

Abstract
Background
The molecular regulation of heart development is regulated by cis- and trans-factors acting on the genome and epigenome. As a class of important regulatory RNAs, the role of long non-coding RNAs (lncRNAs) in human heart development is still poorly understood. Furthermore, factors that interact with lncRNAs in this process are not well characterized.
Methods and Results
Utilizing RNA sequencing, we systematically define the contrasting lncRNA expression patterns between fetal and adult heart. We report that lncRNAs up-regulated in adult versus fetal heart have different sequence features and distributions. For example, the adult heart expresses more sense lncRNAs compared to fetal heart. We also report the co-expression of lncRNAs and neighboring coding genes that have important functions in heart development. Importantly, the regulation of lncRNA expression during fetal to adult heart development appears to be due in part to the coordination of specific developmental epigenetic modifications such as H3K4me1 and H3k4me3. The expression of promoter-associated lncRNAs in adult and fetal heart also appears to be related to these epigenetic states. Finally, transcription factor binding analysis suggests that lncRNAs are directly regulating cardiac gene expression during development.
Conclusions
We provide a systematic analysis of lncRNA control of heart development that gives clues to the roles that specific lncRNAs play in fetal and adult hearts.
Keywords: cardiac development, genomics, gene regulation, heart development, long noncoding RNAs, epigenetic modification
Introduction
Heart development is a dynamic process that involves transcriptome expression changes and cis- (enhancer and promoter activities) and trans- (transcription factor binding) regulation. Much previous work has established the regulatory mechanisms in this process.1–3 As the importance of non-coding RNAs in regulating expression and epigenetic modifications is gaining recognition in heart development, researchers have discovered long non-coding RNA (lncRNA) that directly regulate heart development and disease.4, 5 For example, the lncRNA Fendrr binds to the PRC2 and TrxG/MLL complex and acts as a modulator of chromatin signatures to regulate gene activity in mouse heart.6 The lncRNA Bvht also interacts with a component of the PRC2 complex during cardiac commitment.7 Interestingly, lncRNAs have been implicated in common cardiac congenital abnormalities such as ventricular septal defects.8 They have also been implicated in aberrant chromatin conformations in the region of enhancer, silencer, and insulators during mouse heart development, as has been shown for the antisense lncRNA KCNQ1OT1.9 However, although many lncRNAs are expressed during heart development, their precise functions are largely unknown.
In addition to lncRNAs, cardiac development is also influenced by epigenetic modifications of cis-elements such as enhancers and promoters. Chromatin status in particular contributes to the coordination of downstream gene expression changes.10 Several groups have proposed integrated methods for distinguishing different chromatin states that change the activities of enhancers and promoters at different developmental stages. For example, work in mouse embryonic stem cells (ESCs) and adult tissues suggest that histone H3K4me1 exists primarily in enhancer regions, whereas H3K27ac differentiates active from inactive/poised enhancers.11 Similar findings have also been shown in human ESCs.12 Other work has defined the enhancer features important for mouse ESC differentiation, such as active enhancers with H3K4me1, H3K27ac, and H3K36me3, as well as poised enhancers defined by the presence or absence of H3K27me3 and H3K9me3.13
Perhaps not surprisingly, lncRNA expression is also coordinated by chromatin modification status14 similar to protein-coding genes. In analyzing RNA bound to chromatin-modifying complexes, thousands of strongly conserved lncRNAs have been identified in K4-K36 domains in human and mouse that regulate ESC pluripotency and cell proliferation.15, 16 This suggests that lncRNAs might be organizing enhancer/promoter activities and chromatin modifications, which in turn regulate gene expression during processes such as heart development. In fact, recent work with mouse ESCs and heart has shown that lncRNAs influence cardiac development via enhancer regulatory activities.14 However, the exact transcriptional regulatory mechanisms utilized by lncRNAs, including the coordination of epigenetic modifications, during human development between the fetal and adult heart stages has not been systematically studied.
In this study, to acquire a complete map of contrasting lncRNA expression and potential target gene networks between human fetal and adult heart, we utilize RNA sequencing to determine the transcriptome expression changes between the two states, with a focus on lncRNA activities. Next, we identify potential target genes of these lncRNAs through a bioinformatic correlation network that clusters lncRNAs into specific functional pathways. To further explore the correlation of lncRNA expression and epigenetic modification, we subsequently classify lncRNAs into enhancer- (elncRNA) and promoter-associated (plncRNA) groups according to the chromatin status in putative regulatory regions for each lncRNA. Our analysis defines the repertoire of cardiac-specific non-coding transcripts that are unique to the human fetal and adult heart stages, and enhances our understanding of the importance that lncRNAs play in heart development. Finally, we have created a database, Heart Development-Associated LncRNA Database (HDALD), which tabulates the differentially expressed lncRNAs in our analysis and is available at http://210.42.113.162/Heart/index.php.
Methods
Sample acquisition
All human heart samples were obtained following the guidelines of the Stanford University Institutional Review Board (IRB). Two fetal heart tissues (16 weeks and 17 weeks) were acquired from Stem Express (Placerville, CA). Cardiac tissue was transported in ice-cold UW (University of Wisconsin) solution and cut into small cubes (< 500 mm2), then immediately stored in liquid nitrogen. Two additional human fetal heart RNA-seq datasets (GSM1059494, 17 weeks and GSM1059495, 13 weeks), and three normal adult heart RNA-seq datasets (GSM1101970, GSM1698563, and GSM1698564) were also included in our analysis.
