

Abstract
Adoptive T cell-based immunotherapies can mediate complete and durable regressions in patients with advanced cancer, but current response rates remain inadequate. Maneuvers to improve the fitness and antitumor efficacy of transferred T cells have been under extensive exploration in the field. Small non-coding microRNAs have emerged as critical modulators of immune system homeostasis and T cell immunity. Here, we summarize recent advances in our understanding of the role of microRNAs in regulating T cell activation, differentiation, and function. We also discuss how microRNA therapeutics could be employed to fine-tune T cell receptor signaling and enhance T cell persistence and effector functions, paving the way for the next generation of adoptive immunotherapies.
Keywords: microRNA (miRNA); tumor immunotherapy; adoptive cell transfer; T cell differentiation, CD8+ T cells
Introduction
Adoptive transfer of naturally occurring or genetically redirected tumor-reactive T cells has emerged as one of the most successful immunotherapeutic treatments for patients with advanced hematological malignancies and solid cancers [1, 2]. This therapeutic modality can result in complete and durable responses in a significant fraction of patients with metastases refractory to conventional treatments, but the overall response rate remains unsatisfactory. Advances in our understanding of the biological mechanisms behind the effectiveness of adoptive T cell therapy have underscored the importance of the qualities of transferred T cells [3] and revealed the complexity of the inhibitory barriers posed by the host and tumor cells that need to be overcome for the success of the treatment [4, 5]. Among T cell factors, the avidity of the T cell receptor (TCR) or chimeric antigen receptor (CAR) [6, 7], the proliferative and survival capacities [8–11], and the ability to sustain effector functions within the tumor [12] have been shown to be crucial determinants for triggering the eradication of malignant cells.
Extensive work has been done to develop new strategies to improve these diverse aspects of the T cell infusion product. For instance, in vitro mutagenesis and selection of affinity-matured TCRs or antibodies have been employed to generate high-affinity TCR and CAR [13, 14]. Alternatively, HLA-mismatched donors [15], HLA-transgenic mice [16] and more recently transgenic mice carrying human TCRαβ gene loci and HLA-A2 [17] have also been used to circumvent tolerance and isolate high-avidity TCRs. Gene engineering approaches to overexpress cytokines [18–20], co-stimulatory molecules [21], anti-apoptotic proteins [22, 23] and cytotoxic molecules [24] have been employed to enhance proliferation, survival and effector functions of adoptively transferred T cells. Because these properties are tightly linked with the maturation state of T cells, there has been an increased interest in developing novel approaches to alter T cell differentiation. These maneuvers include the modification of the cytokine milieu used for in vitro cell expansion [25, 26], the manipulation of T cell transcriptional programs [27, 28] and the modulation of T cell metabolism [29–31].
MicroRNA (miRNA) are 21–23 base pair long non-coding RNAs, which mediate post-transcriptional gene silencing [32]. There is now mounting evidence demonstrating that miRNAs are critical players in regulating a wide range of cellular processes including cell proliferation, differentiation, apoptosis, and metabolism [33]. Dysregulation of miRNA expression and activity has been associated with malignant transformation and metastatic behaviors [34]. The past few years have witnessed an explosion of studies aiming at harnessing miRNAs for the treatment of patients with cancer [35, 36]. A largely tumor cell-centric view has led to the development of miRNA therapeutics designed to either block the function of oncogenic miRNAs or to upregulate the expression of tumor-suppressive miRNAs [35, 36]. Here, we propose an entirely different miRNA-based approach for cancer therapy. After summarizing basic aspects of miRNA biology and describing the role of miRNAs in T cell biology, we will discuss how miRNA therapeutics could be employed to enhance the anti-tumor efficacy of adoptively transferred tumor-specific T cells.
miRNA biogenesis and function
MiRNA genes are located in intronic, exonic, or untranslated regions and encoded together with host genes. They are first transcribed by RNA polymerase II into 500–3000 nucleotide pri-miRNAs containing one or multiple stem-loop sequences, and subsequently cleaved by the Drosha-DGCR8 complex to form a 60–100 nucleotide double-stranded pre-miRNA hairpin [37–39]. Pre-miRNAs are then exported into the cytoplasm by Ran GTPase and Exportin 5 and further processed into an imperfect 22-mer miRNA:miRNA duplex by the Dicer protein complex [39, 40]. One of the strands from this duplex – the mature miRNA – binds to Argonaute (AGO) and is incorporated into the RNA-induced silencing complex (RISC) to repress target gene expression [32] (Fig. 1).
Fig. 1. MicroRNA biogenesis.
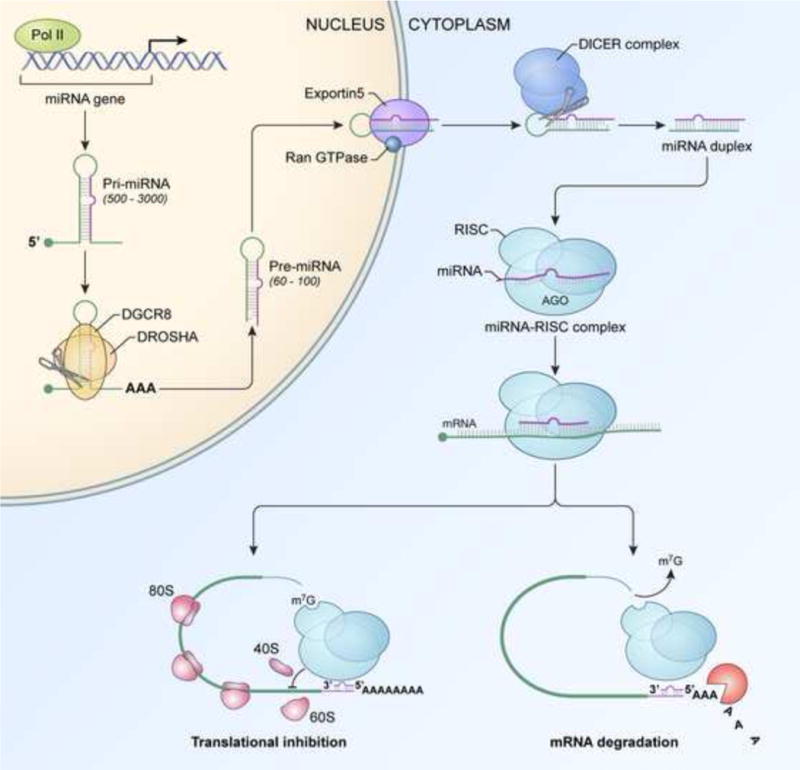
The miRNA gene is transcribed into pri-miRNA by RNA polymerase II (Pol II) within the nucleus and processed into Pre-miRNA by the DROSHA-DGCR8 complex. Pre-miRNA is subsequently transported by Exportin5 and Ran GTPase into the cytoplasm and further processed by the DICER complex into a miRNA:miRNA duplex. Finally, mature miRNA binds to AGO (Argonaute) and is incorporated into the RISC (RNA-induced silencing complex), leading to mRNA degradation and inhibition of protein translation.
