

Abstract
Objectives
Tenofovir disoproxol fumarate (TDF) is increasingly used in HAART regimens of pregnant women, but limited data exist on pregnancy pharmacokinetics of chronically-dosed TDF. This study described tenofovir pharmacokinetics during pregnancy and postpartum.
Methods
IMPAACT P1026s is a prospective, non-blinded pharmacokinetic study of HIV-infected pregnant women that included a cohort receiving 300 mg TDF once daily. Steady-state 24-hour pharmacokinetic profiles were measured at 2nd and 3rd trimester and postpartum, with maternal and umbilical cord samples at delivery. Tenofovir was measured by LC-MS. The target AUC was ≥ 1.99 mcg•hr/mL (non-pregnant historical control 10th percentile).
Results
Median tenofovir AUC was decreased during the 2nd (1.9 mcg•hr/mL) and 3rd (2.4 mcg•hr/mL, p=0.005) trimesters versus postpartum (3.0 mcg•hr/mL). Tenofovir AUC exceeded the target for 2/4 (50%) 2nd trimester; 27/37 (73%; 95% CI: 56%, 86%) 3rd trimester; and 27/32 (84%; 95% CI: 67%, 95%) postpartum women (p>0.05). Median 2nd/3rd trimester troughs were lower (39/54 ng/mL) than postpartum (61 ng/mL). Median 3rd trimester weight was heavier for subjects below target AUC versus those above target (97.9 vs. 74.2 kg, p = 0.006). Median ratio of cord blood to maternal concentrations was 0.88. No infants were HIV infected.
Conclusions
This study found lower tenofovir AUC and troughs during pregnancy. Transplacental passage with chronic TDF use during pregnancy was high. Standard TDF doses appear appropriate for most HIV-infected pregnant women but therapeutic drug monitoring with dose adjustment should be considered in pregnant women with high weight (> 90kg) or inadequate HIV RNA response.
Keywords: HIV, pregnancy, antiretrovirals, tenofovir, prevention of perinatal transmission
INTRODUCTION
HIV-1-infected pregnant women are prescribed antiretroviral regimens to quickly and fully suppress maternal HIV RNA levels in order to prevent perinatal transmission of HIV to the fetus/newborn as well as to optimize maternal health and maintain treatment susceptibility. Common antiretroviral combinations prescribed include nucleoside/tide analogs (NRTIs) with either a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor. While treatment guidelines for HIV in pregnancy are similar to those for non-pregnant adults, treatment success is most commonly achieved with agents that are dosed infrequently and perhaps more importantly have few side effects even in pregnancy1. These attributes make tenofovir disoproxil fumarate (TDF or PMPA, trade name Viread®; Gilead Sciences, Inc., Foster City, CA) an increasingly prescribed component of antepartum regimens.
Physiologic changes in pregnancy can result in lower drug exposure of essentially all classes of antiretroviral agents compared to exposure in non-pregnant persons2. Use of standard adult antiretroviral doses in pregnant women may not be adequate to ensure full suppression of viral load in plasma, which is needed to minimize the risk of fetal HIV exposure and antiretroviral drug resistance. Additionally, agents may have different side effects on the growing fetus compared to infants or children. Therefore, understanding maternal and fetal antiretroviral drug exposure and safety is important to optimize treatment or prevention pregnancy regimens.
TDF is a once daily nucleotide analogue reverse transcriptase inhibitor and is an appealing agent for use as a component of antenatal and peripartum antiretroviral regimens. TDF, the prodrug of tenofovir, is formulated alone as Viread® or in combination with emtricitabine (Truvada®; Gilead Sciences, Inc., Foster City, CA); efavirenz and emtricitabine (Atripla®; Bristol Myers Squibb, New York City, NY and Gilead Sciences, Inc., Foster City, CA); rilpivirine and emtricitabine (Complera®; Gilead Sciences, Inc., Foster City, CA); and elvitagravir, cobicistat and emtricitabine (Stribild®; Gilead Sciences, Inc., Foster City, CA) for treatment of HIV and/or hepatitis B. Adverse events reported have included bone and renal toxicities3, 4.
The primary objectives of this study were to describe tenofovir pharmacokinetics in HIV-infected pregnant and postpartum women and to determine if the standard TDF dose produces equivalent tenofovir exposure during pregnancy to that seen in: 1) historical data from non-pregnant adults; and 2) the same women in the study cohort during the postpartum period. We also evaluated the transplacental passage of tenofovir by comparing concentrations in cord and maternal blood.
METHODS
Study population and design
P1026s (Pharmacokinetic (PK) Properties of Antiretroviral Drugs (ARVs) During Pregnancy) is a multicenter, ongoing, prospective, non-blinded study of the International Maternal Pediatric and Adolescent AIDS Clinical Trials (IMPAACT) group. P1026s subjects were also enrolled in P1025, the Perinatal Core Protocol, a prospective cohort study of HIV-infected pregnant women and their infants receiving care at IMPAACT sites in the U.S. All subjects signed appropriate institutional informed consent prior to participation in P1025 and P1026s. All study procedures were in accordance with the ethical standards of the Helsinki Declaration of 1975, as revised in 2000. This cohort’s P1026s eligibility included receipt of 300 mg TDF daily, either as Viread® or co-formulated with emtricitabine (Truvada®), as part of clinical care for at least two weeks prior to pharmacokinetic sampling and with intent to continue at least 6 weeks postpartum. Exclusion criteria were: multiple gestation or clinical or laboratory toxicity that, in the opinion of the site investigator, might result in a required change to the chosen antiretroviral regimen during the study. Renal dysfunction was not an explicit exclusion criterion. Subjects received medications that were prescribed by their clinical care providers, and remained on study unless a change to a different antiretroviral regimen was desired due to toxicity, lack of viral load suppression or for inability to achieve desired pharmacokinetic targets. Pharmacokinetic sampling of tenofovir was performed between November 2004 and September 2008.