RNA extraction and quantitative RT-PCR analysis
Total RNA was isolated from human cardiac tissues using Qiagen miRNeasy Kit (Qiagen Sciences, Inc, Germantown, MD). Briefly, human cardiac tissues were ground into fine powder in liquid nitrogen with mortar and pestle, and then homogenized in Qiagen miRNeasy lysis buffer in a glass Dounce homogenizer. Afterward, total RNA was extracted according to the manufacturer’s instructions. For reverse transcription, we used the High Capacity RNA-to-cDNA kit (Life Technologies). For quantitative (qPCR) experiments, we used custom primer sets (Table S1) on a StepOne™ Real-Time PCR System (Life Technologies). The experiments were run in triplicate per targeted gene per sample. Expression values were normalized to the average expression level of 18S rRNA.
RNA sequencing
Sonication of cDNA was performed to produce an average fragment size of 280 bps and Illumina sequencing adapters were ligated to 500 ng of cDNA using NEBNext® mRNA Library Prep Reagent Set (New England Biolabs, Ipswich, MA). PCR was performed on the adapter-ligated cDNA using the following conditions: denaturation 98°C 30 seconds, followed by 12 cycles of denaturation 98°C 10 seconds, annealing 65°C 30 seconds, and extension 72°C 30 seconds, and ending with an extension step at 72°C for 5 minutes. Libraries were submitted to the Stanford Stem Cell Institute Genome Center for sequencing using Illumina’s HiSeq2000 platform. Paired end sequencing was performed with an average length of 100 bps (2×100). All RNA-Seq data are uploaded to the GEO database (GSE68279).
RNA sequencing data analysis
All RNA-seq data were mapped to human reference genome hg19 using Tophat17, 18 with default parameters. mRNA and lncRNA information were downloaded from the UCSC and RefSeq databases; multiple transcripts at the same genomic locations were merged into one unique transcript to yield the longest exon possible. A single GTF file was created for use as the reference annotation file. The alignment BAM files were then sorted, converted into SAM files with SAMtools19, and then subjected to read counting using the Python package HTSeq (http://www-huber.embl.de/users/anders/HTSeq/doc/overview.html). We used Bioconductor package DESeq20 for all differential expression (DE) analysis from the raw counts. To eliminate batch effect, we included batch as a factor in our DE analysis. The read counts were converted into RPKM (Reads Per Kilobase of exon model per Million mapped reads) and log2 transformed. The RPKM of lncRNAs were clustered and visualized as principal component analysis (PCA) and heatmaps in R.
Correlation of lncRNA and neighboring coding genes
Neighboring coding genes located 20 kb upstream and downstream of lncRNAs were assessed for co-regulation using Pearson correlation. Significantly correlated coding genes with lncRNAs were treated as potential target pairs. Networks and functional analyses were generated using Ingenuity Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). The P-value was calculated by IPA using the right-tailed Fisher Exact Test.
ChIP-seq data analysis
ChIP-seq data for H3K4me1 and H3K4me3 marks in human fetal heart and adult tissues were downloaded from the NIH Epigenomics Roadmap Project.21 All FASTQ files were aligned to hg19 using Bowtie.22 Peaks and enriched regions were called by MACS23 and annotated by Homer.24 Heatmaps were generated with DeepTools.25 All RNA-seq and ChIP-seq signals were visualized using IGV.26
Classification of enhancer- and promoter- associated lncRNAs
The transcriptional start sites (TSS) of lncRNAs were extracted from the annotation files. The genomic coordination of chromatin modification peaks 2 kb upstream or downstream of each TSS were clustered and used to classify the lncRNAs into enhancer- (elncRNA) or promoter- (plncRNA) associated groups. The differential expression analyses of elncRNA and plncRNA in fetal and adult heart were performed using the Bioconductor package DESeq.
Detection of transcription factor binding sites and human single nucleotide polymorphism sites
Human conserved transcription factor binding sites were downloaded from the UCSC database and mapped to putative lncRNA promoter regions (−2 kb to TSS). Ratios of potential binding events were calculated and displayed as heatmaps using TM4 (www.tm4.org). Human single nucleotide polymorphisms (SNPs) were obtained from the dbSNP142 Database. All genomic coordination analyses were performed by in-house Perl scripts, which are available upon request.
Results
Identification of lncRNAs enriched in fetal or adult heart
By integrating the UCSC gene database and NCBI RefSeq database, we acquired all available lncRNA annotations for putative human lncRNAs. LncRNAs that overlap by one exon or more and are transcribed from the same strand were merged into a single lncRNA that represented the longest possible transcript. Those RNAs shorter than 250 bp, including small nuclear RNAs, were removed from our annotation file. Using this pipeline, we identified 12,180 non-redundant lncRNAs with unique genomic locations. RNA sequencing revealed the expression levels (RPKM) of each lncRNA in three adult and four fetal heart samples (Figure 1A). Of these, 4,918 lncRNAs were expressed in both adult and fetal heart (average RPKM>0.01). Through differential expression analysis, we further identified 277 lncRNAs (Benjamini and Hochberg (BH) adjusted P-value <0.05, fold change (FC) >2.5) that were differentially expressed between adult and fetal heart. Among them, 164 (BH adjusted P-value <0.05, FC >2.5) were up-regulated in fetus and 113 (BH adjusted P-value <0.05, FC >2.5) were up-regulated in adult heart (Figure 1B). The remaining lncRNAs did not exhibit significant differential expression between adult and fetal heart.
Figure 1.