Target identification and inhibition is directed by the miRNA ‘seed’ sequence, which is comprised of nucleotides spanning from position 2 to 7 and forms a perfect or near-perfect complementary pair with a 6–8 bp-long motif located within the 3′UTR of target mRNAs [32, 39]. Once miRNA identifies and binds to the target 3′UTR, the associated ‘miRISC’ complex initiates mRNA degradation by deadenylation, 5-terminal cap removal and direct exonucleolytic cleavage [32]. The miRISC complex can also block protein translation by interfering with 5′cap recognition and 40S and 60S ribosomal subunit recruitment and assembly, resulting in defective formation of the 80S ribosomal complex [41]. Therefore, miRNAs restrain complementary targets at both mRNA and protein levels.
Although their inhibitory effects on individual proteins are subtle – usually less than 2-fold – miRNAs are potent cellular modulators due to their ability to target multiple molecules within a particular pathway or diverse proteins in converging pathways or biological processes [42]. Furthermore, a single protein can be influenced by several individual miRNAs targeting shared or distinct sequences within the target 3′UTR [42]. Thus, miRNA can potently regulate biological networks by cumulatively or cooperatively inhibiting consonant molecules. Conversely, they may fine-tune particular signaling pathways by targeting positive and negative regulatory components. Altogether, these features make miRNA a robust and essential arm for coordinated and precise regulation of cellular processes.
Dynamic changes in miRNA expression upon T cell activation and differentiation
Cumulative evidence has demonstrated that miRNAs play critical roles in the development, differentiation, and function of a variety of immune cells including B and T lymphocytes, dendritic cells, and macrophages [43]. Extensive reports have provided a comprehensive view of the expression and functionality of miRNAs in diverse lymphoid cell lineages [44–47]. Here we will review miRNAs specifically expressed in mature T lymphocytes, with a particular focus on those influencing CD8+ T cell differentiation and function.
Upon activation, T cells undergo global gene expression remodeling to support cell growth, proliferation, and effector functions. These transcriptional changes are also accompanied by substantial and rapid modifications of the T cell miRNAome [48]. Following T cell stimulation, acute proteasomal degradation of AGO results in severe miRNA instability and global depletion [49]. A handful of miRNAs, including miR-17, miR-18a, miR-19a/b, miR-20a, miR-21, miR-31, miR-130, miR-146a, miR-155, miR-214 and miR-301, are instead upregulated in response to T cell activation [49–54]. While miR-19b, miR-31, miR-155 and miR-214 are rapidly upregulated, peaking at 12–24h after activation [49, 51], the induction of miR-21, miR-130, miR-301, and miR-146a exhibits slower kinetics, peaking three to six days after initial stimulation [50, 52, 53]. The strength of T cell stimulation appears to dictate the induction levels for some of these miRNAs. For instance, the magnitude of miR-155 expression strongly correlates with TCR affinity [55]. Costimulation is also critical for proper induction of miRNAs as demonstrated by the observation that blockade of CD28 signaling by CTLA4-Ig mitigates miR-214 expression in activated T cells [51]. Thus, miRNA expression dynamically changes upon T cell activation, suggesting that tight regulation of miRNA expression is essential for precise and effective immune responses.
Depending on the strength and quality of stimulatory signals received during priming, naive T cells differentiate into a variety of memory and effector cell subsets characterized by distinct phenotypic and functional profiles [3, 56, 57]. Despite the considerable wealth of information available regarding the transcriptional [56–58], epigenetic [57, 59] and metabolic changes [60, 61] that accompany T cell differentiation, it is surprising how limited the existing data on miRNA expression in different T cell subsets is. Naïve T cells express the large majority of miRNAs, including miR-15b, miR-16, miR-142-3p and -5p, miR-150, and the let-7 family [62, 63]. These miRNAs are progressively downregulated during T cell differentiation, showing intermediate levels of expression in memory cells and the lowest levels in effector cells [62, 63]. By contrast, a limited number of miRNAs, such as miR-155, miR-21, miR-146a and the miR-17~92 cluster, are highly expressed in effector T cells compared to naïve cells [62–64]. This expression pattern is strikingly conserved in mouse and human CD8+ T cells, suggesting that global modulation of miRNA levels during T cell differentiation is a functionally relevant event.
CD8+ T cell activation and differentiation must be tightly regulated to induce productive immune responses and establish long-term immunological memory [56, 57]. Alterations in the nature, duration and setting of antigen stimulations can result in dysfunctional T cell states such as anergy, tolerance and exhaustion [65, 66]. Schietinger et al. [67] have recently shed light on miRNAs potentially contributing to CD8+ T cell tolerance. Unexpectedly, only three miRNAs – miR-21, miR-184, and miR-181a – were differentially expressed in tolerant cells compared to cells that were functionally rescued under lymphopenic conditions [67]. MiR-181a was highly expressed in tolerant cells, whereas miR-21 and miR-184 were overexpressed in rescued cells, possibly as a result of functional reactivation and subsequent proliferation. Notably, known and predicted targets of miR-181a were reduced in tolerant cells, suggesting a key role for this miRNA in the establishment and maintenance of tolerance [67]. Exploration of the miRNA landscape in mature CD8+ T cells has just begun; future studies are warranted to comprehensively dissect miRNA expression across all physiological and dysfunctional T cell states and subsets.
Critical miRNAs regulating T cell effector differentiation
MiRNAs have emerged as pivotal regulators of T cell development and effector function. Evidence stems from the observation that Dicer, a molecule essential for the processing and generation of mature miRNAs, is required for proper thymocyte development, T cell differentiation, and effector cytokine production [50, 68]. In the absence of Dicer, T cell development is compromised resulting in defective numbers of peripheral T lymphocytes [50, 68]. Both CD4+ and CD8+ T cells deficient of Dicer exhibit altered proliferation and are prone to apoptosis upon TCR stimulation [50, 68]. Moreover, T cells lacking Dicer experience more pronounced activation and preferentially differentiate into short-lived effector cells [50, 68]. For instance, Dicer-deficient CD4+ T cells exhibit overt Th1 programming characterized by increased production of IFN-γ [68]. Likewise, CD8+ T cells lacking Dicer are hyper-activated following stimulation and display rapid and sustained expression of the activation marker CD69 [50]. Dicer deficient CD8+ T cells also express higher levels of perforin and granzyme B and show moderate increases in the secretion of effector cytokines IFN-γ and TNF-α, demonstrating a propensity to differentiate into cytotoxic effector T cells [69]. Reminiscent of the Dicer knockout phenotype, depletion of miRNAs in AGO2-deficient T cells resulted in enhanced differentiation and effector cytokine production [49].