Clinical and laboratory monitoring
Maternal and infant clinical, laboratory, demographic and historical data were accessed from the P1025 and P1026s databases. On each pharmacokinetic sampling visit and at delivery, subjects had an interval medical history, physical examination and laboratory studies [complete blood count, alanine aminotransferase (ALT), aspartate aminotransferase (AST), bilirubin, creatinine, blood urea nitrogen (BUN), and albumin]. The P1026s study team reviewed toxicity reports on monthly conference calls and assigned causality and grade for all toxicities according to the Division of AIDS (DAIDS) standardized Toxicity Table for Grading Severity of Adult Adverse Experiences (August 1992); http://rcc.tech-res-intl.com). All toxicities were followed through resolution; and the subject’s clinical care provider was responsible for subject toxicity management.
Sample collection and measurement
Pharmacokinetic sampling was performed during the second trimester (20–28 weeks gestation), the third trimester (30–38 weeks gestation) and postpartum (6–12 weeks after delivery). Subjects were asked to take TDF at the same time for three days prior to and on the day of assessment when plasma samples were collected immediately before the witnessed dose (C0) and 1, 2, 4, 6, 8, 12 and 24 hours post-dose. Single umbilical cord blood and maternal plasma samples were collected at delivery. Tenofovir concentrations were measured by a previously described validated, liquid chromatography-mass spectrometry (LC-MS) method5,6. The linear range was 10 ng/mL to 1500 ng/mL, with a lower limit of detection for tenofovir of 10 ng/mL. Accuracy and precision were within ± 20% at 10 ng/mL and ± 15% at other quality control concentrations6.
Pharmacokinetic analyses
The pre-dose concentration (C0), the maximum plasma concentration (Cmax), the corresponding time (Tmax), and the last measurable concentration (C24) were identified by direct inspection. The area under the concentration versus time curve from time 0 to 24 hours post dose (AUC) for tenofovir was estimated using the trapezoidal rule up to the last measurable concentration.
Apparent oral clearance (CL/F) from plasma was calculated as the dose (135.6 mg of tenofovir contained in a 300 TDF mg tablet) divided by AUC. The apparent volume of distribution (Vd/F) was determined as CL/F divided by λz, where λz was the terminal slope of the log concentration versus time curve. The half-life (t½) was calculated as 0.693/λz. Because the log concentration versus time curve of the observed data shows two distinct slopes, CL/F, distribution volumes and elimination rate constants were also computed using a two-compartment model in WinNonlin, Version 6.2.1 (Pharsight Corp., St Louis, MO).
Statistical analyses
The study design incorporated a two-stage analysis approach. Each individual woman’s tenofovir exposure during pregnancy was determined in real time, compared with the average and estimated 10th percentile target (1.99 mcg•hr/mL) AUCs for non-pregnant, adult HIV-1-infected historical controls, and reported to each subject and her care provider. The control population included HIV-infected and healthy subjects reported in the 2003 Viread Package Insert; co-administered medications including other antiretrovirals in these subjects were not described at that time7.Based on the subject’s pharmacokinetics, clinical and laboratory status, and the study pharmacologist’s recommendations (if any), the subject and care provider could decide whether or not to modify dosing. If the latter option was chosen, pharmacokinetic assessment after at least one week on the new dose was offered. A study stopping criterion to trigger an evaluation of the adequacy of drug exposure was predefined as six of 25 women (24%; exact 80% confidence limits: 13%, 38%) falling below the target AUC. The goal was to prevent excess accrual to a cohort with known inadequate anti-retroviral exposure.
After all pharmacokinetic sampling was complete for all subjects, a repeated measures design was used to assess antepartum and postpartum tenofovir exposure for each woman. Third-trimester tenofovir exposure was compared at the within-subject level to postpartum exposure, using 90% confidence limits for the geometric mean ratios of antepartum to postpartum pharmacokinetic parameters. When the true geometric mean of the ratio (the antilog of the true mean of the log ratios) of the pharmacokinetic parameters for pregnant and non-pregnant conditions has a value of 1, this indicates equal geometric mean pharmacokinetic parameters for the pregnant and non-pregnant conditions. If the 90% confidence intervals (CIs) are entirely outside the limits (0.8 and 1.25), the pharmacokinetic exposure parameters for the pregnant and non-pregnant conditions are considered different. If the 90% CIs are entirely within the limits, the drug exposures are considered equivalent. If, however, the 90% CIs overlap the 0.8 or 1.25 limits, these data alone do not support any conclusions. The magnitudes of the differences in the median values of pharmacokinetic parameters antepartum and postpartum were assessed with the Wilcoxon signed-rank test. Differences in weight and serum creatinine between groups meeting and not meeting AUC targets were explored with univariate Wilcoxon rank-sum tests and Spearman’s correlation.
RESULTS
Subject characteristics
Thirty-seven women enrolled and completed third trimester pharmacokinetic sampling. Of these, 4 also completed second trimester sampling and 32 completed postpartum sampling. Table 1 summarizes the clinical characteristics of the subjects. Women received TDF as part of a combination antiretroviral regimen, which most commonly included emtricitabine, atazanavir, fosamprenavir and/or ritonavir. Four women were taking non-nucleoside based regimens, and 33 were on protease inhibitor containing regimens. Median gestational age at delivery was 38 weeks. All infants had birth weights that were appropriate for gestational age. Median CD4+ cell count at delivery was 469 cells/mm3. Viral load at delivery was available for 34 subjects, and was ≤ 400 copies/mL for 33 subjects and 9733 copies/mL for the remaining subject.
