in cells that were labeled with the macrophage marker CD68 (Fig. 2B). These results demonstrate that C. burnetii specifically targets alveolar macrophages for replication in human lung tissue.
FIG 2.

C. burnetii replicates specifically within alveolar macrophages in human PCLS. PCLS were incubated with avirulent C. burnetii for 72 h and then processed for confocal fluorescence microscopy using antibodies directed against the PV marker CD63 (A) or the macrophage marker CD68 (B) and C. burnetii (red). Arrowheads indicate macrophages that do not harbor C. burnetii. DAPI (4′,6′-diamidino-2-phenylindole) was used to stain DNA (blue), and colors are shown in the images. Bar, 10 μm. C. burnetii replicates in a CD63-positive, lysosome-like PV only in CD68-positive alveolar macrophages.
Avirulent C. burnetii triggers prolonged IL-1β production in hAMs and PCLS.
We previously discovered that avirulent, but not virulent, C. burnetii triggers robust IL-1β production by primary hAMs (8), a response that is critical for clearing many bacterial pathogens (20). To further characterize the human innate response to C. burnetii, we monitored IL-1β production and secretion during infection of PCLS or isolated hAMs. Avirulent C. burnetii triggered a robust and prolonged IL-1β response, as evidenced by increased mRNA transcripts from 2 to 24 hpi (data not shown), increased production of pro-IL-1β and mature IL-1β in whole-cell lysates from 24 to 48 hpi (Fig. 3A and B) using immunoblot analysis, and increased levels of secreted IL-1β in supernatants of infected hAMs from 24 to 72 hpi (Fig. 3C) using an ELISA. Additionally, IL-18 transcripts increased similarly to those of IL-1β (data not shown). Furthermore, IL-1β production required live C. burnetii, as heat-killed bacteria did not trigger cytokine production (data not shown). We next assessed the IL-1β response during infection of PCLS to determine if C. burnetii elicits a proinflammatory response during replication in the lung environment. Confirming the hAM results, we observed similar responses to avirulent C. burnetii in PCLS from 24 to 96 hpi (Fig. 3D), indicating that this sustained proinflammatory innate response to avirulent organisms occurs in a context relevant to human disease.
FIG 3.

Avirulent C. burnetii triggers caspase-dependent IL-1β production in hAMs and PCLS. (A and B) hAMs were infected with avirulent C. burnetii for the indicated times in the absence (A) or presence (B) of the caspase-1 inhibitor YVAD-CHO. Samples were subjected to immunoblot analysis using an IL-1β antibody, and the results shown are representative of three individual experiments. UI, uninfected cells; +, cells treated with LPS and ATP. Mature IL-1β production does not occur in the presence of YVAD-CHO, suggesting that caspase-1 activity is required. (C and D) hAMs (C) or PCLS (D) were left uninfected (UI), treated with LPS and ATP (+), or infected for the indicated times, and supernatants were harvested. YVAD-CHO was added where indicated, and IL-1β was detected in supernatants by sandwich ELISA. Avirulent C. burnetii triggers secretion of IL-1β by infected hAMs and PCLS, and this response is inhibited by YVAD-CHO. Error bars indicate the standard deviation from the mean; *, P < 0.05 according to two-way ANOVA comparing infected cells to uninfected and YVAD-CHO-treated cells. These results indicate that avirulent C. burnetii triggers IL-1β production and secretion through a caspase-dependent mechanism in hAMs and PCLS.
Macrophage recognition of intracellular bacteria involves activation of a specific inflammasome in response to bacterial products such as LPS, flagellin, and secreted proteins (12, 21). Inflammasome complexes are composed of a NOD-like protein, a caspase, and, depending on the inflammasome, an adapter molecule (21, 22). Following activation, caspase-1 processes pro-IL-1β to mature IL-1β that is secreted from the cell to trigger an immune response (13). To determine if the mechanism of IL-1β production in response to avirulent C. burnetii involves canonical activation of caspase-1 (23), we used the established caspase-1 peptide inhibitor AC-YVAD-CHO during infection (24). When caspase-1 activity was inhibited, mature IL-1β production and secretion were prevented in hAMs and PCLS (Fig. 3B to D). However, using an established FLICA assay, no significant caspase-1 activation was observed (Fig. 4A). Additionally, using a lactate dehydrogenase release assay (Fig. 4B), levels of infected-cell death did not significantly increase compared to those for uninfected cells, similar to the results of our previous apoptosis studies (25–27). This is unexpected if caspase-1 is activated, as this event typically triggers pyroptosis, an inflammatory form of cell death (23). Thus, C. burnetii may activate a noncanonical caspase-1-independent inflammasome or may prevent caspase-1-mediated death.
