

Abstract
The effect of CYP2D6 genotype on the dose-exposure relationship for atomoxetine has not been well characterized in children. Children 6–17 years of age diagnosed with ADHD were stratified by CYP2D6 genotype into groups with 0 (PM, n=4), 0.5 (IM, n=3), one (EM1, n=8) or two (EM2, n=8) functional alleles) and administered a single 0.5 mg/kg oral dose of atomoxetine. Plasma and urine samples were collected for 24 (IM, EM1 and EM2) or 72 hours (PMs). Dose-corrected atomoxetine systemic exposure (AUC0-∞) varied 29.6-fold across the study cohort, ranging from 4.4±2.7 μM*h in EM2s to 5.8±1.7 μM*h, 16.3±2.9 μM*h and 50.2±7.3 μM*h in EM1s, IMs and PMs, respectively (p<0.0001). Simulated steady state profiles at the maximum FDA-recommended dose suggest that most patients are unlikely to attain adequate ATX exposures. These data support the need for individualized dosing strategies for more effective use of the medication.
Keywords: Pediatrics, pharmacokinetics, pharmacogenetics, CYP, Psychiatric
Introduction
Atomoxetine (ATX) is a norepinephrine reuptake inhibitor utilized as an alternative to stimulant medications in the treatment of pediatric attention deficit/hyperactivity disorder (ADHD). The highly polymorphic drug metabolizing enzyme cytochrome P450 2D6 (CYP2D6) is responsible for the formation of therapeutically active 4-hydroxyatomoxetine (4-OH-ATX), the primary metabolite detected in all individuals, which is then rapidly conjugated to the inactive 4-hydroxyatomoxetine glucuronide. To a lesser extent, ATX is also metabolized to the inactive N-desmethylatomoxetine (NDA) via CYP2C19, which is further metabolized to N-desmethyl 4-OH-ATX, presumably through CYP2D6.1 Although less well described, secondary metabolites of an initial 2-methylhydroxylation, 4-hydroxycarboxy ATX (4-OH-carboxy-ATX) and a hydroxycarboxy ATX metabolite (for which the actual site of hydroxylation has not been definitively characterized) are also formed.2 The functional consequence of the predominant role of CYP2D6 in ATX disposition is the observation of considerable variability in drug exposure depending on an individual’s CYP2D6 activity.
Pharmacokinetic studies in adults demonstrate that the apparent oral clearance of ATX at steady state is approximately 10-fold lower in CYP2D6 poor metabolizers (PMs) compared to extensive metabolizers (EMs), resulting in greater systemic exposure.2 Formal pharmacokinetic data in children are limited to CYP2D6 EMs;3 although methodological details are not provided, the difference in ATX exposures between pediatric PMs and EMs is consistent with the 8–10-fold differences observed in adults.4 However, the EM groups in these studies are not well characterized and should more accurately be described as "non-PMs". Non-PM CYP2D6 alleles range from partial activity alleles to multiple copies of fully functional alleles, and the concept of "activity score" (AS) has been proposed to simplify prediction of an individual’s phenotype based upon their specific CYP2D6 diplotype.5 The relationship between a broader range of CYP2D6 dipotypes present in a population (as defined by activity score) and total exposure to the therapeutically-active analytes (ATX and 4-OH-ATX) has not been determined, except for the CYP2D6*10 allele in Asian adults.6, 7
Despite established differences in ATX exposure, dosing recommendations from the product label advise a starting dose of 0.5 mg/kg in all children less than 70 kg, regardless of CYP2D6 genotype. Although ATX has been shown to have a wide therapeutic index, delays in therapeutic response and an increased frequency of adverse effects have been shown in PMs as compared to EMs.8, 9 As suggested by de Leon, a more important consequence of genetic variation for drugs like ATX is inadequate drug exposure, especially for CYP2D6 ultrarapid metabolizers (UMs).10, 11 The objective of this study was to determine the magnitude of effect of CYP2D6 genotype (activity score) on the dose-exposure relationship for ATX and its metabolites in children and adolescents with ADHD.
Results
Demographics
Written permission/assent was obtained for 24 children and 23 completed all study procedures. One participant withdrew from the study following failure to obtain intravenous access. Participants were between 9.5–17.8 years of age, predominantly male (87%), and self-identified as Caucasian (52%), African-American (30%), mixed ethnicity (13%), or Native Hawaiian/Pacific Islander (4%). The average ATX dose was 0.43±0.07 mg/kg. Two participants were overweight and eight were obese. Detailed demographic information is provided in Table 1.
Table 1.