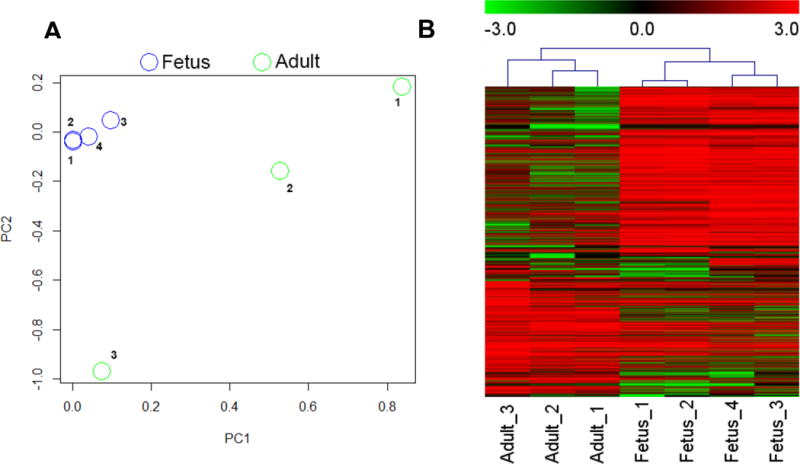
Transcriptome patterns in fetal and adult heart. (A) Principal component analyses of 4 fetal (blue) and 3 adult (green) heart samples based on transcriptome expression. Fetus_1 (GSM1059494), Fetus_2 (GSM1059495), Fetus_3 and Fetus_4 (both in-house acquired fetal heart samples); Adult_1 (GSM1698563), Adult_2 (GSM1698564), and Adult_3 (GSM1101970). (B) Hierarchical clustering of differentially expressed transcripts in fetal and adult heart shows a distinct expression pattern that is unique to each developmental stage.
In order to understand the functional differences between lncRNAs up-regulated in adult heart compared to those in fetal heart, we first analyzed their respective sequence features. We found that 52% of fetal heart up-regulated lncRNAs have 2 to 4 exons, which is more than adult up-regulated lncRNAs (37%). Ten percent of fetal-up-regulated lncRNAs and 16% of adult-up-regulated lncRNAs have one exon (Figure 2A). Reviewing the length of differential expressed (DE) lncRNAs, we calculated that 85% of fetal and 72% of adult up-regulated lncRNAs ranged from 1 to 10 kbp (Figure 2B). Eight percent of fetal-up-regulated lncRNAs are between 250 bp to 1 kbp in length, a lower percentage than in adult-up-regulated lncRNAs (24%). In total, 7% of fetal heart up-regulated lncRNAs were longer than 10 kbps, which is slightly higher than the 4% seen in adult heart. Some interesting specific examples of annotated lncRNAs emerged from this analysis. For example, the 21 kb lncRNA MEG3 was up-regulated in fetal heart and is a known tumor suppressor.27 A second 382 bp lncRNA, BCYRN1, was up-regulated in adult heart and has been reported to be up-regulated and targeted by c-MYC in non-small-cell lung cancer.28
Figure 2.
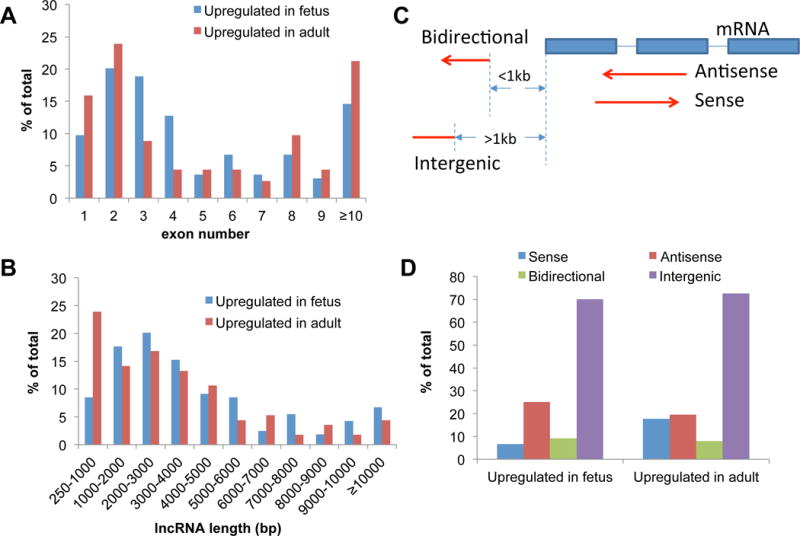
Characterization of lncRNAs up-regulated in fetal and adult heart. (A) The distribution of exon number in fetal and adult heart up-regulated lncRNAs. The x-axis represents the exon number of detected lncRNAs and y-axis represents the total percentage for the indicated group. (B) LncRNA length distribution in fetal and adult heart. The x-axis represents length of detected lncRNAs. (C) Schematic showing the four categories of lncRNAs based on their relative genomic locations to neighboring coding genes (mRNAs): Sense, Antisense, Bidirectional, and Intergenic. (D) Expression differences of the four types of lncRNAs in fetal and adult heart.
Based on the genomic positions of lncRNAs in relation to neighboring coding genes, we clustered significantly expressed lncRNAs into four types: sense (overlap with coding genes on sense strand), antisense (overlap with coding genes on antisense strand), bidirectional (distance to coding genes is less than 1 kb on reserve strand), and intergenic (located between coding genes) (Figure 2C). We observed that adult and fetal hearts have similar expression levels of antisense and intergenic lncRNAs (Figure 2D). However, the percentage of sense lncRNAs are more than two times greater in adult (18%) compared to fetal heart (7%). In contrast, the number of antisense lncRNAs is somewhat increased in fetal (25%) compared to adult heart (19%). Specific examples include the lncRNA LOC728407, which is located in the PARG transcript body and is up-regulated in adult heart. The host gene PARG is known to be associated with cell death.29 In a second example, the fetal heart up-regulated lncRNA SLC8A1-AS1 is located on the opposite strand of the protein coding gene SLC8A1, a protein involved in congenital heart disease.30
lncRNAs are involved in multiple networks during cardiac development
To determine how differentially expressed lncRNAs might function in cardiac development, we performed correlation analysis to screen for potential targets. As lncRNAs are often co-expressed with neighboring coding genes31, we first identified coding genes within 20 kb of lncRNAs and used Pearson correlations to detect significant correlations (Tables S2 and S3). In 164 fetal up-regulated lncRNAs, we identified 54 lncRNA-mRNA pairs that had significant expression correlations (|r|>0.7, P-value<0.05). Among them, 49 lncRNA-mRNA pairs showed positive correlations, with only 5 pairs showing negative correlation. Of 113 adult up-regulated lncRNAs, 5 and 15 lncRNA-mRNA pairs showed positive and negative correlations (|r|>0.7, P-value<0.05), respectively. For comparison, of the 4,641 non-significant DE lncRNAs, 317 showed positive correlation pairs and 121 showed negative correlation pairs. qPCR results validated one up-regulated lncRNA-mRNA pair in adult heart (TCONS_00018783/ANKRD1) and two pairs that were up-regulated in fetal heart (TCONS_00020859/TMTC2 and TCONS_00007979/WDR1) (Figure 3).