Table 1.
Characteristics of the Study Population and Pregnancy Outcomes
| Median (Range)a or Number of Subjects (%)b | |
|---|---|
|
| |
| Maternal Demographics | |
|
| |
| Age (years) at 3rd trimestera | 30.8 (13.5 – 39.2) |
|
| |
| Weight (kilograms) at 3rd trimestera | 80.6 (50.8 – 121.9) |
|
| |
| Race/Ethnicityb | |
| Hispanic, Latina | 15 (41) |
| Black, non-Hispanic | 10 (27) |
| White, non-Hispanic | 9 (24) |
| Asian, Pacific Islander | 1 (3) |
| More than one race | 1 (3) |
| Unknown/missing | 1 (3) |
|
| |
| CD4+ count (cells/mm3) at deliverya | 469 (9 – 1494) |
|
| |
| Women with plasma HIV RNA at:b | |
| 2nd Trimester | |
| Undetectable (< 400 copies/mL)* | 4/4 (100) |
| Detectable (≥ 400 copies/mL) | 0/4 (0) |
| 3rd Trimester | |
| Undetectable (< 400 copies/mL)† | 33/35 (94) |
| Detectable (≥ 400 copies/mL) | 2/35 (6) |
| Delivery | |
| Undetectable (< 400 copies/mL)‡ | 33/34 (97) |
| Detectable (≥ 400 copies/mL) | 1/34 (3) |
| Postpartum | |
| Undetectable (< 400 copies/mL)§ | 22/26 (85) |
| Detectable (≥ 400 copies/mL) | 11/26 (15) |
|
| |
| Concomitant antiretrovirals | |
| Emtricitabine | 25 |
| Atazanavir (with rtv) | 16 |
| Lopinavir/ritonavir | 12 |
| Lamivudine | 10 |
| Zidovudine | 7 |
| Nelfinavir | 4 |
| Abacavir | 3 |
| Saquinavir (with rtv) | 2 |
| Didanosine, efavirenz, fosamprenavir (with rtv), nevirapine | 1 each |
|
| |
| Pregnancy Outcomes | |
|
| |
| Gestational age at delivery (weeks)a | 38.1 (34.7 – 41.7) |
|
| |
| Infant birth weight (kilograms) | 2.93 (2.23 – 4.05) |
Median (range)
Number of subjects (percent)
3/4 (75%) subjects were < 50 copies per milliliter.
22/35 (63%) subjects were < 50 copies per milliliter.
20/34 (59%) subjects were < 50 copies per milliliter.
11/26 (42%) subjects were < 50 copies per milliliter.
Six women had Grade 3 elevated total bilirubin. Six infants had anomalies as follows, 1) bilateral hip clicks, 2) lacrimal duct stenosis, 3) Mongolian spots on shoulder and buttocks, 4) vesicoureteral reflux, 5) parvovirus positive, and 6) bilateral preauricular skin tags. Investigators were unable to judge whether the parvovirus result and the vesicoureteral reflex were related to maternal use of tenofovir. The other four anomalies were deemed unrelated to maternal use of tenofovir. None of the infants were HIV infected.
Tenofovir exposure
The target tenofovir exposure was AUC ≥ 1.99 mcg•hr/mL, the 10th percentile of the average exposure for non-pregnant historical controls. This target was met for 27/37 (73%) in 3rd trimester and 2/4 (50%) in 2nd trimester. The 10 subjects with AUC’s below the 1.99 mcg•hr/mL target remained on the standard dose of 300 mg TDF once daily. Figure 1 shows the antepartum concentration versus time curves for each subject. Figure 2 shows the postpartum concentration versus time curves for each subject, of whom 27/32 (84%) met or surpassed the tenofovir target AUC.
Figure 1.
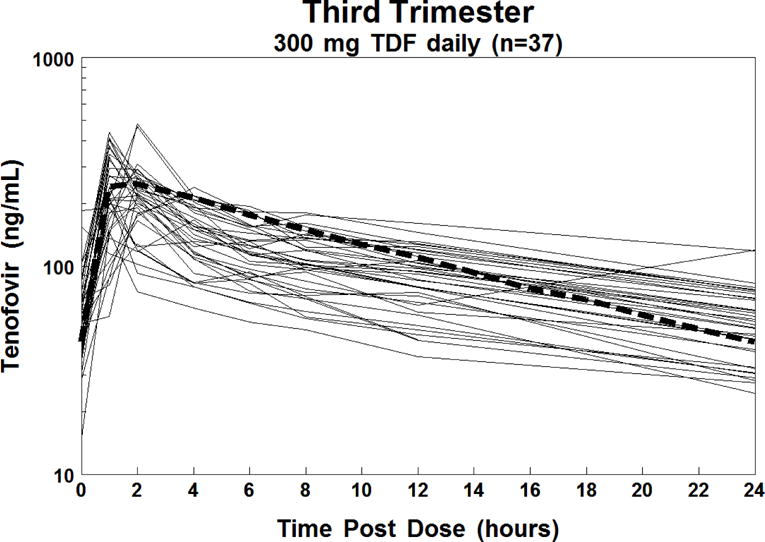
Individual plasma concentration-time curves of tenofovir in 37 HIV-1 infected pregnant women in the 3rd trimester (solid lines) and the estimated 50th percentile concentration-time curve for non-pregnant HIV-infected historical adult controls (thick dashed line).