FIG 4.

C. burnetii infection does not elicit elevated caspase-1 activity or promote cell lysis indicative of pyroptosis. hAMs were left uninfected (UI), treated with LPS and ATP (+), or infected with avirulent C. burnetii for the indicated times. (A) Active caspase-1 was detected with a fluorescent FAM-FLICA probe. Avirulent C. burnetii does not trigger significant caspase-1 activity. (B) LDH present in the supernatant was measured as an indicator of cell lysis. Values for experimental samples were calculated as percentages of the positive-control value (set to 100%). Data are representative of at least three separate experiments using cells from at least two separate donors, and error bars represent the standard deviation from the mean. n.s., not significantly different from value for uninfected cells. C. burnetii-infected cells do not release significantly more LDH than uninfected hAMs.
Caspase-1 and ASC colocalize with IL-1β at the PV membrane.
Inflammasome formation can be assessed by microscopy to detect formation of characteristic specks containing caspase-1, IL-1β, a NOD-like protein, and the adapter protein ASC (23). To assess inflammasome formation, we used confocal fluorescence microscopy to observe caspase-1, IL-1β, and ASC localization during infection. In avirulent C. burnetii-infected hAMs, caspase-1 colocalized with IL-1β in characteristic specks that were in close proximity to intracellular bacteria (Fig. 5A). Specks containing ASC and IL-1β were also evident in C. burnetii-infected hAMs (Fig. 5B). Interestingly, a portion of ASC-containing specks were present within the C. burnetii PV, suggesting that some inflammasomes are trafficked into the degradative lysosome-like vacuole. Combined with the caspase inhibitor data, these results suggest that avirulent C. burnetii triggers ASC-dependent inflammasome activation and caspase-dependent production of IL-1β in primary hAMs and PCLS.
FIG 5.
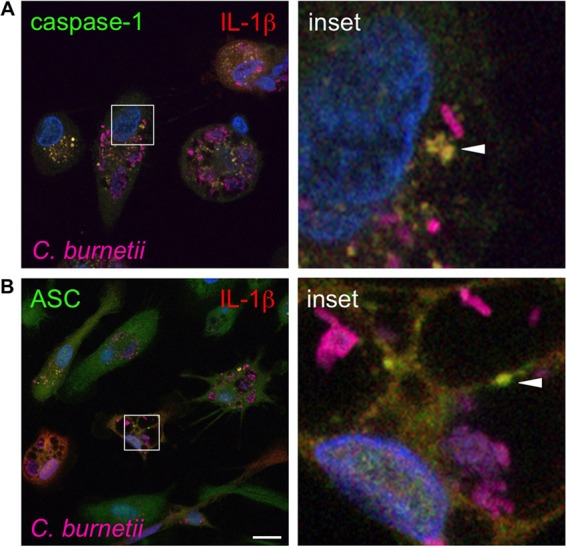
Caspase-1 and ASC colocalize with IL-1β near avirulent C. burnetii. hAMs were infected with avirulent C. burnetii for 72 h and then processed for confocal fluorescence microscopy using antibodies directed against caspase-1 (A), ASC (B), and IL-1β and C. burnetii. DAPI was used to stain DNA (blue), and colors are shown in images. Bar, 10 μm. Caspase-1 and ASC colocalize with IL-1β (yellow; arrowheads) near C. burnetii, suggesting that inflammasomes are located near the PV for detection of avirulent bacteria.
C. burnetii triggers increased expression of nlrp3, nod2, and casp5.
To probe the type of inflammasome activated in response to avirulent C. burnetii, we used an RT-PCR array to determine inflammasome- and caspase-related gene expression. Inflammasome activation is not typically controlled at the transcriptional level; however, this analysis provides clues about increased expression of products potentially involved in bacterial recognition. As shown in Fig. 6A, expression of nlrp3 and nod2 increased significantly during avirulent C. burnetii infection compared to that in uninfected hAMs, suggesting that an NLRP3 inflammasome is involved in detecting avirulent organisms.
FIG 6.