Figure 3.
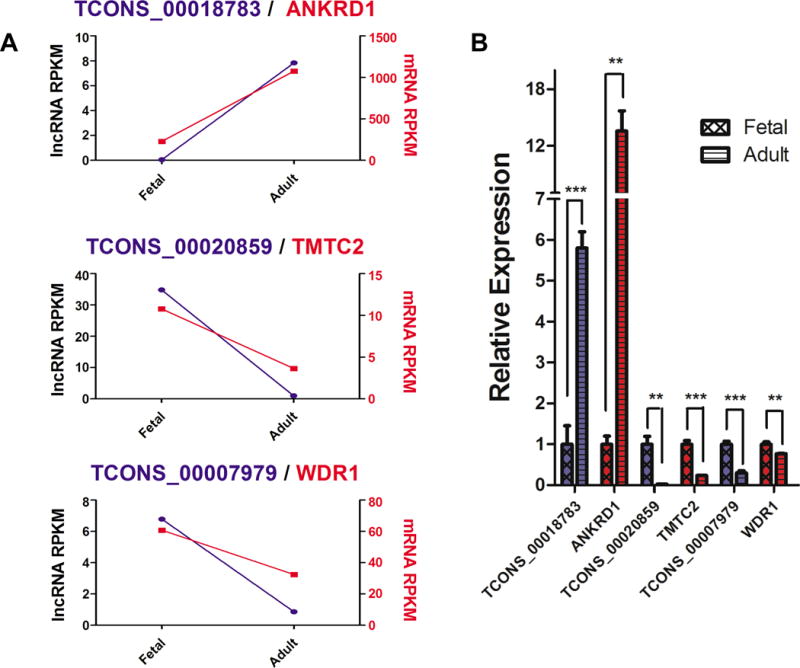
Co-expression of lncRNA-mRNA pairs between human fetal and adult heart. (A) Expression level of 3 pairs of lncRNA-mRNA from RNA-seq datasets. RPKM is used to define the expression level. Left y-axis represents lncRNA expression and right y-axis represents neighboring mRNA (protein-coding) gene expression. (B) qPCR validation of the co-expression of lncRNA-mRNA pairs. *P<0.05, **P<0.01, and ***P<0.001. Blue bars represent lncRNA expression and red bars represent neighboring protein-coding gene expression.
To explore the putative function of lncRNAs that are up-regulated in fetus or adult heart, we utilized Ingenuity Pathway Analysis (IPA) to classify lncRNAs into functional categories according to neighboring significantly correlated coding genes. In total, 54 coding genes were correlated to up-regulated lncRNAs in fetal heart, and 20 coding genes were correlated to up-regulated lncRNAs in adult heart. The top regulation networks for these co-expressed genes in adult and fetal heart are depicted in Figures S1 and S2, respectively. In fetal heart, these correlated genes have several known disease and biological functions: intrauterine growth retardation (−log P-value = 2.7), dilated cardiomyopathy type 1T (−log P-value = 2.6), and hypoplasia of ventricle (−log P-value = 2.61) (Figure 4A). In contrast, adult heart expresses correlated genes involved in familial hypertrophic cardiomyopathy type 9 (−log P-value = 3.01), tachyarrhythmia (−log P-value = 1.93), and cardiac output (−log P-value = 2.02).
Figure 4.
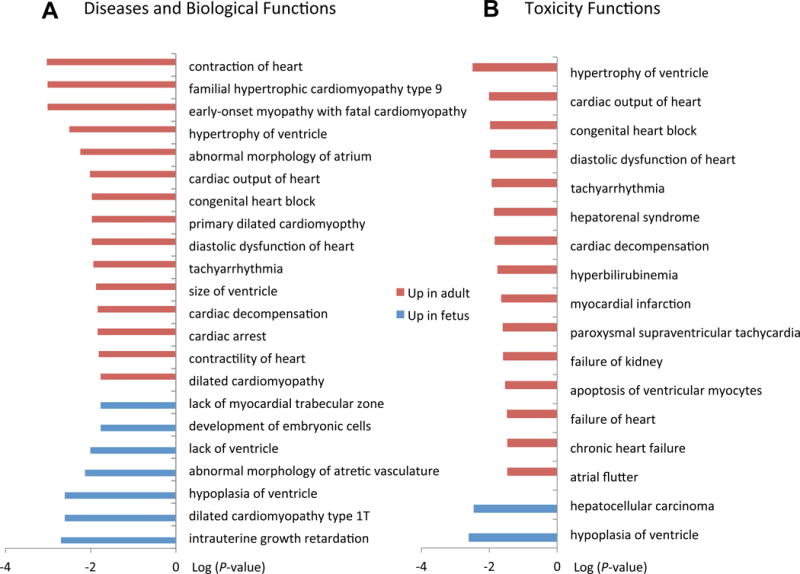
Functional clustering of lncRNAs in fetal and adult heart based on protein-coding gene co-expression. (A) IPA pathway analysis of diseases and biological functions of protein coding genes exhibiting co-expression with lncRNAs. (B) IPA Pathway analysis of toxicity functions. Pathways are ranked based on significance (−log P-value).