Figure 2.
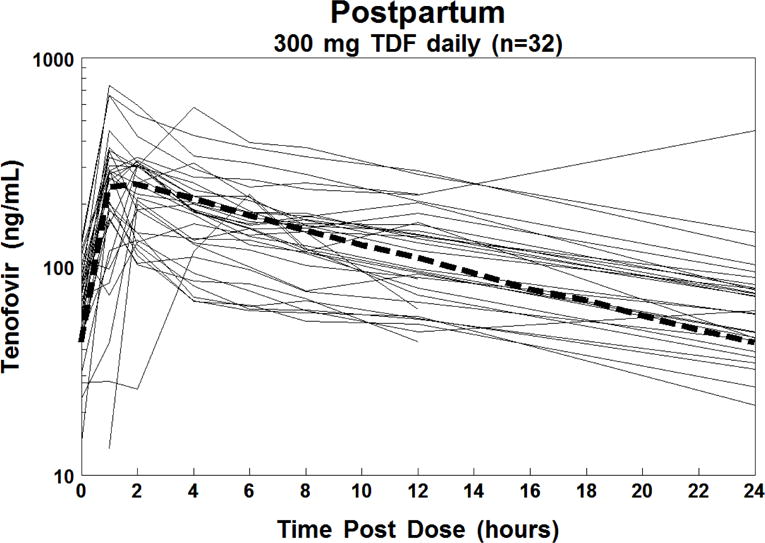
Individual plasma concentration-time curves of tenofovir in 32 HIV-1-infected women postpartum (solid lines) and the estimated 50th percentile concentration-time curve for non-pregnant HIV-infected historical adult controls (thick dashed line).
Table 2 summarizes the tenofovir pharmacokinetic parameters from the noncompartmental analysis, and Figure 3 outlines the median antepartum and postpartum concentration-time curves. The median AUC was significantly higher postpartum compared to 3rd trimester; and was lowest in the 2nd trimester. For the four women with 2nd trimester data, their corresponding median 2nd trimester, 3rd trimester and postpartum AUCs were 1.9, 2.1 and 2.6 mcg•hr/mL. For the entire population, CL/F, Vd/F, and half-life were significantly higher in the 3rd trimester compared to postpartum. C24 and C0 were significantly lower in the 3rd trimester compared to postpartum, but Cmax was not different antepartum compared to postpartum. Only one pharmacokinetic parameter was deemed different by the pre-specified geometric mean ratio and 90% CI criterion; Vd/F was higher in the 3rd trimester than postpartum. For all other parameters, the geometric mean ratios and 90% CI were inconclusive (the 90% CI all crossed one of the limits, 0.8 or 1.25).
Table 2.
Pharmacokinetic Parameters by Gestational Age
| PK Parameter Median (Interquartile Range) | Second trimester (20–28 weeks gestation) (n=4) | Third Trimester (30–38 weeks gestation) (n=37) | Postpartum (6–12 weeks after delivery) (n=32) | 3rd Trimester/Postpartum Ratio Geometric Mean (90% CI) | P Value* |
|---|---|---|---|---|---|
| AUC (mg·h/L) | 1.9 (1.7, 2.5) |
2.4 (1.9, 3.1) |
3.0 (2.2, 3.7) |
0.80 (0.72, 0.89) |
0.0046 |
| CL/F (L/hr) | 72 (58, 82) |
57 (44, 71) |
46 (36, 62) |
1.25 (1.12, 1.40) |
0.0122 |
| Vd/F (L) | 1570 (941, 2501) |
1155 (1027, 1613) |
899 (652, 1236) |
1.59 (1.29, 1.97) |
0.0014 |
| Cmax (mcg/L) | 250 (202, 355) |
245 (207, 334) |
298 (200, 341) |
0.89 (0.79, 1.01) |
0.1636 |
| Tmax (hours) | 1 (1, 1.25) |
1 (1, 2) |
1 (1, 2) |
– | 0.3302 |
| T1/2 (hours) | 16.9 (15.2, 20) |
16.1 (14.4, 18.2) |
12.4 (11.7, 15.9) |
1.28 (1.07, 1.53) |
0.0213 |
| C0 (mcg/L) | 38 (35, 50) |
54 (44, 69) |
64 (42, 86) |
0.83 (0.70, 0.98) |
0.01 |
| C24 (mcg/L) | 39 (34, 49) |
54 (40, 70) |
61 (45, 79) |
0.82 (0.71, 0.96) |
0.0325 |
3rd trimester compared to postpartum
Figure 3.
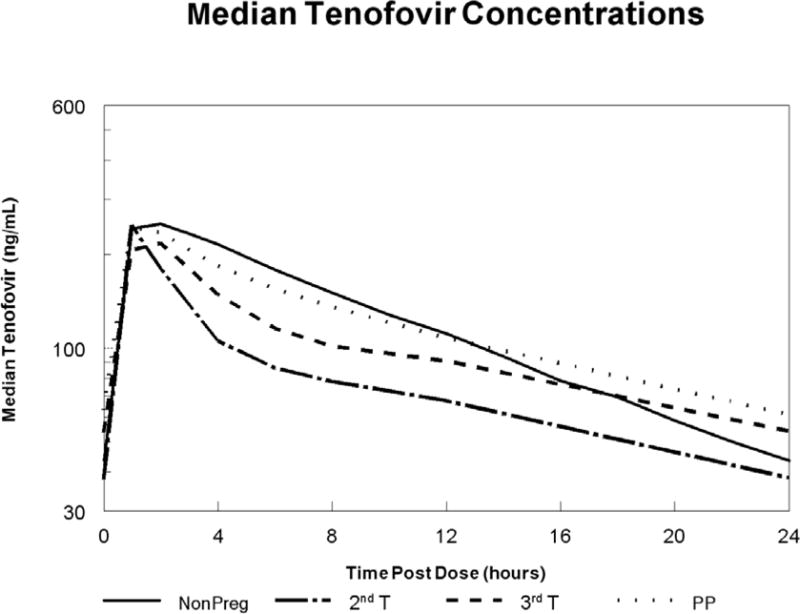
Median tenofovir concentration-time curves during the second trimester (dash/dot line; n = 4); third trimester (dashed line; n = 37) and postpartum (dotted line; n = 32), and the estimated 50th percentile concentration-time curve for non-pregnant HIV-infected historical adult controls (solid line). IQR, interquartile range.