Findings from studies employing mice with defective miRNA processing machinery have demonstrated that the dominant function of miRNAs in T cells is to restrain their differentiation. Physiologically, global depletion of miRNAs in activated T cells is achieved by multiple mechanisms, including acute proteasomal degradation of AGO [49] and progressive downregulation of Dicer protein in differentiating cells [69]. Recently, key miRNAs responsible for the maintenance of T cell naivety have begun to be revealed. MiR-139 and miR-342 were found to target and inhibit essential components of the transcriptional and cytotoxic machinery of effector cells such as Eomesodermin (EOMES) and perforin [69]. In addition, miR-29 has been shown to repress effector T cell programming by simultaneously targeting non-redundant master regulators of cytotoxic T cell differentiation T-BET and EOMES, as well as IFN-γ [70, 71]. MiR-150 has also been proposed to inhibit the development of effector T cells by limiting the pro-differentiating action of IL-2 through down-regulation of IL-2 receptor α [69]. However, recent findings have revealed that deletion of miR-150 in CD8+ T cells impaired effector immune responses, indicating that this miRNA rather promotes T cell proliferation and differentiation [72]. While experiments using Dicer and AGO2-deficient mice have proven to be fundamental, studies focusing on manipulation of individual miRNAs or miRNA clusters provide a deeper understanding of their role in specific biological contexts.
As discussed above, miR-155 is one of the few miRNAs upregulated upon T cell activation [49]. This miRNA is considered an indispensable immuno-miRNA as it is required for appropriate immune responses [73]. Mice lacking B-cell integration cluster (BIC), the gene encoding miR-155, display impaired immune cell development and function that is not restricted to T lymphocytes but also involves other immune components such as B cells, NK cells and dendritic cells [73–75]. A number of recent studies have demonstrated that miR-155 is essential for mounting CD8+ T cell effector responses against intracellular pathogens and cancer [55, 76–79]. Impaired effector responses are especially pronounced under conditions in which CD8+ T cells face continuous antigenic stimulation such as chronic infections and tumors, leading to uncontrolled viral replication [55, 77, 79] and unrestrained tumor growth [55, 76]. Regardless of the disease model, miR-155-deficient CD8+ T cells uniformly showed impaired expansion, poor survival, and decreased production of effector cytokines such as IFN-γ and TNF-α [55, 76–79]. The underlying biology is multifaceted, but it appears to converge on a few signaling pathways that are severely dysregulated in the absence of miR-155. T cells deficient of miR-155 exhibit defective PI3K-AKT signaling [79] due to heightened levels of the inositol 5-phosphatase SHIP-1 [76, 80]. The signal transducer and activator of transcription (STAT) pathways are also influenced by miR-155 activity [55, 77, 81]. For instance, the accumulation of the miR-155 targets Suppressor of Cytokine Signaling 1 (SOCS-1) [55, 81] and the protein tyrosine phosphatase PTPN2 [81] in miR-155 deficient cells dampens STAT5 signaling. Deficiency of miR-155 has also been associated with increased activity of STAT1 and other type I interferon–associated transcription factors such as IRF7 [77], although the biological mechanism underlying this observation has not been determined. Recent findings using a transgenic mouse model in which the regulation of SOCS-1 by miR-155 is specifically abolished have underscored how miR-155 biology in T cells is largely attributable to the repression of this molecule, especially in settings of chronic antigenic stimulation [82]. The failed physiological suppression of SOCS-1 by miR-155 resulted in severely impaired expansion and survival of viral-specific CD8+ T cells following chronic LCMV infection [82]. Consistent with these observations, enforced expression of SOCS-1 recapitulated the miR-155 knockout phenotype [55].
Another series of studies have recently focused on the activity of the polycistronic miR-17~92 cluster, which is also highly expressed in effector CD8+ T cells [64]. This cluster encodes six miRNAs – miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92 – which are generated via differential processing from a common pri-miRNA transcript [83]. Conditional deletion of miR-17~92 in viral-specific cells impaired cell proliferation and differentiation, resulting in lower numbers of antigen-specific cells with reduced effector molecule expression [64]. Mechanistically, miR-17~92 enhances mammalian target of rapamycin (mTOR) signaling by targeting upstream inhibitory phosphatases such as Phosphatase and tensin homolog (PTEN) [64, 84] and PH Domain And Leucine Rich Repeat Protein Phosphatase 2 (PHLPP2) [84]. Consistently, miR-17~92 overexpression enhanced mTOR activity in CD8+ T cells and promoted the development of terminal effectors at the expense of memory T cells [64]. Notably, genetic manipulation of miR-17~92 levels was associated with opposing changes in the expression of the DNA-binding inhibitor Id3, a key transcriptional regulator of memory differentiation, further supporting the notion that miR-17~92 levels control the balance of effector and memory formation [28, 64, 85, 86].
Similar to miR-155 and the miR-17~92 cluster, miR-146a is also upregulated in T cells following TCR engagement. However, contrary to these miRNAs, miR-146a functions as a negative regulator preventing excessive and unrestrained T cell activity [52]. In the absence of miR-146a, CD8+ T cells are hyper-responsive, mounting more potent primary and secondary immune responses [52]. In a model of chronic inflammation, uncontrolled T cell activation resulted in the accumulation of effectors in peripheral tissues and the development of T cell-mediated autoimmunity [52]. MiR-146a was found to act in a negative feedback loop to dampen TCR-induced NF-κB activity by targeting the NF-κB signaling transducers TRAF6 and IRAK1 [52, 87]. Altogether, studies from mice deficient of specific miRNAs strengthen the notion that miRNAs are indispensable players regulating proper T cell immunity.
Harnessing miRNA biology to enhance adoptive T cell immunotherapy
The capacity of miRNAs to target multiple proteins in converging pathways and their involvement in regulating various aspects of T cell biology make them attractive molecules to exploit in the pursuit of more effective adoptive T cell therapies. Here, we describe methods to enhance T cell sensitivity to tumor antigens, fitness, and effector functions by harnessing miRNA biology (Fig. 2).
Fig. 2. Harnessing miRNA biology to enhance adoptive T cell immunotherapy.