C. burnetii infection triggers increased expression of specific inflammasome-related genes. hAMs were infected with avirulent C. burnetii for 24 h and total RNA isolated. The resulting cDNA was subjected to analysis using an RT-PCR array specific for relevant inflammasome-related genes (A) or caspase-related genes (B). Transcript levels were compared to uninfected hAMs (UI), and the dashed line in each graph denotes the 2-fold cutoff. Data represent two separate experiments using cells from two separate donors, and error bars represent the standard error. Avirulent C. burnetii triggers increased transcription of nlrp3, nod2, and casp5.
Caspase-1 peptide inhibitors such as YVAD-CHO target the catalytic site of caspase-1, which is homologous to the catalytic sites of caspase-4 and caspase-5 (24). Due to low levels of caspase-1 activity and cell death during avirulent C. burnetii infection, we sought to determine if other caspases are involved in intracellular detection of the pathogen. Indeed, the closely related Legionella pneumophila and intracellular Salmonella are detected by an inflammasome requiring murine caspase-11, the murine homolog to human caspase-4 and caspase-5 (28, 29). To assess caspase involvement in infection, we monitored expression of caspase-related genes during avirulent C. burnetii infection using an RT-PCR array. As shown in Fig. 6B, avirulent C. burnetii infection of hAMs elicited increased expression of casp5, but not casp1, casp8, card6, or card18. Together, these results suggest that a novel caspase-5-containing noncanonical NLRP3 inflammasome is activated in response to avirulent C. burnetii.
C. burnetii elicits caspase-4 and caspase-5 processing in hAMs.
Similar to other caspases, caspase-4 and caspase-5 are processed from a pro form (∼50 kDa) to an active protein (∼10 to 20 kDa) to control cellular events. Because casp5 expression increased during avirulent C. burnetii infection, activation was monitored by immunoblotting in hAMs. As shown in Fig. 7, caspase-4 processing was observed at 48 hpi, a time following IL-1β production in avirulent C. burnetii-infected hAMs. However, caspase-5 processing showed kinetics similar to those for IL-1β production, with processed caspase apparent by 24 hpi. Unfortunately, attempts to silence expression of casp5 were unsuccessful due to poor hAM transfection efficiency (data not shown). However, these results indicate that caspase-5 activation corresponds to IL-1β secretion during avirulent C. burnetii infection and may be involved in detection of the pathogen.
FIG 7.
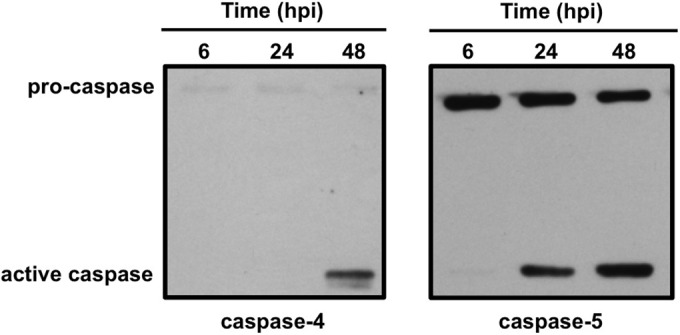
Avirulent C. burnetii triggers caspase-4 and caspase-5 processing. hAMs were infected with avirulent C. burnetii for 6 to 48 h and samples subjected to immunoblot analysis using antibodies directed against caspase-4 or caspase-5. Caspase-5 processing is evident from 24 to 48 hpi, and active caspase-4 is present at 48 hpi, suggesting noncanonical inflammasome activation during C. burnetii infection involving caspase-5.
DISCUSSION
Here, we show that primary hAMs and PCLS mount a proinflammatory IL-1β response following exposure to avirulent C. burnetii. We previously showed that this response is absent when hAMs are infected with virulent C. burnetii (8), demonstrating a major difference in the cellular response to disease-causing and attenuated bacteria. In over 70 years since its discovery as the causative agent of Q fever (30–32), the human immune response to C. burnetii has remained difficult to understand. This difficulty is due, in part, to the fact that ∼50% of infected individuals remain asymptomatic, suggesting that an intact immune system can effectively combat the pathogen. Unfortunately, we do not understand how C. burnetii avoids the immune response to cause acute or chronic disease in symptomatic cases.