At the 3rd trimester visit, the median (range) weight for the 10 women who did not meet the target AUC was 97.9 (68.1 – 121.9) kg. In contrast, the median (range) weight for the 27 women who met the AUC target was 74.2 (50.8 – 93.5) kg (p=0.006). Third trimester weight and tenofovir AUC were inversely correlated (spearman’s rho = −0.48, p=0.004). Three of these 10 women were also below the AUC target at their postpartum visit; all three weighed > 90 kg both ante- and postpartum. For the five women who did not meet the AUC target at the postpartum visit (the three mentioned above plus two who had met the target during their 3rd trimester visit), their median (range) postpartum weight was 83.7 (59.8 – 101.4) kg. The women who met the AUC target at the postpartum visit weighed 71.2 (45.2 – 108.4) kg, which was not significantly different (p=0.282). Again, however, the postpartum tenofovir AUC and subject weights were inversely correlated (spearman’s rho = −0.51, p = 0.005).
Serum creatinine was a median (range) 20% (−38% to 60%) higher postpartum than during the 3rd trimester (p=0.002 for within-subject comparison). At the 3rd trimester visit, the median (range) serum creatinine for the 10 women who did not meet the target AUC was 0.5 (0.4 – 0.8) mg/dL. The median (range) serum creatinine for the 27 women who met the AUC target was 0.6 (0.4 – 0.8) mg/dL (p=0.209). Third trimester serum creatinine and tenofovir AUC were directly correlated (spearman’s rho = 0.39, p=0.024). For the five women who did not meet the AUC target at the postpartum visit, their median (range) postpartum serum creatinine was 0.6 (0.6 – 0.8) mg/dL. The women who met the AUC target at the postpartum visit had a median serum creatinine of 0.7 (0.5 – 1.1) mg/dL, which was also not significantly different (p=0.612). Again, however, the postpartum tenofovir AUC and subjects’ serum creatinine were positively correlated (spearman’s rho = 0.41, p = 0.025).
For the two-compartment analysis, the model successfully estimated the pharmacokinetic parameters for all four subjects (100%) in the 2nd trimester, for 22 (63%) of the 3rd trimester subjects, and for 18 (69%) of the postpartum subjects. The median (interquartile range; IQR) CL/F for the 2nd trimester, 3rd trimester, and postpartum were 35 (25, 45) L/hr, 35 (16, 44) L/hr, and 28 (18, 32) L/hr. The central compartment apparent volumes were 260 (178, 339) L, 256 (191, 321) L and 211 (149, 278) L and the peripheral compartment apparent volumes were 750 (567, 1315) L, 705 (549, 1045) L, and 433 (304, 738) L in the 2nd trimester, 3rd trimester and postpartum, respectively. The terminal half-lives in the three study periods were 17 (15, 40) hours, 20 (16, 35) hours and 18 (15, 28) hours.
The median (IQR) concentration of tenofovir in 31 maternal plasma samples collected at delivery was 62.4 (44.1, 73.4) ng/mL, and in 32 umbilical cord blood samples was 56.7 (37.0, 76.5) ng/mL. Cord blood to maternal plasma concentration ratio was 0.88 (0.76, 1.03) for 28 mother/infant pairs.
DISCUSSION
In this study of 37 pregnant women with 584 plasma tenofovir samples, tenofovir overall exposure was 20% lower in the 3rd trimester compared to postpartum in the same women. Similarly, CL/F was 25% higher and C24 and C0 were about 20% lower during pregnancy. Apparent volume of distribution was nearly 60% larger in the 3rd trimester compared to postpartum. Even though CL/F was faster during pregnancy, the terminal half-life was actually longer in pregnancy compared to postpartum, presumably due to a larger magnitude increase in distribution volume compared to CL/F (half-life is directly proportional to volume of distribution, but inversely proportional to clearance). The time of maximum concentration post dose was 1 hour and was not influenced by pregnancy, nor was maximum concentration significantly different between pregnancy and postpartum. The plasma concentration just before an observed dose did not differ from the plasma concentration 24 hours after the observed dose, suggesting that women had been consistently taking prescribed TDF.
The postpartum tenofovir AUC found in this study of 3.0 mcg•hr/mL is similar to the average AUC in non-pregnant adults, 3.3 mcg•hr/mL. The 10th percentile target was met for 2/4 (50%) women in 2nd trimester, for 27/37 (73%) in 3rd trimester and 27/32 (84%) postpartum. Ninety-four percent of subjects had an HIV RNA ≤ 400 copies/mL in the 3rd trimester and none discontinued their regimen or altered their TDF dose based on their reported lower-than-target AUC. At delivery, 97% of women were virally suppressed under 400 copies/mL. In a study of tenofovir pharmacokinetics in Caucasian and Black European pregnant women from the PANNA network, 27 of 34 (79%) women had HIV RNA ≤ 50 copies/mL at delivery8.