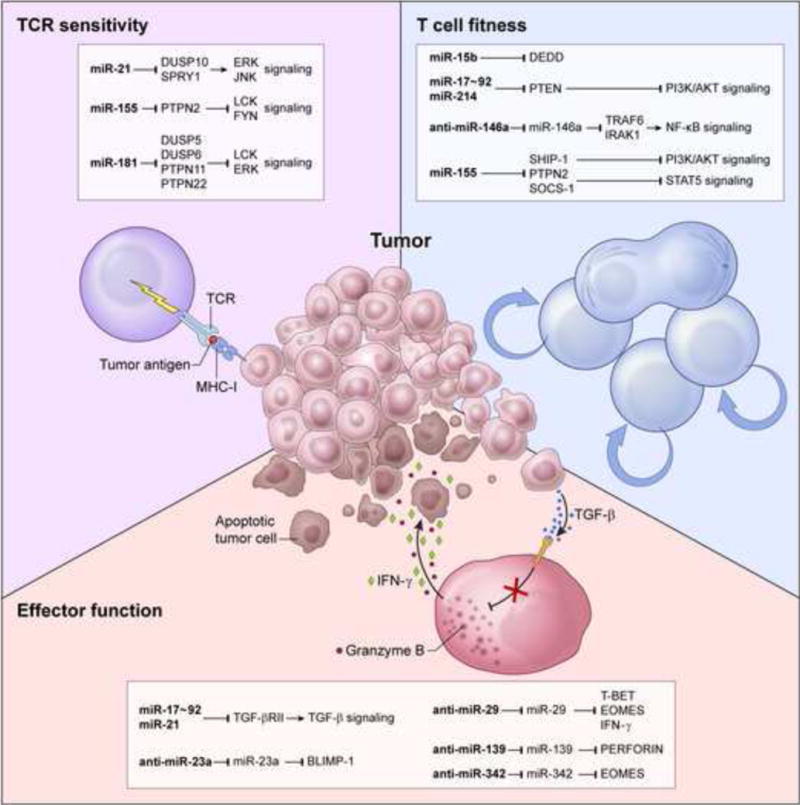
miRNA, anti-miRNA (bold font) and their downstream targets could be employed to augment tumor destruction by enhancing T cell receptor (TCR) sensitivity (upper left), T cell fitness (upper right), and effector functions (lower panel). DUSP, Dual Specificity Phosphatase; SPRY1, Sprouty Homolog 1; PTPN, Protein Tyrosine Phosphatase, Non-Receptor Type; JNK, JUN N-Terminal Kinase; DEDD, Death Effector Domain Containing; PTEN, Phosphatase And Tensin Homolog; TRAF6, TNF Receptor-Associated Factor 6; IRAK1, Interleukin-1 Receptor-Associated Kinase 1; SHIP-1, SH2 Domain-Containing Inositol 5-Phosphatase 1; SOCS-1, Suppressor Of Cytokine Signaling 1; PI3K, Phosphatidylinositol 3-Kinase; STAT5, Signal Transducer And Activator Of Transcription 5; TGF-β, Transforming Growth Factor, Beta; TGF-βRII, Transforming Growth Factor, Beta Receptor II; BLIMP1, B lymphocyte-induced maturation protein-1; EOMES, Eomesodermin; IFN-γ, interferon gamma.
Enhancing TCR sensitivity
T cell sensitivity to antigen is intrinsically regulated to ensure balance between immunity and tolerance [88]. The increased accessibility of high throughput deep-sequencing technology has recently revived interest in targeting tumor-specific neoantigens, based on the notion that TCR recognizing this class of antigens are highly avid [89, 90]. However, because immunogenic neoantigens typically arise on the basis of individual cancers, therapies that target them must be tailored to individual patients. By contrast, the shared nature of non-mutated, tumor-associated self antigens has made them an attractive target for T cell-based immunotherapy, but intrinsic tolerance mechanisms limit the avidity and effectiveness of the vast majority of T cells targeting this type of antigen [91].
Extensive efforts to enhance antigen reactivity and circumvent tolerance have focused on identifying rare, high avidity self/tumor reactive TCR [90, 91]. Alternatively, modulation of TCR signaling thresholds to enhance T cell activation and function has also been explored [12, 92, 93]. Because several miRNAs have been found to influence TCR signaling by targeting key inhibitory phosphatases [47], they may be promising candidates to manipulate for this purpose. For instance, the tyrosine phosphatase PTPN2, which negatively regulates TCR signaling by repressing LCK Proto-Oncogene, Src Family Tyrosine Kinase (LCK) and FYN Proto-Oncogene, Src Family Tyrosine Kinase (FYN) [94], can be efficiently repressed by miR-155 [81]. Another promising candidate, miR-21, promotes T cell activation in a positive feedback loop by down-regulating the serine/threonine phosphatases DUSP10 [95, 96] and Sprouty 1 [97, 98], which suppress TCR-induced JNK and ERK phosphorylation [98, 99]. Consistently, miR-21 overexpressing T cells have been shown to induce severe autoimmune arthritis upon adoptive transfer [100], supporting the idea that enforced expression of this miRNA can be utilized to boost T cell immunity. The capacity of miR-181a to simultaneously target multiple serine/threonine and tyrosine phosphatases may make it the most attractive candidate in this category. This miRNA has been shown to enhance LCK and ERK activity by inhibiting DUSP5, DUSP6, PTPN11 (also known as SHP2), and PTPN22 [101] and to govern central and peripheral tolerance [102, 103]. Indeed, overexpression of miR-181a in T cells increased TCR sensitivity to cognate antigen and enhanced intracellular calcium flux upon TCR triggering, resulting in more pronounced release of IL-2 [101]. However, the recent observation that miR-181a is enriched in tolerant tumor-reactive T cells [67] warrants in-depth characterization of this miRNA in the tumor immunotherapy setting.
Improving T cell fitness
The ability of T cells to engraft and persist, here referred to as ‘T cell fitness’, is a critical determinant of the success of adoptive T cell therapies [3]. Indeed, T cell persistence has been shown to correlate strongly with the induction of objective tumor responses in numerous clinical studies [10, 11, 104, 105] and preclinical animal models [8, 9, 27, 29–31, 106]. The survival capacity of adoptively transferred T cells is dependent upon both their responsiveness to homeostatic cytokines and their resistance to cell death [107]. To support T cell engraftment and enhance fitness, adoptive immunotherapies are currently performed in conjunction with the provision of exogenous cytokines and lymphodepletion conditioning, which removes endogenous lymphocyte populations competing for access to limited amounts of homeostatic cytokines [108, 109]. We have recently reported that miR-155 overexpression can be employed to enhance T cell responsiveness to homeostatic γc cytokines in lymphoreplete hosts, resulting in enhanced T cell survival and superior antitumor responses [81]. MiR-155 facilitates cytokine signaling by concurrently targeting multiple negative regulators upstream of the PI3K/AKT and STAT pathways [81]. This cell-intrinsic strategy has the potential to reduce the severe toxicities associated with non-specific maneuvers such as lymphodepletion [110] and exogenous cytokine administration [111]. Furthermore, it also provides the possibility to deliver multiple rounds of T cell infusions in short succession, an approach that is currently prohibited by the toxicities associated with preconditioning treatments.