We next compared toxicity-related (Tox) functions between fetal and adult heart. LncRNAs up-regulated in fetal heart were again highly associated with genes regulating hypoplasia of ventricle (−log P-value = 2.6), suggesting a developmental role. LncRNAs up-regulated in adult heart were again highly associated with hypertrophy, cardiac output and tachyarrhythmias (Figure 4B). Finally, comparison of canonical pathways of lncRNA-neighboring genes revealed that actin cytoskeleton signaling (−log P-value = 1.74) is up-regulated in adult heart, whereas granzyme A signaling (−log P-value = 1.3) is up-regulated in fetal heart (Figure S3). The development and disease categories classified here suggest that the lncRNAs exhibiting correlated expression with neighboring genes may likewise be involved in processes of fetal heart development and adult heart physiology.
lncRNAs are associated with epigenetic modifications in fetal and adult hearts
Previous work has shown that lncRNAs function as epigenetic regulators of gene expression.14 In order to explore how lncRNA expression is regulated during cardiac development, we determined the relationship between lncRNA expression and histone modification in gene enhancers and promoters. Using ChIP-seq data from the NIH Roadmap Epigenomics Project21, we acquired chromatin modification information for histone 3 (H3k4me1 and H3k4me3) in human adult and fetal hearts. Each locus is treated as a potential poised or active enhancer (H3k4me1) or promoter (H3k4me3).12, 14 Across the entire genome, we identified 108,926 H3k4me1 and 24,818 H3k4me3 loci in adult heart, and 83,799 H3k4me1 and 32,356 H3k4me3 loci in fetal heart. We then classified lncRNAs into elncRNA or plncRNA groups by determining the distribution of H3k4me1 and H3k4me3 marks within 2 kb from the TSS for each lncRNA, similar to previous work using primary erythroid cells (Figure 5A).32 We identified 1,277 lncRNAs located near adult enhancer loci (adult elncRNAs), 1,090 lncRNAs located near fetal enhancer loci (fetal elncRNAs), and 1,935 lncRNAs located near both adult and fetus enhancers (overlap elncRNAs) (Figure 5B). Using the same method for analysis of promoter lncRNA associations, we identified 1,193 adult plncRNAs, 184 fetal plncRNAs, and 1,699 overlap plncRNAs.
Figure 5.
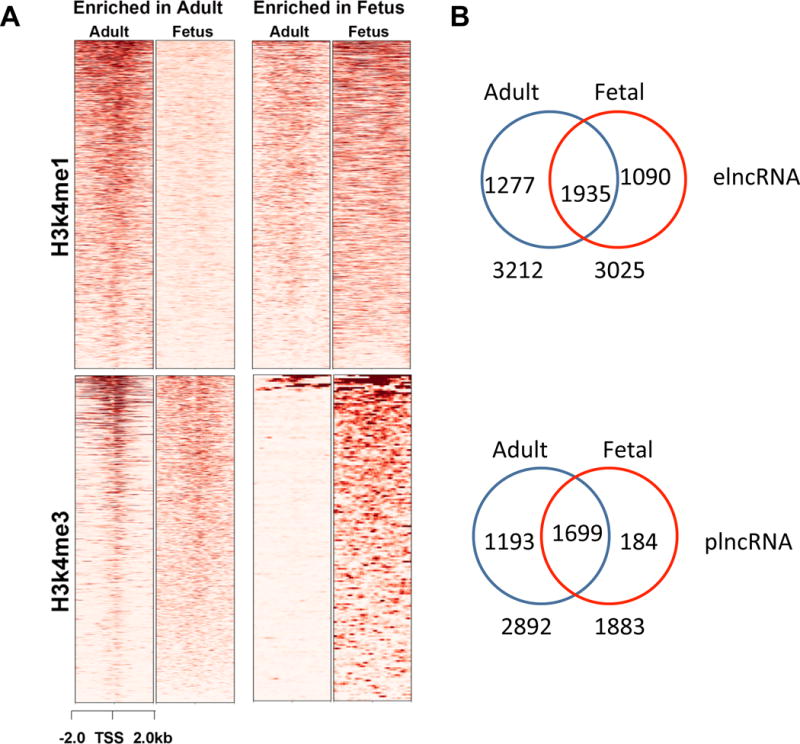
Identification of enhancer- and promoter-asociated lncRNAs in adult and fetal heart. (A) Heatmap of chromatin modifications in putative lncRNA regulatory regions. ChIP signals are shown for H3k4me1 (enhancer) and H3k4me3 (promoter) histone modifications within 2 kb of the TSS for each lncRNA. (B) Distribution of elncRNAs and plncRNAs between adult and fetal heart. Regions of overlap in Venn diagrams indicate lncRNAs that are up-regulated in both adult and fetal hearts.
To determine whether these epigenetic modifications in enhancer regions may be related to lncRNA expression, we performed differential expression analysis of our identified promoter- and enhancer-associated lncRNAs in adult and fetal heart. Our results show significantly different expression of plncRNAs in adult and fetal hearts (Figure 6A). In particular, adult plncRNAs show increased up-regulation (59%) compared to fetal heart (41%). By contrast, fetal plncRNAs show increased up-regulation in fetal (64%) compared to adult heart (36%), suggesting that the chromatin status of promoter regions is a predictor of lncRNA expression status. However, we did not find significantly different expression of elncRNAs between adult and fetal heart: 54% of adult elncRNAs were up-regulated in adult and 46% were up-regulated in fetus, while 49% and 51% of fetal elncRNAs were up-regulated in fetal and adult heart, respectively (Figure 6B). As our analysis used a 4 kb window surrounding the TSS of lncRNAs to identify putative regulatory enhancers, we excluded potential enhancers further upstream or downstream that may be regulating lncRNA expression.33 In contrast to enhancers, promoters are more often located near the TSS34, possibly explaining why we observed a more obvious correlation between plncRNA expression and promoter chromatin status.