Animal models have previously been used to provide information about the importance of specific cell types and cytokines in the anti-C. burnetii response. Indeed, using various species, researchers have shown that tumor necrosis factor alpha (TNF-α), gamma interferon (IFN-γ), and T cell activation play a role in the anti-C. burnetii response (33). However, most mice are refractory to infection, and few reagents are available to study infection of guinea pigs, the most disease-relevant small animal model of Q fever. To study the human lung response to C. burnetii, we developed human PCLS as a novel infection platform. Our findings indicate that PCLS infection is a disease-relevant approach to studying C. burnetii innate interactions with humans. Using PCLS, we show, for the first time, that hAMs are the only cell type that supports C. burnetii intracellular replication in the lung. Interestingly, the pathogen enters other alveolar cells but does not replicate. In contrast, C. burnetii replicates in most cell types in vitro, including alveolar epithelial cells. It is possible that infected hAMs alert bystander cells to the presence of C. burnetii, allowing other cells to mount effective intracellular defenses against the pathogen, and this possibility is under investigation.
Primary hAMs and PCLS allowed characterization of the IL-1β response to avirulent C. burnetii, which is not triggered by virulent isolates. Inflammasomes are intracellular sensors of microbes and control responses that alert other immune cells. Multiple inflammasomes have been described, and intracellular pathogens typically activate a specific type. For example, L. pneumophila flagellin is recognized by an NLRC4 inflammasome. NLRC4 coordinates caspase-1 activation and resultant IL-1β production (34, 35). In addition to caspase-1, caspase-11 recognizes L. pneumophila-containing vacuoles and restricts growth of the pathogen (29). Furthermore, caspase-4 is activated in cells infected with Salmonella spp., leading to clearance of the pathogen from the intestinal epithelium (28, 29). Caspase-1 activation is typically triggered by stimulation of an intracellular NOD-like receptor that promotes assembly of an inflammasome that recruits and activates the caspase (14, 21, 22). Although we do not know the specific inflammasome that responds to C. burnetii, TLR ligation is the first step in the IL-1β response, and avirulent C. burnetii likely triggers TLR-2 signaling, as the organism does not activate TLR-4 (36, 37).
C. burnetii recognition by hAMs alters inflammasome-related gene expression, including increased expression of nlrp3, nod2, and casp5. These data support NLRP3-dependent detection of C. burnetii. Furthermore, caspase-1 and ASC colocalize with IL-1β close to the PV. It is currently not known whether C. burnetii actively recruits caspase-1 and ASC to the PV; however, a subset of inflammasome specks appears in the PV lumen, suggesting that they are delivered into the vacuole. The PV is a degradative environment (38) and, C. burnetii may recruit inflammasomes to counteract intracellular detection and the proinflammatory response. Indeed, we recently demonstrated that C. burnetii uses T4SS effectors to recruit autophagosomes to the PV (39), and inflammasome turnover is controlled by autophagy in other systems (15).
The mechanism of virulent C. burnetii suppression of IL-1β production is under investigation and represents a major difference in the hAM response to virulent and avirulent bacteria. The best characterized difference between isolates lies in their LPS, with avirulent bacteria producing severely truncated LPS that stimulates TLR signaling (40, 41). However, LPS differences alone are not entirely responsible for promoting IL-1β production, as infection of hAMs with virulent C. burnetii triggers initial production of pro-IL-1β and barely detectable mature IL-1β (8), suggesting that the pathogen combats the response after activation. It is possible that virulent C. burnetii produces a subset of proteins not present in avirulent C. burnetii. Indeed, a genome study by Beare et al. found a group of genes containing single nucleotide polymorphisms (SNPs) present in NMII (avirulent) compared to NMI (virulent) C. burnetii (42, 43). If these SNPs alter protein production, NMI may produce proteins that prevent inflammasome activation. Genes containing SNPs are under investigation to determine if they alter inflammasome-mediated detection of virulent C. burnetii.
To our knowledge, this is the first study to implicate caspase-5 in macrophage detection of intracellular bacteria. Notably, this discovery would not have been possible without our hAM infection platform, because caspase-4 and caspase-5 are human-specific proteins. Additionally, caspase-5 is undetectable in a THP-1 human macrophage-like cell line model (data not shown). casp5 expression increases substantially during avirulent C. burnetii infection. Previous studies proposed that caspase-4 and caspase-5 are homologs of murine caspase-11 that promote noncanonical inflammasome activation (23, 44–46). However, our results suggest that these proteins function independently, as the timing of caspase-5 processing (24 hpi) corresponds to IL-1β production in avirulent C. burnetii-infected hAMs, while cleaved caspase-4 is not seen until 48 hpi. These findings open a new avenue of study to define the function of human caspase-5 in the host response to C. burnetii.