The fitness of transferred cells might be also boosted by engineering T cells to express the miR-17~92 cluster. By suppressing the PI3K/AKT inhibitor PTEN and the pro-apoptotic molecule BIM, miR-17~92 has been shown to support extensive proliferation and accumulation of effector T cells [64, 85]. However, sustained miR-17~92 and AKT activity can also lead to defective memory cell development [64, 112], which may be detrimental to long-lasting anti-tumor responses. Recently Ohno et al. have provided evidence of the potential benefit of overexpressing miR-17~92 in tumor-specific T cells [113]. Enforced expression of the miR-17~92 cluster in anti-EGFRvIII CAR-modified T cells provided prolonged protection against tumor re-challenge in a xenograft model of human glioblastoma [113]. Like miR-17~92, miR-214 also targets PTEN [51] and might be harnessed to enhance AKT-driven T cell expansion, although its effect in models of adoptive immunotherapy has yet to be evaluated.
The overexpression of miRNAs that suppress proapoptotic molecules represents another therapeutic opportunity. For example, miR-15b, which has been shown to target and inhibit Death Effector Domain-containing DNA binding protein (DEDD) [114], might be used to enhance the survival of adoptively transferred T cells. However, overexpression of this miRNA can also inhibit T cell activation and lessen IL-2 and IFN-γ secretion, potentially damaging antitumor effector responses [114]. Whether the beneficial effect on T cell fitness exceeds the negative impact on T cell activation remains to be addressed.
Antagonizing miRNAs that negatively regulate T cell immune responses is another theoretically appealing approach to increase T cell fitness and antitumor function. As discussed previously, miR-146a is a major suppressor of NF-κB signaling that is upregulated in response to T cell activation in order to dampen effector responses [52]. AntagomiRs, miRNA decoys, or miRNA sponges targeting miR-146a could be employed to augment NF-κB activity in adoptively transferred cells, potentially enhancing the potency of their anti-tumor responses. Because increased expression of several of the miRNA discussed here, including mir17~92, miR-21 and miR-155, has been associated with the development or aggressiveness of malignancies, their overexpression in tumor-specific T cells warrants some precautions. Safety could be ensured by introducing suicide genes [115, 116] or alternatively by employing inducible promoters to tightly control miRNA expression [117].
Augmenting effector functions
To successfully eradicate tumors, adoptively transferred T cells must ultimately migrate into tumor deposits, release substantial amounts of pro-inflammatory cytokines, and exert potent cytotoxicity. Productive effector responses, however, are often impeded by multiple cellular and molecular immunosuppressive hurdles within the tumor microenvironment [4]. Transforming growth factor-beta (TGF-β) is a key immunosuppressive cytokine that profoundly shapes the tumor milieu [118]. There have been extensive efforts to inhibit TGF-β signaling in patients with cancer, but the broad involvement of this signaling pathway in numerous homeostatic processes and the potential adverse effects of such treatments in normal tissues have hampered the development of anti-TGF-β therapies [118].
Adoptive T cell therapy offers the advantage of targeting TGF-β activity in transferred cells without affecting the entire host. Genetic engineering of TGF-β dominant negative receptors in tumor-specific T cells has proven to be an effective approach in preclinical mouse studies [119–121]. As TGF-β signaling is intimately involved with miRNA biogenesis [122], an alternative strategy is to counter the TGF-β-induced program in T cells by artificially modulating miRNA levels. It was recently reported that release of TGF-β within the tumor microenvironment increased miR-23a expression in CD8+ T cells, resulting in suppression of its target Blimp1, a master regulator of effector T cell differentiation [123–126]. Accordingly, granzyme B and IFN-γ levels negatively correlated with the amount of miR-23a in T cells [123]. Overexpression of a miR-23a decoy attenuated the immunosuppressive effects of TGF-β, increased granzyme B and IFN-γ expression, and enhanced the antitumor effectiveness of adoptively transferred CD8+ T cells in mouse models of established tumors [123]. The miR-17~92 cluster might also be employed to mitigate the inhibitory influence of TGF-β signaling on transferred T cells. By inhibiting the TGF-β type II receptor (TGF-βRII), miR-17~92 overexpression has been shown to promote CD8+ T cell proliferation and enhance IFN-γ production and cytotoxicity following cognate antigen stimulation [127]. Finally, enforced expression of miR-21, a key miRNA involved in the TGF-β pathway, might be utilized to overcome TGF-β tolerogenic effects. This miRNA is induced by TGF-β and is thought to act in a negative feedback loop that limits the intensity and duration of TGF-β signaling by down regulating TGF-βRII directly [128]. Interestingly, upregulation of miR-21 was associated with exit from tolerance and functional T cell reactivation [67]. In agreement with these findings, a recent report showed that miR-21 overexpressing T cells displayed enhanced IFN-γ and TNF-α production upon stimulation [129]. Altogether, these studies indicate the potential for miRNA therapeutics to mitigate the effects of TGF-β-mediated immunosuppression on adoptively transferred tumor-reactive T cells.
Several miRNAs involved in silencing molecules that are essential for T cell effector functions have been recently identified, and represent another exciting class of targets to antagonize using miRNA inhibitors. For example, inhibition of miR-139 and miR-342, which downregulate perforin and EOMES [69], could be employed to boost T cell cytotoxicity. In addition, IFN-γ release could be potentiated by targeting miR-29. This miRNA has emerged as a major negative regulator of IFN-γ synthesis that functions both by directly binding to the Ifng 3-UTR and suppressing transcriptional factors that are required for optimal IFN-γ production, such as T-BET and EOMES [70, 71]. Recently, Ma et al. demonstrated that transgenic mice expressing a ‘sponge’ to deplete endogenous miR-29 exhibit high circulating levels of IFN-γ in response to bacterial challenge and enhanced bacterial clearance [69]. Although miR-29 sponges have not been evaluated in the context of tumor-bearing hosts, it is conceivable that suppression of miR-29 activity might lead to improved antitumor immunity.
A potential drawback of this strategy is that the enforcement of the effector program might lead to terminal differentiation and senescence [124–126, 130], thus impairing T cell fitness and anti-tumor responses. Therefore, it would be desirable to engineer miRNA and miRNA inhibitors potentiating effector differentiation under the control of inducible promoters to spatially and temporally regulate their expression. To this end, the NFAT promoter could be employed to boost effector functions within tumor masses, as it will drive miRNA expression only upon cognate antigen stimulation [117].