Figure 6.
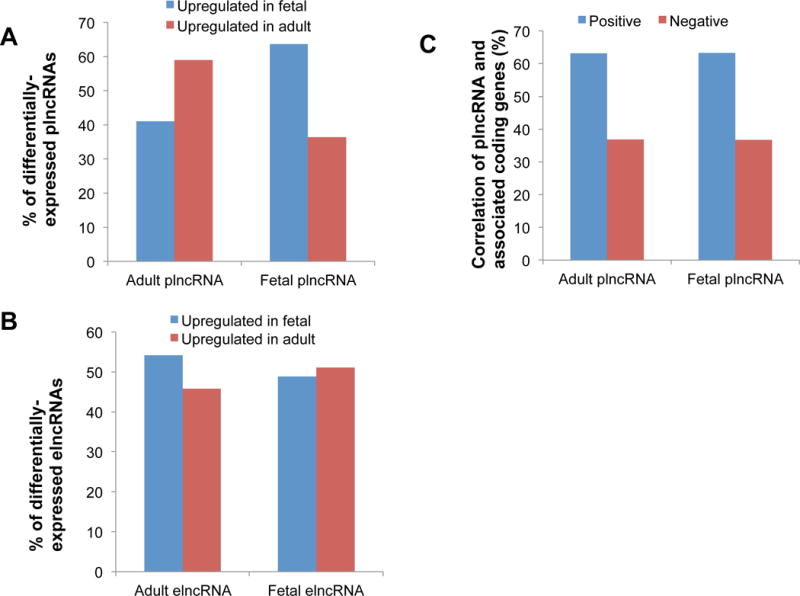
Expression patterns of enhancer- and promoter- associated lncRNAs in adult and fetus. (A) The total percentage of plncRNAs expressed in adult and fetal hearts. (B) The total percentage of elncRNAs expressed in adult and fetal hearts. (C) Percentage of co-expression of plncRNAs and associated coding genes that share the same putative promoter.
Finally, we detected a positive correlation between plncRNAs and coding genes that share the same promoter (promoter loci are located ±2 kb from the TSS in both lncRNA and coding gene). 63% of plncRNAs were positively correlated (r>0.7, P-value <0.05) and 37% of plncRNAs were negatively correlated (r<−0.7, P-value <0.05) with associated coding genes (Table S4, S5 and Figure 6C). We also analyzed human homologous Braveheart (Bvht) expression, an lncRNA that has been shown to be important in mouse heart commitment.7 We found that Bvht is highly expressed in fetal heart (Figure S4A) and is correlated with a number of coding genes (Figure S4B).
Transcription factor binding and SNPs in development-associated lncRNAs
One of the functions of lncRNAs is to act as chaperones for transporting and localizing transcription factors to specific genomic loci.35 Understanding the transcription factor binding sites (TFBS) in heart development that have associated lncRNAs may help elucidate the interplay between them in regulating cardiac development. Toward this goal, conserved TFBS (total of 81 factors) were downloaded from the UCSC Table Browser. We detected 651 and 486 TFBS upstream of fetal and adult heart up-regulated lncRNAs, respectively (Table S6 and Figure 7). In fetal up-regulated lncRNAs, transcription factors ATF (2.3%) and RREB-1 (2.2%) had more than two times the number of binding sites compared to adult up-regulated lncRNAs (0.6% and 0.8%, respectively). In contrast, adult heart up-regulated lncRNAs demonstrated a higher percentage of upstream binding with RORalpha1 (2.1%), Pbx1a (1.9%), and LCR-F1 (2.3%) compared to fetal heart (1.4%, 0.5%, and 1.7%, respectively) (Figure 7). Among these transcription factors, ATF plays a role in heart failure36 and LCR-F1 is involved in transcription activation in cardiomyocytes.37
Figure 7.
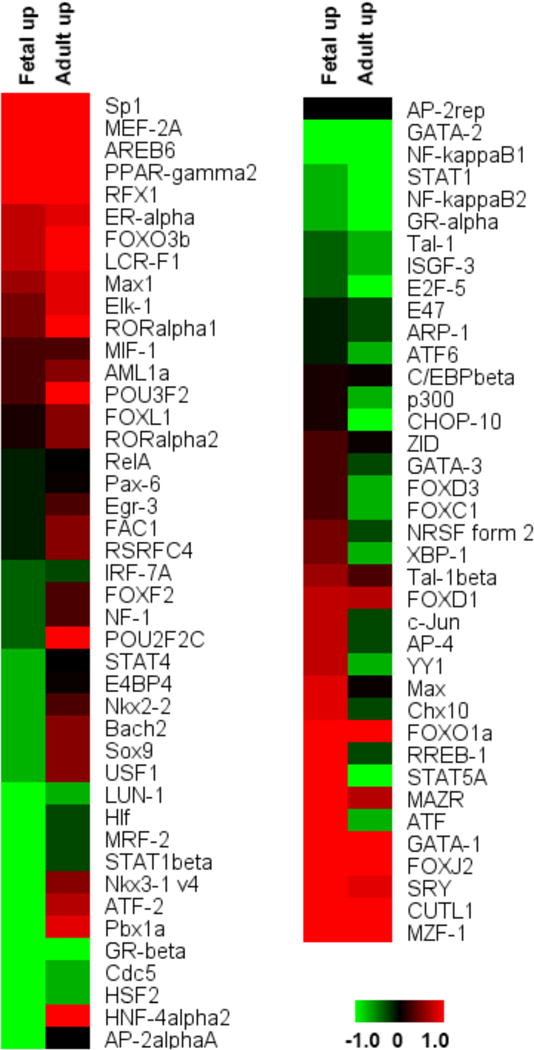
Conserved transcription factor binding sites in putative cardiac development lncRNA regulatory regions. Each row represents one potential TFBS for the indicated transcription factor in fetal and adult hearts, and colors indicate the relative downstream lncRNA expression (within 2 kb) of this putative regulatory region. Eighty-one human conserved transcription factors were used in this analysis. The range represents the log2 percentage of TFBS.