A recent study using avirulent C. burnetii showed that the organism does not trigger IL-1β secretion from mouse macrophages (47). That study presents an intriguing difference between murine and human C. burnetii infection and suggests that the two cell models can be used to define unique intracellular pathogen activities. Additionally, that study showed that C. burnetii secretes an effector, termed IcaA, that prevents caspase-11 activity. It is possible that avirulent C. burnetii uses IcaA to prevent activity of human caspase-4 (as a caspase-11 homolog) yet fails to inhibit caspase-5 activity that controls mature IL-1β secretion. Nonetheless, that study, combined with our results, indicates that C. burnetii must confront caspase activity and inflammasome-mediated detection during intracellular growth in macrophages.
Collectively, the current study further defines the primary hAM IL-1β response to C. burnetii and establishes human PCLS as a new infection platform. Due to the extensive use of avirulent C. burnetii in the field, these results should be considered when planning host response experiments. Numerous studies, including our own, have shown that avirulent and virulent bacteria display nearly identical intracellular behavior, including development of similar PVs and activation of host signaling. The current study supports the existence of important differences in the host cell response to differing pathotypes and promotes enhanced appreciation for using primary hAMs and ex vivo human lung tissue to characterize the innate immune response to C. burnetii.
ACKNOWLEDGMENTS
J.G.G. was supported by a supplement to R01AI087669 to promote diversity in research.
We thank Punsiri M. Colonne and Frances I. Onyilagha for critical reading of the manuscript.
REFERENCES
- 1.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Madariaga MG, Rezai K, Trenholme GM, Weinstein RA. 2003. Q fever: a biological weapon in your backyard. Lancet Infect Dis 3:709–721. doi: 10.1016/S1473-3099(03)00804-1. [DOI] [PubMed] [Google Scholar]
- 3.Akporiaye ET, Rowatt JD, Aragon AA, Baca OG. 1983. Lysosomal response of a murine macrophage-like cell line persistently infected with Coxiella burnetii. Infect Immun 40:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carey KL, Newton HJ, Luhrmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voth DE, Heinzen RA. 2009. Coxiella type IV secretion and cellular microbiology. Curr Opin Microbiol 12:74–80. doi: 10.1016/j.mib.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khavkin T, Tabibzadeh SS. 1988. Histologic, immunofluorescence, and electron microscopic study of infectious process in mouse lung after intranasal challenge with Coxiella burnetii. Infect Immun 56:1792–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Medzhitov R. 2001. Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 10.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cunha LD, Zamboni DS. 2013. Subversion of inflammasome activation and pyroptosis by pathogenic bacteria. Front Cell Infect Microbiol 3:76. doi: 10.3389/fcimb.2013.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariathasan S, Monack DM. 2007. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 13.Raupach B, Peuschel SK, Monack DM, Zychlinsky A. 2006. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun 74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns K, Martinon F, Tschopp J. 2003. New insights into the mechanism of IL-1beta maturation. Curr Opin Immunol 15:26–30. doi: 10.1016/S0952-7915(02)00017-1. [DOI] [PubMed] [Google Scholar]
- 15.Schroder K, Tschopp J. 2010. The inflammasomes. Cell 140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Rathinam VA, Vanaja SK, Fitzgerald KA. 2012. Regulation of inflammasome signaling. Nat Immunol 13:333–342. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooper PR, Kurten RC, Zhang J, Nicholls DJ, Dainty IA, Panettieri RA. 2011. Formoterol and salmeterol induce a similar degree of beta2-adrenoceptor tolerance in human small airways but via different mechanisms. Br J Pharmacol 163:521–532. doi: 10.1111/j.1476-5381.2011.01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Currie BJ. 2003. Melioidosis: an important cause of pneumonia in residents of and travellers returned from endemic regions. Eur Respir J 22:542–550. doi: 10.1183/09031936.03.00006203. [DOI] [PubMed] [Google Scholar]
- 19.Orme IM, Robinson RT, Cooper AM. 2015. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol 16:57–63. doi: 10.1038/ni.3048. [DOI] [PubMed] [Google Scholar]