Concluding remarks
It is now well established that miRNAs are pivotal regulators of T cell activation, proliferation, survival, differentiation, and effector functions, which are all key factors affecting the therapeutic outcome of adoptive immunotherapies. An ever-growing understanding of the role that miRNAs play in these processes is bringing us closer to the prospect of safer and more effective T cell therapies based on miRNA therapeutics. Harnessing miRNAs has multiple advantages over other gene engineering strategies. Due to their ability to target multiple molecules simultaneously, manipulation of a single miRNA can radically change T cell fate and behavior [42]. Modulation of miRNA levels also allows for fine-tuning of cellular processes without the deleterious effects that can result from overexpression of constitutively activated transcription factors or enzymes [112]. Lastly, thanks to their small size, miRNA genes can be easily integrated into existing multicistronic TCR and CAR platforms. In summary, miRNA gene engineering of tumor-specific T cells is a powerful and versatile approach that has the potential to revolutionize the next generation of adoptive T cell-based immunotherapies.
Highlights.
miRNA expression is dynamically regulated following CD8+ T cell activation.
miRNAs critically regulate CD8+ T cell differentiation and function.
miRNA therapeutics can be used to enhance T cell-based immunotherapies.
Acknowledgments
This work was supported by the Intramural Research Program of the National Cancer Institute, Center for Cancer Research, National Institutes of Health (ZIA-BC011480).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annual review of immunology. 2014;32:189–225. doi: 10.1146/annurev-immunol-032713-120136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nature reviews Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi: 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- 5.Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature reviews Cancer. 2005;5:263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 6.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. The Journal of clinical investigation. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9571–9576. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. Journal of immunology. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer DC, Guittard GC, Franco Z, Crompton JG, Eil RL, Patel SJ, et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. The Journal of experimental medicine. 2015;212:2095–2113. doi: 10.1084/jem.20150304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holler PD, Chlewicki LK, Kranz DM. TCRs with high affinity for foreign pMHC show self-reactivity. Nature immunology. 2003;4:55–62. doi: 10.1038/ni863. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Q, Ahmed M, Tassev DV, Hasan A, Kuo TY, Guo HF, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015 doi: 10.1038/leu.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilde S, Geiger C, Milosevic S, Mosetter B, Eichenlaub S, Schendel DJ. Generation of allo-restricted peptide-specific T cells using RNA-pulsed dendritic cells: A three phase experimental procedure. Oncoimmunology. 2012;1:129–140. doi: 10.4161/onci.1.2.18216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanislawski T, Voss RH, Lotz C, Sadovnikova E, Willemsen RA, Kuball J, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nature immunology. 2001;2:962–970. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 17.Li LP, Lampert JC, Chen X, Leitao C, Popovic J, Muller W, et al. Transgenic mice with a diverse human T cell antigen receptor repertoire. Nature medicine. 2010;16:1029–1034. doi: 10.1038/nm.2197. [DOI] [PubMed] [Google Scholar]
- 18.Liu K, Rosenberg SA. Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. Journal of immunology. 2001;167:6356–6365. doi: 10.4049/jimmunol.167.11.6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu C, Jones SA, Cohen CJ, Zheng Z, Kerstann K, Zhou J, et al. Cytokine-independent growth and clonal expansion of a primary human CD8+ T-cell clone following retroviral transduction with the IL-15 gene. Blood. 2007;109:5168–5177. doi: 10.1182/blood-2006-06-029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28-CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. The Journal of experimental medicine. 2003;198:947–955. doi: 10.1084/jem.20021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charo J, Finkelstein SE, Grewal N, Restifo NP, Robbins PF, Rosenberg SA. Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer research. 2005;65:2001–2008. doi: 10.1158/0008-5472.CAN-04-2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton D, Gilham DE, O’Neill A, Hawkins RE. Retroviral transduction of human peripheral blood lymphocytes with Bcl-X(L) promotes in vitro lymphocyte survival in pro-apoptotic conditions. Gene therapy. 2002;9:527–535. doi: 10.1038/sj.gt.3301685. [DOI] [PubMed] [Google Scholar]
- 24.Hwu P, Yannelli J, Kriegler M, Anderson WF, Perez C, Chiang Y, et al. Functional and molecular characterization of tumor-infiltrating lymphocytes transduced with tumor necrosis factor-alpha cDNA for the gene therapy of cancer in humans. Journal of immunology. 1993;150:4104–4115. [PubMed] [Google Scholar]
- 25.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. doi: 10.1182/blood-2007-09-113050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature medicine. 2009;15:808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji Y, Pos Z, Rao M, Klebanoff CA, Yu Z, Sukumar M, et al. Repression of the DNA-binding inhibitor Id3 by Blimp-1 limits the formation of memory CD8+ T cells. Nature immunology. 2011;12:1230–1237. doi: 10.1038/ni.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. The Journal of clinical investigation. 2013;123:4479–4488. doi: 10.1172/JCI69589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer research. 2015;75:296–305. doi: 10.1158/0008-5472.CAN-14-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Waart AB, van de Weem NM, Maas F, Kramer CS, Kester MG, Falkenburg JH, et al. Inhibition of Akt signaling promotes the generation of superior tumor-reactive T cells for adoptive immunotherapy. Blood. 2014;124:3490–3500. doi: 10.1182/blood-2014-05-578583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiological reviews. 2011;91:827–887. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 34.Lujambio A, Lowe SW. The microcosmos of cancer. Nature. 2012;482:347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nature reviews Drug discovery. 2013;12:847–865. doi: 10.1038/nrd4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nature reviews Drug discovery. 2010;9:775–789. doi: 10.1038/nrd3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nature genetics. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nature reviews Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–98. doi: 10.1126/science.1090599. [DOI] [PubMed] [Google Scholar]
- 41.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annual review of biochemistry. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 42.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baltimore D, Boldin MP, O’Connell RM, Rao DS, Taganov KD. MicroRNAs: new regulators of immune cell development and function. Nature immunology. 2008;9:839–845. doi: 10.1038/ni.f.209. [DOI] [PubMed] [Google Scholar]