Because sequence mutations in the promoters of lncRNAs may negatively affect transcription factor binding,35 we next analyzed SNPs located in putative TFBS upstream of lncRNAs. We downloaded human dbSNP142 data from the UCSC browser and mapped them to the predicted TFBS. In promoters of adult up-regulated lncRNAs, we identified 68 TFBS in 50 lncRNAs that might be affected by 193 SNPs (Table S7). For example, the transcription factor POU2F2C had a higher percentage (2.1%) of binding in adult up-regulated lncRNAs than in fetal heart (0.8%), and this TFBS contains the SNP rs562165402 (Minor Allele Frequency (MAF) = 0.02%). In promoters of fetal up-regulated lncRNAs, we also identified 65 TFBS in 56 lncRNAs that could be affected by 219 SNPs (Table S8). RREB-1, which binds preferentially to fetal up-regulated lncRNAs, might be affected by SNP rs146951114 (MAF = 0.04%). We believe these sequence variants in lncRNA promoters provided a well-annotated repository for transcription factor binding analysis in cardiac development.
Discussion
In this study, we performed a genome-wide analysis of lncRNAs associated with heart development and their co-expressed protein coding genes. Building on previous studies that detected differentially expressed lncRNAs during mouse heart development,6, 7, 9, 31, 38 cardiac cell differentiation,39 and heart disease,8, 40 we report here the contrasting “lncRNA-omes” between human fetal and adult hearts. Furthermore, through expression correlations between lncRNAs and protein coding mRNAs based on genomic proximity, we construct a co-expression network that enables identification of important coding genes and lncRNAs in human heart development. This allows prediction of functional categories for the correlated lncRNAs, including various cardiac development pathways such as cardiomyocyte differentiation, apoptosis, and heart formation and failure. We have also included analysis of the sequence features and genomic positions of significant fetal and adult heart up-regulated lncRNAs that exhibit striking differences between the two stages. Until future research elucidates the function of these lncRNA sub-classes, we can only hypothesize their specific biological roles during human development. Finally, our HDALD database can be used to search the genomic locations of differentially expressed lncRNAs and view their expression levels from RNA-seq data. We believe these data provide a new useful resource for exploring novel markers of heart disease and development disorders.
Based on public ChIP-seq data, our analysis also suggests that lncRNAs maybe important regulators of protein-coding gene expression. These findings confirm previous work that proposed lncRNAs as epigenetic regulators.31 Here we further observe the possible effect of epigenetic modifications acting on lncRNA expression between human fetal and adult heart. We determined specific histone modifications that were different across both heart stages, and observed that promoter activities (defined by H3K4me3 histone modifications) were associated with lncRNA expression differences in fetal and adult heart. Although experimental validation is needed, our results suggest that specific chromatin modifications are correlated with lncRNA expression in the human heart. Finally, we detected potential transcription factor binding sites in heart development-associated lncRNAs. We found a clear distinction between TFBS in fetal vs. adult heart and which may be related to important cardiac developmental processes. DNA sequence variants in these TFBS may affect transcription factor binding events, and our results revealed numerous SNPs associated with TFBS in adult and fetal heart. Further exploration of these SNPs through experimental studies may yield important DNA mutations that affect fetal-to-adult heart development.
In conclusion, we performed a genome-wide exploration of lncRNA changes in human heart development from the fetus to adult stage, and compared the sequence features of differentially expressed lncRNAs. We found that lncRNA changes were associated with protein-coding genes that play important functions in cardiac development and disease. Our results show that lncRNAs are frequently co-expressed with neighboring coding genes, the latter of which are involved in important heart development processes. Furthermore, we also inspected the chromatin modifications in this process and classified lncRNAs into enhancer- and promoter-associated types. The expression differences in promoter-associated lncRNAs were correlated with the chromatin modifications. These results provide a noncoding genomic landscape for human cardiac development, and demonstrate the interplay between epigenetic modifications and transcription factor binding.
Supplementary Material
Clinical Perspective.
Heart development is a dynamic process that requires the interplay of gene expression with cis- (enhancer and promoter activities) and trans- (transcription factor binding) regulation. Interestingly, a relatively new class of non-coding RNAs, long non-coding RNAs (lncRNA), has recently been shown to also regulate gene expression in pathways related to heart development and disease. However, the molecular mechanism(s) by which lncRNAs exert their effect on the heart are largely unknown. In this study, we use RNA-seq and associated epigenetic and transcription factor data from fetal and adult hearts to perform genome-wide analyses of differentially expressed lncRNAs. We report significant changes in the “lncRNA-ome” between these two important stages of the heart. Through expression correlation studies of lncRNAs and nearby protein coding mRNAs, we then construct a co-expression network that enables identification of important coding and non-coding gene pairs in heart development. This network enables prediction of the functional categories of differentially expressed lncRNAs in fetal and adult heart, which include critical cardiac development pathways such as cardiomyocyte differentiation, apoptosis, heart formation and failure. We also analyze the epigenetic modifications in putative promoter and enhancer regions for important lncRNAs and provide in silico evidence of critical transcription factor binding events at these regulatory regions. In conclusion, our analysis defines the repertoire of cardiac-specific non-coding transcripts that are unique to the human fetal and adult heart stages, and enhances our mechanistic understanding of these lncRNAs’ functional role in human heart development.