The PANNA study found 3rd trimester and postpartum AUCs of 2.5 and 3.2 mcg•hr/mL8, comparable to the values found in the current study, a US cohort, of 2.4 and 3.0 mcg•hr/mL. The PANNA study also reported approximately 20% reductions in maximum and trough concentrations in the 3rd trimester compared to postpartum. The current study found similar reductions of about 17% in maximum and trough concentrations during pregnancy, though the findings were not statistically different from postpartum for Cmax. CL/F was significantly increased by 30% during the 3rd trimester in the PANNA study, similar to our observed 25% increase. One potential explanation for the increased clearance during pregnancy is that tenofovir is being more rapidly eliminated by the kidney. The subjects in this study did have lower serum creatinine values during 3rd trimester compared to postpartum, and serum creatinine was positively correlated with tenofovir AUC at both time points. Women who met AUC targets had a serum creatinine that was a median of 0.1 mg/dL higher than the value in those who did not meet AUC targets both in the 3rd trimester and postpartum. However, these differences were not statistically significant, possibly due to lack of power with small numbers of subjects not meeting AUC targets. Pregnancy causes an increase in renal plasma flow by 25 – 50% due to increased cardiac output9, and tenofovir undergoes renal elimination. In contrast to this study, the PANNA study reported identical terminal half-lives during third trimester and postpartum, 15 hours, while this study found significantly longer half-lives (16 hours versus 12 hours) and larger distribution volumes during pregnancy compared to postpartum. However, as the women in this study were assessed at steady-state with multiple dosing, a half-life associated with the terminal elimination phase may not have been reliably estimated. Because intravenous tenofovir formulations have not been studied during pregnancy, possible alterations in oral absorption during pregnancy cannot be teased apart from changes in clearance and distribution volume. The European study did not have 2nd trimester data for comparison.
In this study, the subject’s weight was inversely significantly correlated with tenofovir AUC during the 3rd trimester and postpartum. During the 3rd trimester, the 10 women who were below the target AUC were significantly heavier, by about 23 kg, than the 27 women who met the AUC target. One potential explanation for the lower tenofovir concentrations is that the larger subjects have a larger distribution volume of tenofovir. Tenofovir is distributed to most tissues, with highest concentrations in kidney, liver and intestinal contents7. Obese women may have higher distribution volumes and lower tenofovir exposure in general, which can then be compounded by the ~20–25% decrease in tenofovir exposure seen during pregnancy.
Benaboud et al. reported a population pharmacokinetic study of tenofovir chronic use in French pregnant and non-pregnant women10. The retrospective study dataset included 52 samples collected clinically from 46 pregnant women (6 in the 1st trimester, 18 in the 2nd trimester and 28 in the 3rd trimester) for therapeutic drug monitoring; doses were not observed. The data were best described by a two-compartment model with linear absorption and elimination. Pregnancy significantly increased oral clearance of tenofovir, by 39% at delivery specifically. The model was not able to discriminate changes in clearance by gestational age due to the small number of samples, especially early in pregnancy. The estimated AUC and Cmin during pregnancy from this population analysis were 1.6 mcg•hr/mL and 39 ng/mL, similar to the values reported in this study in the 2nd trimester of pregnancy, 1.9 mcg•hr/mL and 38 ng/mL. Age also had a significant impact on the overall population pharmacokinetic model, while weight did not. A measure of renal function was not included.
The pharmacokinetics, including intracellular concentrations of tenofovir metabolites have been described by Hirt et al. at delivery and 1 week postpartum11,12, and by Flynn et al.13 for single dose maternal and neonatal or 7 days of maternal peripartum dosing. With single doses at delivery, tenofovir appears to cross the placenta well, achieving 73% of the maternal level in the cord blood13. With chronic TDF dosing during pregnancy, the ratio of cord to maternal blood concentrations was 0.82 in the PANNA study8 and 0.88 in this study. Even so, infants must be dosed almost immediately after birth to maintain appropriate levels11.
This study was not powered for a full safety analysis, or to understand the single contribution of tenofovir exposure to the antiretroviral regimen in prevention of perinatal HIV transmission. However, adverse effects and transmission data were collected. The maternal subjects appeared to tolerate TDF well during pregnancy. None of the infants was small for gestational age or infected. Measurements of bone toxicities, kidney function or long-term growth of the infants were not available in this study.
One limitation of this study is that the active moiety, intracellular tenofovir diphosphate, was not collected or measured. While plasma tenofovir concentrations are being used as a surrogate marker for active drug exposure at the site of action, the intracellular active metabolite pharmacokinetics may or may not be affected by and altered to the same extent during pregnancy as plasma pharmacokinetics. Another limitation is that measurements were not collected in the early stages of pregnancy in these women, so the timing of the pregnancy-induced changes cannot be well characterized.