- 44.Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32:828–839. doi: 10.1016/j.immuni.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeker LT, Bluestone JA. MicroRNA regulation of T-cell differentiation and function. Immunological reviews. 2013;253:65–81. doi: 10.1111/imr.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monticelli S. MicroRNAs in T helper cell differentiation and plasticity. Seminars in immunology. 2013;25:291–298. doi: 10.1016/j.smim.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 47.Simpson LJ, Ansel KM. MicroRNA regulation of lymphocyte tolerance and autoimmunity. The Journal of clinical investigation. 2015;125:2242–2249. doi: 10.1172/JCI78090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bronevetsky Y, Ansel KM. Regulation of miRNA biogenesis and turnover in the immune system. Immunological reviews. 2013;253:304–316. doi: 10.1111/imr.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bronevetsky Y, Villarino AV, Eisley CJ, Barbeau R, Barczak AJ, Heinz GA, et al. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. The Journal of experimental medicine. 2013;210:417–432. doi: 10.1084/jem.20111717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang N, Bevan MJ. Dicer controls CD8+ T-cell activation, migration, and survival. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21629–21634. doi: 10.1073/pnas.1016299107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jindra PT, Bagley J, Godwin JG, Iacomini J. Costimulation-dependent expression of microRNA-214 increases the ability of T cells to proliferate by targeting Pten. Journal of immunology. 2010;185:990–997. doi: 10.4049/jimmunol.1000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a controls the resolution of T cell responses in mice. The Journal of experimental medicine. 2012;209:1655–1670. doi: 10.1084/jem.20112218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carissimi C, Carucci N, Colombo T, Piconese S, Azzalin G, Cipolletta E, et al. miR-21 is a negative modulator of T-cell activation. Biochimie. 2014;107(Pt B):319–326. doi: 10.1016/j.biochi.2014.09.021. [DOI] [PubMed] [Google Scholar]
- 54.Teteloshvili N, Smigielska-Czepiel K, Kroesen BJ, Brouwer E, Kluiver J, Boots AM, et al. T-cell Activation Induces Dynamic Changes in miRNA Expression Patterns in CD4 and CD8 T-cell Subsets. MicroRNA. 2015;4:117–122. doi: 10.2174/2211536604666150819194636. [DOI] [PubMed] [Google Scholar]
- 55.Dudda JC, Salaun B, Ji Y, Palmer DC, Monnot GC, Merck E, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. 2013;38:742–753. doi: 10.1016/j.immuni.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nature reviews Immunology. 2012;12:749–761. doi: 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nature immunology. 2014;15:1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaventhiran JE, Fearon DT, Gattinoni L. Transcriptional regulation of effector and memory CD8+ T cell fates. Current opinion in immunology. 2013;25:321–328. doi: 10.1016/j.coi.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory CD8(+) T-cell differentiation. Immunological reviews. 2014;261:157–168. doi: 10.1111/imr.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342:1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Man K, Kallies A. Synchronizing transcriptional control of T cell metabolism and function. Nature reviews Immunology. 2015;15:574–584. doi: 10.1038/nri3874. [DOI] [PubMed] [Google Scholar]
- 62.Wu H, Neilson JR, Kumar P, Manocha M, Shankar P, Sharp PA, et al. miRNA profiling of naive, effector and memory CD8 T cells. PloS one. 2007;2:e1020. doi: 10.1371/journal.pone.0001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salaun B, Yamamoto T, Badran B, Tsunetsugu-Yokota Y, Roux A, Baitsch L, et al. Differentiation associated regulation of microRNA expression in vivo in human CD8+ T cell subsets. Journal of translational medicine. 2011;9:44. doi: 10.1186/1479-5876-9-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu T, Wieland A, Araki K, Davis CW, Ye L, Hale JS, et al. Temporal expression of microRNA cluster miR-17-92 regulates effector and memory CD8+ T-cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9965–9970. doi: 10.1073/pnas.1207327109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends in immunology. 2014;35:51–60. doi: 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature reviews Immunology. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science. 2012;335:723–727. doi: 10.1126/science.1214277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. The Journal of experimental medicine. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trifari S, Pipkin ME, Bandukwala HS, Aijo T, Bassein J, Chen R, et al. MicroRNA-directed program of cytotoxic CD8+ T-cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18608–18613. doi: 10.1073/pnas.1317191110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nature immunology. 2011;12:861–869. doi: 10.1038/ni.2073. [DOI] [PubMed] [Google Scholar]
- 71.Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, et al. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity. 2011;35:169–181. doi: 10.1016/j.immuni.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith NL, Wissink EM, Grimson A, Rudd BD. miR-150 Regulates Differentiation and Cytolytic Effector Function in CD8+ T cells. Scientific reports. 2015;5:16399. doi: 10.1038/srep16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316:608–611. doi: 10.1126/science.1139253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316:604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- 76.Huffaker TB, Hu R, Runtsch MC, Bake E, Chen X, Zhao J, et al. Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell reports. 2012;2:1697–1709. doi: 10.1016/j.celrep.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gracias DT, Stelekati E, Hope JL, Boesteanu AC, Doering TA, Norton J, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nature immunology. 2013;14:593–602. doi: 10.1038/ni.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsai CY, Allie SR, Zhang W, Usherwood EJ. MicroRNA miR-155 affects antiviral effector and effector Memory CD8 T cell differentiation. Journal of virology. 2013;87:2348–2351. doi: 10.1128/JVI.01742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lind EF, Elford AR, Ohashi PS. Micro-RNA 155 is required for optimal CD8+ T cell responses to acute viral and intracellular bacterial challenges. Journal of immunology. 2013;190:1210–1216. doi: 10.4049/jimmunol.1202700. [DOI] [PubMed] [Google Scholar]
- 80.Rouquette-Jazdanian AK, Kortum RL, Li W, Merrill RK, Nguyen PH, Samelson LE, et al. miR-155 Controls Lymphoproliferation in LAT Mutant Mice by Restraining T-Cell Apoptosis via SHIP-1/mTOR and PAK1/FOXO3/BIM Pathways. PloS one. 2015;10:e0131823. doi: 10.1371/journal.pone.0131823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ji Y, Wrzesinski C, Yu Z, Hu J, Gautam S, Hawk NV, et al. miR-155 augments CD8+ T-cell antitumor activity in lymphoreplete hosts by enhancing responsiveness to homeostatic gammac cytokines. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:476–481. doi: 10.1073/pnas.1422916112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lu LF, Gasteiger G, Yu IS, Chaudhry A, Hsin JP, Lu Y, et al. A Single miRNA-mRNA Interaction Affects the Immune Response in a Context- and Cell-Type-Specific Manner. Immunity. 2015;43:52–64. doi: 10.1016/j.immuni.2015.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jiang P, Rao EY, Meng N, Zhao Y, Wang JJ. MicroRNA-17-92 significantly enhances radioresistance in human mantle cell lymphoma cells. Radiation oncology. 2010;5:100. doi: 10.1186/1748-717X-5-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Khan AA, Penny LA, Yuzefpolskiy Y, Sarkar S, Kalia V. MicroRNA-17~92 regulates effector and memory CD8 T-cell fates by modulating proliferation in response to infections. Blood. 2013;121:4473–4483. doi: 10.1182/blood-2012-06-435412. [DOI] [PubMed] [Google Scholar]