Acknowledgments
The authors would like to thank the Stanford Stem Cell Institute Genome Center for their sequencing expertise, and Blake Wu for help in editing the manuscript.
Funding Sources: This study was funded by the American Heart Association Established Investigator Award, the National Institutes of Health R01 HL130020, R01 HL123968, and R01 HL126527 (JCW).
Footnotes
Journal Subject Terms: Gene Expression and Regulation; Epigenetics; Developmental biology; Myocardial Biology
Conflict of Interest Disclosures: None.
References
- 1.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–1927. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruneau BG. Transcriptional regulation of vertebrate cardiac morphogenesis. Circ Res. 2002;90:509–519. doi: 10.1161/01.res.0000013072.51957.b7. [DOI] [PubMed] [Google Scholar]
- 3.Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schonrock N, Harvey RP, Mattick JS. Long noncoding RNAs in cardiac development and pathophysiology. Circ Res. 2012;111:1349–1362. doi: 10.1161/CIRCRESAHA.112.268953. [DOI] [PubMed] [Google Scholar]
- 5.Papait R, Kunderfranco P, Stirparo GG, Latronico MV, Condorelli G. Long noncoding RNA: A new player of heart failure? J Cardiovasc Transl Res. 2013;6:876–883. doi: 10.1007/s12265-013-9488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song G, Shen Y, Zhu J, Liu H, Liu M, Shen YQ, et al. Integrated analysis of dysregulated lncRNA expression in fetal cardiac tissues with ventricular septal defect. PloS One. 2013;8:e77492. doi: 10.1371/journal.pone.0077492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korostowski L, Sedlak N, Engel N. The KCNQ1OT1 long non-coding RNA affects chromatin conformation and expression of KCNQ1, but does not regulate its imprinting in the developing heart. PLoS Genet. 2012;8:e1002956. doi: 10.1371/journal.pgen.1002956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27AC separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ounzain S, Pezzuto I, Micheletti R, Burdet F, Sheta R, Nemir M, et al. Functional importance of cardiac enhancer-associated noncoding RNAs in heart development and disease. J Mol Cell Cardiol. 2014;76:55–70. doi: 10.1016/j.yjmcc.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trapnell C, Pachter L, Salzberg SL. Tophat: Discovering splice junctions with RNA-seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. Deeptools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balik V, Srovnal J, Sulla I, Kalita O, Foltanova T, Vaverka M, et al. Meg3: A novel long noncoding potentially tumour-suppressing RNA in meningiomas. J Neurooncol. 2013;112:1–8. doi: 10.1007/s11060-012-1038-6. [DOI] [PubMed] [Google Scholar]
- 28.Hu T, Lu YR. BCYRN1, a c-MYC-activated long non-coding RNA, regulates cell metastasis of non-small-cell lung cancer. Cancer Cell Int. 2015;15:36. doi: 10.1186/s12935-015-0183-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirai H, Poetsch AR, Gunji A, Maeda D, Fujimori H, Fujihara H, et al. PARG dysfunction enhances DNA double strand break formation in S-phase after alkylation DNA damage and augments different cell death pathways. Cell Death Dis. 2013;4:e656. doi: 10.1038/cddis.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costain G, Lionel AC, Ogura L, Marshall CR, Scherer SW, Silversides CK, et al. Genome-wide rare copy number variations contribute to genetic risk for transposition of the great arteries. Int J Cardiol. 2015;204:115–121. doi: 10.1016/j.ijcard.2015.11.127. [DOI] [PubMed] [Google Scholar]
- 31.Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C, Dorn GW., 2nd Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci U S A. 2014;111:12264–12269. doi: 10.1073/pnas.1410622111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marques AC, Hughes J, Graham B, Kowalczyk MS, Higgs DR, Ponting CP. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biol. 2013;14:R131. doi: 10.1186/gb-2013-14-11-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 34.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 35.Dorn GW, 2nd, Matkovich SJ. Epitranscriptional regulation of cardiovascular development and disease. J Physiol. 2015;593:1799–1808. doi: 10.1113/jphysiol.2014.283234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks AC, DeMartino AM, Brainard RE, Brittian KR, Bhatnagar A, Jones SP. Induction of activating transcription factor 3 limits survival following infarct-induced heart failure in mice. Am J Physiol Heart Circ Physiol. 2015;309:H1326–1335. doi: 10.1152/ajpheart.00513.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang F, Wagner M, Siddiqui MA. Structure, expression, and functional characterization of the mouse CLP-1 gene. Gene. 2002;292:245–259. doi: 10.1016/s0378-1119(02)00596-6. [DOI] [PubMed] [Google Scholar]
- 38.Zhu JG, Shen YH, Liu HL, Liu M, Shen YQ, Kong XQ, et al. Long noncoding RNAs expression profile of the developing mouse heart. J Cell Biochem. 2014;115:910–918. doi: 10.1002/jcb.24733. [DOI] [PubMed] [Google Scholar]
- 39.Zhu S, Hu X, Han S, Yu Z, Peng Y, Zhu J, et al. Differential expression profile of long non-coding RNAs during differentiation of cardiomyocytes. Int J Med Sci. 2014;11:500–507. doi: 10.7150/ijms.7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, et al. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J. 2015;36:353–368a. doi: 10.1093/eurheartj/ehu180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.