- 20.Kim B, Lee Y, Kim E, Kwak A, Ryoo S, Bae SH, Azam T, Kim S, Dinarello CA. 2013. The interleukin-1alpha precursor is biologically active and is likely a key alarmin in the IL-1 family of cytokines. Front Immunol 4:391. doi: 10.3389/fimmu.2013.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. 2009. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 23.Broz P, Monack DM. 2013. Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog 9:e1003144. doi: 10.1371/journal.ppat.1003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. 1998. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 25.Voth DE, Howe D, Heinzen RA. 2007. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun 75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voth DE, Heinzen RA. 2009. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun 77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macdonald LJ, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii exploits host cAMP-dependent protein kinase signalling to promote macrophage survival. Cell Microbiol 16:146–159. doi: 10.1111/cmi.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA. 2014. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16:249–256. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, Zamboni DS, Roy CR. 2013. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A 110:1851–1856. doi: 10.1073/pnas.1211521110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox HR. 1941. Cultivation of rickettsiae of the Rocky Mountain spotted fever, typhus and Q fever groups in the embryonic tissues of developing chicks. Science 94:399–403. doi: 10.1126/science.94.2444.399. [DOI] [PubMed] [Google Scholar]
- 31.Derrick EH. 1937. “Q” fever, a new fever entity: clinical features, diagnosis, and laboratory investigation. Med J Aust 2:281–299. [DOI] [PubMed] [Google Scholar]
- 32.Burnet FM, Freeman M. 1937. Experimental studies on the virus of “Q” fever. Med J Aust 2:299–305. [Google Scholar]
- 33.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Case CL, Roy CR. 2011. Asc modulates the function of NLRC4 in response to infection of macrophages by Legionella pneumophila. mBio 2:e00117. doi: 10.1128/mBio.00117-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 36.Shannon JG, Howe D, Heinzen RA. 2005. Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A 102:8722–8727. doi: 10.1073/pnas.0501863102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ammerdorffer A, Schoffelen T, Gresnigt MS, Oosting M, den Brok MH, Abdollahi-Roodsaz S, Kanneganti TD, de Jong DJ, van Deuren M, Roest HJ, Rebel JM, Netea MG, Joosten LA, Sprong T. 2015. Recognition of Coxiella burnetii by Toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J Infect Dis 211:978–987. doi: 10.1093/infdis/jiu526. [DOI] [PubMed] [Google Scholar]
- 38.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winchell CG, Graham JG, Kurten RC, Voth DE. 2014. Coxiella burnetii type IV secretion-dependent recruitment of macrophage autophagosomes. Infect Immun 82:2229–2238. doi: 10.1128/IAI.01236-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meghari S, Honstettre A, Lepidi H, Ryffel B, Raoult D, Mege JL. 2005. TLR2 is necessary to inflammatory response in Coxiella burnetii infection. Ann N Y Acad Sci 1063:161–166. doi: 10.1196/annals.1355.025. [DOI] [PubMed] [Google Scholar]
- 41.Zamboni DS, Campos MA, Torrecilhas AC, Kiss K, Samuel JE, Golenbock DT, Lauw FN, Roy CR, Almeida IC, Gazzinelli RT. 2004. Stimulation of Toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J Biol Chem 279:54405–54415. doi: 10.1074/jbc.M410340200. [DOI] [PubMed] [Google Scholar]
- 42.Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, Gilk SD, Williams KP, Sobral BW, Kupko JJ III, Porcella SF, Samuel JE, Heinzen RA. 2009. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect Immun 77:642–656. doi: 10.1128/IAI.01141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beare PA, Samuel JE, Howe D, Virtaneva K, Porcella SF, Heinzen RA. 2006. Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J Bacteriol 188:2309–2324. doi: 10.1128/JB.188.7.2309-2324.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 45.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, Monack DM. 2012. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490:288–291. doi: 10.1038/nature11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 47.Cunha LD, Ribeiro JM, Fernandes TD, Massis LM, Khoo CA, Moffatt JH, Newton HJ, Roy CR, Zamboni DS. 2015. Inhibition of inflammasome activation by Coxiella burnetii type IV secretion system effector IcaA. Nat Commun 6:10205. doi: 10.1038/ncomms10205. [DOI] [PMC free article] [PubMed] [Google Scholar]