This is the largest study to date of tenofovir pharmacokinetics in pregnant women, and provides the first published data available on intensive 2nd trimester pharmacokinetic profiles and the potential impact of higher weight in pregnancy on tenofovir exposure. This and prior studies have shown that tenofovir exposure is decreased during pregnancy compared to postpartum8 and non-pregnant adults10. CL/F is increased by 25 – 30% during pregnancy, with corresponding 15 – 25% decreases in AUC and minimum concentrations. The clinical impact of these exposure changes remains unclear. Colbers et al. (the PANNA study) concluded that the 25% lower exposure was not associated with virologic failure or infant transmission. Other nucleoside reverse transcriptase inhibitors have also shown decreased exposure during pregnancy, but with small enough magnitudes that the standard dose is still thought to be therapeutic14–16. On the other hand, Benaboud et al. suggested that women should consider taking increased tenofovir doses in the second and third trimesters to achieve exposure similar to those of non-pregnant women. The population pharmacokinetic model predicted an AUC of 3.4 mcg•hr/mL and a Cmin of 78 ng/mL with two tablets of TDF (600 mg dose of TDF = 272 mg of tenofovir). In our study, the majority of women achieved an AUC greater than the 10th percentile of expected in non-pregnant adults. HIV RNA was ≤ 400 copies/mL in all but one subject taking the standard dose, and no infants were infected. However, more sensitive viral load measurements (between 20 – 400 copies/mL) were not available for all study subjects, and high weight was associated with lower than expected tenofovir exposure. Given the currently available evidence, standard doses appear appropriate for a majority of HIV-infected pregnant women. HIV RNA should be closely monitored in pregnant women taking TDF. Therapeutic drug monitoring with TDF dose adjustment should be considered in women with high weight (> 90kg), or in those with detectable HIV RNA during pregnancy or with a less than expected decline in HIV RNA if initiating therapy during pregnancy.
Acknowledgments
Sources of Funding: The authors wish to thank the women that participated in the protocol, the staff of the participating IMPAACT centers, P1026s Protocol Team Members, and Lane R. Bushman and Brian L. Robbins from the Antiviral Pharmacology Laboratory for performing the tenofovir plasma concentration assays. In addition to the co-authors above, P1026s Protocol Team members include: Francesca Aweeka, Pharm.D.; Emily Barr, C.P.N.P., C.N.M., M.S.; Nantasak Chotivanich, M.D.; Tim Roy Cressey, Ph.D.; Lisa M. Frenkel, M.D.; Amita Gupta, M.D., M.H.S.; Amy Jennings, B.S.; Gonzague Jourdain, M.D. ; Regis Kreitchmann, M.D, PhD; Rita Patel; Kittipong Rungruengthanakit, M.Sc.; David Shapiro, Ph.D.; and Pra-ornsuda Sukrakanchana, B.S.N.
Participating sites and site personnel include: 5048 USC LA NICHD CRS (Andrea Kovacs, MD; James Homans, MD; LaShonda Spencer, MD; Francoise Kramer, MD); 3801 Texas Children’s Hospital CRS (Shelley Buschur, RN, CNW; Hunter Hammill II, MD; Mary E. Paul, MD; Chivon McMullen-Jackson, RN, BSN, AND); 5017 Seattle Children’s Hospital CRS (Ann Melvin, MD, MPH; Corry Venema-Weiss, ARNP; Jenna Lane, ARNP; Jane Hitti, MD, MPH; This publication was supported by the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000423); 6501 St. Jude/UTHSC CRS (Katherine Knapp, MD; Edwin Thorpe, Jr, MD; L. Jill Utech, RN, MSN; Nina Sublette, RN, MSN, PhD); 5091 University of California, San Francisco NICHD CRS (Diane Wara, MD; Deborah Cohan, MD; Nicole Tilton, PNP); 6901 Bronx-Lebanon Hospital IMPAACT CRS (Mary Elizabeth Vachon, MPH; Mirza Mahboobullah Baig, MD; Murli Udharam Purswani, MD; Jenny Gutierrez, MD); 7301 WNE Maternal Pediatric Adolescent AIDS CRS (Katherine Luzuriaga, MD; Sharon Cormier, RN; Margaret McManus); 4601 University of California San Diego Maternal, Child, and Adolescent HIV CRS (Andrew Hull, BMedSCi; Mary Caffery, RN, MSN; Kimberly Norris, RN, BSN; Stephen A. Spector, MD); 5045 Harbor UCLA Medical Center NICHD CRS (Margaret A Keller, MD; Susan Ballagh, MD; Judy Hayes, RN; Yolanda Gonzalez, RN; The project described was supported by the National Center for Advancing Translational Sciences through UCLA CTSI Grant UL1TR000124. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH); 5052 University of Colorado Denver NICHD CRS (Emily Barr, CPNP, CNM, MSN; Tara Kennedy, MSN, FNP-BC; Alisa Katai, MHA; Jenna Wallace MSW; NIH/NCATS Colorado CTSI Grant Number UL1 TR000154); 5083 Rush University Cook County Hospital, Chicago NICHD CRS (James B McAuley, MD, MPH; Helen Cejtin, MD; Maureen McNichols, RN, MSN, CCRC; Julie Schmidt, MD); 5094 University of Maryland Baltimore NICHD CRS (Douglas Watson, MD; Judy Ference, BSN; Corinda Hilyard; This work supported by the University of Maryland Clinical Translational Science Institute and the University of Maryland General Clinical Research Center); 5096 University of Alabama Birmingham NICHD CRS (Marilyn Crain, MPH, MD; Tina Y. Simpson, MD, MPH, PhD; Alan T.N. Tita, MD, MPH, PhD); 6601 University of Puerto Rico Pediatric HIV/AIDS Research Program CRS (Irma L Febo, MD; Carmen D Zorrilla, MD; Vivian Tamayo-Agrait, MD; Ruth Santos, RN, MPH).