- 86.Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nature immunology. 2011;12:1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:12481–12486. doi: 10.1073/pnas.0605298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hogquist KA, Jameson SC. The self-obsession of T cells: how TCR signaling thresholds affect fate ‘decisions’ and effector function. Nature immunology. 2014;15:815–823. doi: 10.1038/ni.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 90.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Obenaus M, Leitao C, Leisegang M, Chen X, Gavvovidis I, van der Bruggen P, et al. Identification of human T-cell receptors with optimal affinity to cancer antigens using antigen-negative humanized mice. Nature biotechnology. 2015;33:402–407. doi: 10.1038/nbt.3147. [DOI] [PubMed] [Google Scholar]
- 92.Stromnes IM, Fowler C, Casamina CC, Georgopolos CM, McAfee MS, Schmitt TM, et al. Abrogation of SRC homology region 2 domain-containing phosphatase 1 in tumor-specific T cells improves efficacy of adoptive immunotherapy by enhancing the effector function and accumulation of short-lived effector T cells in vivo. Journal of immunology. 2012;189:1812–1825. doi: 10.4049/jimmunol.1200552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stromnes IM, Blattman JN, Tan X, Jeevanjee S, Gu H, Greenberg PD. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. The Journal of clinical investigation. 2010;120:3722–3734. doi: 10.1172/JCI41991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. The Journal of clinical investigation. 2011;121:4758–4774. doi: 10.1172/JCI59492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chang CC, Zhang QY, Liu Z, Clynes RA, Suciu-Foca N, Vlad G. Downregulation of inflammatory microRNAs by Ig-like transcript 3 is essential for the differentiation of human CD8(+) T suppressor cells. Journal of immunology. 2012;188:3042–3052. doi: 10.4049/jimmunol.1102899. [DOI] [PubMed] [Google Scholar]
- 96.Chen L, Xu Z, Chang C, Ho S, Liu Z, Vlad G, et al. Allospecific CD8 T suppressor cells induced by multiple MLC stimulation or priming in the presence of ILT3.Fc have similar gene expression profiles. Human immunology. 2014;75:190–196. doi: 10.1016/j.humimm.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 97.Jin XL, Sun QS, Liu F, Yang HW, Liu M, Liu HX, et al. microRNA 21-mediated suppression of Sprouty1 by Pokemon affects liver cancer cell growth and proliferation. Journal of cellular biochemistry. 2013;114:1625–1633. doi: 10.1002/jcb.24504. [DOI] [PubMed] [Google Scholar]
- 98.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y, Blattman JN, Kennedy NJ, Duong J, Nguyen T, Wang Y, et al. Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature. 2004;430:793–797. doi: 10.1038/nature02764. [DOI] [PubMed] [Google Scholar]
- 100.Iliopoulos D, Kavousanaki M, Ioannou M, Boumpas D, Verginis P. The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. European journal of immunology. 2011;41:1754–1763. doi: 10.1002/eji.201040646. [DOI] [PubMed] [Google Scholar]
- 101.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell. 2007;129:147–161. doi: 10.1016/j.cell.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 102.Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nature immunology. 2009;10:1162–1169. doi: 10.1038/ni.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schaffert SA, Loh C, Wang S, Arnold CP, Axtell RC, Newell EW, et al. mir-181a-1/b-1 Modulates Tolerance through Opposing Activities in Selection and Peripheral T Cell Function. Journal of immunology. 2015;195:1470–1479. doi: 10.4049/jimmunol.1401587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nature medicine. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nature medicine. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nature immunology. 2003;4:355–360. doi: 10.1038/ni908. [DOI] [PubMed] [Google Scholar]
- 108.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nature reviews Immunology. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. The Journal of experimental medicine. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Muranski P, Boni A, Wrzesinski C, Citrin DE, Rosenberg SA, Childs R, et al. Increased intensity lymphodepletion and adoptive immunotherapy–how far can we go? Nature clinical practice Oncology. 2006;3:668–681. doi: 10.1038/ncponc0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Acquavella N, Kluger H, Rhee J, Farber L, Tara H, Ariyan S, et al. Toxicity and activity of a twice daily high-dose bolus interleukin 2 regimen in patients with metastatic melanoma and metastatic renal cell cancer. Journal of immunotherapy. 2008;31:569–576. doi: 10.1097/CJI.0b013e318177a4ba. [DOI] [PubMed] [Google Scholar]
- 112.Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ohno M, Ohkuri T, Kosaka A, Tanahashi K, June CH, Natsume A, et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. Journal for immunotherapy of cancer. 2013;1:21. doi: 10.1186/2051-1426-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhong G, Cheng X, Long H, He L, Qi W, Xiang T, et al. Dynamically expressed microRNA-15b modulates the activities of CD8+ T lymphocytes in mice with Lewis lung carcinoma. Journal of translational medicine. 2013;11:71. doi: 10.1186/1479-5876-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 116.Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nature reviews Immunology. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bollard CM, Rossig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 120.Quatromoni JG, Suzuki E, Okusanya O, Judy BF, Bhojnagarwala P, Venegas O, et al. The timing of TGF-beta inhibition affects the generation of antigen-specific CD8+ T cells. BMC immunology. 2013;14:30. doi: 10.1186/1471-2172-14-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-beta signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene therapy. 2013;20:575–580. doi: 10.1038/gt.2012.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Blahna MT, Hata A. Regulation of miRNA biogenesis as an integrated component of growth factor signaling. Current opinion in cell biology. 2013;25:233–240. doi: 10.1016/j.ceb.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lin R, Chen L, Chen G, Hu C, Jiang S, Sevilla J, et al. Targeting miR-23a in CD8+ cytotoxic T lymphocytes prevents tumor-dependent immunosuppression. The Journal of clinical investigation. 2014;124:5352–5367. doi: 10.1172/JCI76561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, et al. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity. 2009;31:309–320. doi: 10.1016/j.immuni.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rutishauser RL, Martins GA, Kalachikov S, Chandele A, Parish IA, Meffre E, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity. 2009;31:283–295. doi: 10.1016/j.immuni.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 127.Kosaka A, Ohkuri T, Ikeura M, Kohanbash G, Okada H. Transgene-derived overexpression of miR-17-92 in CD8+ T-cells confers enhanced cytotoxic activity. Biochemical and biophysical research communications. 2015;458:549–554. doi: 10.1016/j.bbrc.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yu Y, Kanwar SS, Patel BB, Oh PS, Nautiyal J, Sarkar FH, et al. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (TGFbetaR2) in colon cancer cells. Carcinogenesis. 2012;33:68–76. doi: 10.1093/carcin/bgr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ando Y, Yang GX, Kenny TP, Kawata K, Zhang W, Huang W, et al. Overexpression of microRNA-21 is associated with elevated pro-inflammatory cytokines in dominant-negative TGF-beta receptor type II mouse. Journal of autoimmunity. 2013;41:111–119. doi: 10.1016/j.jaut.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]