Overall support for the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) was provided by the National Institute of Allergy and Infectious Diseases (NIAID) [U01 AI068632], the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Mental Health (NIMH) [AI068632]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was supported by the Statistical and Data Analysis Center at Harvard School of Public Health, under the National Institute of Allergy and Infectious Diseases cooperative agreement #5 U01 AI41110 with the Pediatric AIDS Clinical Trials Group (PACTG) and #1 U01 AI068616 with the IMPAACT Group. Support of the sites was provided by the National Institute of Allergy and Infectious Diseases (NIAID) and the NICHD International and Domestic Pediatric and Maternal HIV Clinical Trials Network funded by NICHD (contract number N01-DK-9-001/HHSN267200800001C).
EV Cappparelli has consulted for Abbott Laboratories, Cerexa Pharmaceuticals, Theravance, and Trius Pharmaceuticals. M Mirochnick has consulted for Abbott Laboratories and Farmanguinhos/Fiocruz (the official laboratory of the Ministry of Health of Brazil). C Hu consults for the Harvard School of Public Health. BM Best serves on Data and Safety Monitoring Boards for PPD Development, LP and Vertex Pharmaceuticals.
Footnotes
Presented in part at the: 14th Conference on Retroviruses and Opportunistic Infections (February 25–28, 2007; Los Angeles, California); abstract L-1005.
Conflicts of Interest: None of the other authors have a conflict of interest to declare.
References
- 1.Panel on Treatment of HIV-Infected Pregnant Women and Prevention of Perinatal Transmission. Recommendations for Use of Antiretroviral Drugs in Pregnant HIV-1-Infected Women for Maternal Health and Interventions to Reduce Perinatal HIV Transmission in the United States. Available at http://aidsinfo.nih.gov/contentfiles/lvguidelines/PerinatalGL.pdf. Accessed July 9, 2013.
- 2.Buckoreelall K, Cressey TR, King JR. Pharmacokinetic optimization of antiretroviral therapy in pregnancy. Clin Pharmacokinet. 2012;51:639–59. doi: 10.1007/s40262-012-0002-0. [DOI] [PubMed] [Google Scholar]
- 3.Cao Y, Han Y, Xie J, et al. Impact of a tenofovir disoproxil fumarate plus ritonavir-boosted protease inhibitor-based regimen on renal function in HIV-infected individuals: a prospective, multicenter study. BMC Infect Dis. 2013;13:301. doi: 10.1186/1471-2334-13-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen TA, Jensen D, Tolstrup M, et al. Comparison of bone and renal effects in HIV-infected adults switching to abacavir or tenofovir based therapy in a randomized trial. PLoS One. 2012;7:e32445. doi: 10.1371/journal.pone.0032445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delahunty T, Bushman L, Fletcher CV. Sensitive assay for determining plasma tenofovir concentrations by LC/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830:6–12. doi: 10.1016/j.jchromb.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 6.Delahunty T, Bushman L, Robbins B, et al. The simultaneous assay of tenofovir and emtricitabine in plasma using LC/MS/MS and isotopically labeled internal standards. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:1907–14. doi: 10.1016/j.jchromb.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gilead Sciences. Viread® [package insert] 2003 [Google Scholar]
- 8.Colbers AP, Hawkins DA, Gingelmaier A, et al. The pharmacokinetics, safety and efficacy of tenofovir and emtricitabine in HIV-1-infected pregnant women. AIDS. 2012;27:739–48. doi: 10.1097/QAD.0b013e32835c208b. [DOI] [PubMed] [Google Scholar]
- 9.Krauer B, Krauer F, Hytten FE. Drug disposition and pharmacokinetics in the maternal-placental-fetal unit. Pharmacol Ther. 1980;10:301–28. doi: 10.1016/0163-7258(80)90085-6. [DOI] [PubMed] [Google Scholar]
- 10.Benaboud S, Hirt D, Launay O, et al. Pregnancy-related effects on tenofovir pharmacokinetics: a population study with 186 women. Antimicrob Agents Chemother. 2012;56:857–62. doi: 10.1128/AAC.05244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirt D, Ekouevi DK, Pruvost A, et al. Plasma and intracellular tenofovir pharmacokinetics in the neonate (ANRS 12109 trial, step 2) Antimicrob Agents Chemother. 2011;55:2961–7. doi: 10.1128/AAC.01377-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirt D, Urien S, Ekouevi DK, et al. Population pharmacokinetics of tenofovir in HIV-1-infected pregnant women and their neonates (ANRS 12109) Clin Pharmacol Ther. 2009;85:182–9. doi: 10.1038/clpt.2008.201. [DOI] [PubMed] [Google Scholar]
- 13.Flynn PM, Mirochnick M, Shapiro DE, et al. Pharmacokinetics and safety of single-dose tenofovir disoproxil fumarate and emtricitabine in HIV-1-infected pregnant women and their infants. Antimicrob Agents Chemother. 2011;55:5914–22. doi: 10.1128/AAC.00544-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Best BM, Mirochnick M, Capparelli EV, et al. Impact of pregnancy on abacavir pharmacokinetics. AIDS. 2006;20:553–60. doi: 10.1097/01.aids.0000210609.52836.d1. [DOI] [PubMed] [Google Scholar]
- 15.Moodley J, Moodley D, Pillay K, et al. Pharmacokinetics and antiretroviral activity of lamivudine alone or when coadministered with zidovudine in human immunodeficiency virus type 1-infected pregnant women and their offspring. J Infect Dis. 1998;178:1327–33. doi: 10.1086/314431. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Livingston E, Patil S, et al. Pharmacokinetics of didanosine in antepartum and postpartum human immunodeficiency virus–infected pregnant women and their neonates: an AIDS clinical trials group study. J Infect Dis. 1999;180:1536–41. doi: 10.1086/315067. [DOI] [PubMed] [Google Scholar]