Demographic characteristics for participants completing all study procedures
| Subject ID | Age (y) | Weight (kg) | BMI (kg/m2) | BMI1 (pct) | CYP2D6 Genotype | CYP2D6 Act Score | CYP2C19 Genotype | Gender | Ethnicity2 | Dose (mg) | Dose (mg/kg) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AT01 | 16.7 | 57.0 | 19.8 | 32.1 | *4/*4×N | 0 | *1*2 | Male | CA | 25 | 0.44 |
| AT13 | 17.4 | 101.5 | 32.4 | 98.4 | *4/*4[+*68×4] | 0 | *1*17 | Male | CA | 40 | 0.39 |
| AT14 | 15.4 | 67.8 | 24.4 | 88.2 | *4/*4[+*68×4] | 0 | *1*17 | Male | CA | 40 | 0.59 |
| AT15 | 13.3 | 74.3 | 26.0 | 95.8 | **4/*4[+*68×4] | 0 | *1*17 | Male | CA | 40 | 0.54 |
| AT20 | 11.4 | 42.7 | 19.5 | 78.1 | *5/*17 | 0.5 | *1*17 | Male | AA | 18 | 0.42 |
| AT22 | 15.3 | 73.1 | 22.1 | 74.6 | *4/*9 | 0.5 | *1*17 | Male | CA | 40 | 0.55 |
| AT23 | 15.6 | 65.2 | 22.6 | 76.9 | *5/*17 | 0.5 | *1*2 | Male | AA | 25 | 0.38 |
| AT02 | 11.6 | 43.2 | 17.7 | 52.2 | *2/*5 | 1 | *1*17 | Male | AA | 18 | 0.42 |
| AT04 | 17.9 | 64.6 | 20.6 | 33.3 | *1/*3 | 1 | *1*2 | Male | CA | 30 | 0.46 |
| AT05 | 15.9 | 110.4 | 32.7 | 98.8 | *1/*3 | 1 | *1*17 | Male | CA | 40 | 0.36 |
| AT06 | 17.5 | 93.8 | 26.4 | 89.7 | *2/*4 | 1 | *1*1 | Male | CA | 40 | 0.43 |
| AT17 | 11.0 | 40.7 | 17.6 | 62.5 | *10/*41 | 1 | *1*1 | Male | CA | 18 | 0.44 |
| AT18 | 11.5 | 60.4 | 25.3 | 96.9 | *9/*29 | 1 | *1*1 | Male | AA/CA | 25 | 0.41 |
| AT19 | 16.8 | 118.3 | 32.5 | 98.6 | *1/*4 | 1 | *1*1 | Male | AA/CA | 40 | 0.34 |
| AT21 | 10.8 | 25.5 | 15.7 | 21.7 | *2/*4 | 1 | *1*2 | Female | NH/PI | 10 | 0.39 |
| AT03 | 13.9 | 110.1 | 37.7 | 99.5 | *1/*2 | 2 | *1*17 | Male | AA | 40 | 0.36 |
| AT07 | 15.9 | 63.4 | 20.1 | 44.9 | *1/*1 | 2 | *1*1 | Male | CA | 25 | 0.39 |
| AT09 | 15.0 | 46.9 | 19.8 | 48.4 | *1/*1 | 2 | *1*1 | Male | CA | 25 | 0.53 |
| AT10 | 17.8 | 67.6 | 23.9 | 74.6 | *1/*2 | 2 | *1*2 | Female | AA | 25 | 0.37 |
| AT11 | 16.3 | 111.4 | 36.4 | 99.4 | *1/*1 | 2 | *1*1 | Male | AA | 40 | 0.36 |
| AT12 | 9.5 | 44.9 | 22.9 | 95.8 | *1/*2 | 2 | *1*1 | Female | AA/CA | 18 | 0.4 |
| AT16 | 14.2 | 47.9 | 18.1 | 30.1 | *1/*1 | 2 | *17*17 | Male | CA | 25 | 0.52 |
| AT25 | 11.3 | 29.8 | 16.0 | 24.9 | *1/*2×N | 3 | *2*2 | Male | AA | 10 | 0.34 |
BMI percentile for age and sex
AA, African American; CA, Caucasian; AA/CA, mixed African American and Caucasian; NH/PI, Native Hawaiian or Pacific Islander
Pharmacokinetic analysis
ATX was rapidly absorbed in the EM2 and EM1 groups, reaching maximum concentrations of 0.7±0.2 μM and 1.0±0.3 μM, respectively, approximately 1.5 hrs after dosing (Figure 1A). In PMs, Cmax (2.5±0.3 μM) and Tmax (4.5±1.0 h) were significantly increased relative to the IM, EM1, and EM2 groups (p<0.0001) (Table 2). The Cmax observed for total 4-OH-ATX (i.e. aglycone plus glucuronide metabolite) was genotype-dependent (Figure 1B), being significantly lower in PMs (0.1±0.01 μM) as compared to EM1s (1.9±0.2 μM; p<0.001) and EM2s (1.6±0.5 μM*; p=0.002), but was not significantly different relative to IMs (0.7±0.2 μM; p=0.20). The majority (98.6±1.4%) of plasma 4-OH-ATX was present as the glucuronide conjugate and was unaffected by CYP2D6 genotype (98.5±0.8%, 99.0±0.3%, 98.7±1.3% and 98.5±1.9% for PMs, IMs, EM1s and EM2s), respectively. Peak concentrations of the therapeutically active non-conjugated 4-OH-ATX were considerably lower: 0.0017±0.004 μM in PMs, 0.0063±0.0044 μM in IMs, 0.0126±0.0053 μM in EM1s, and 0.0123±0.0064 μM in EM2s such that the contribution of unconjugated 4-OH-ATX to total therapeutically active ATX was <1%.
Figure 1.
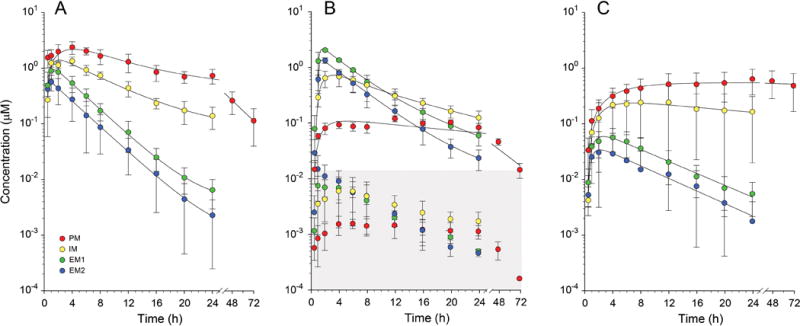
Effect of CYP2D6 genotype on mean ± SD plasma concentration time profiles for ATX (panel A), 4-OH-ATX (panel B), and its N-desmethyl metabolite NDA (panel C). In panel B, lines of best fit are provided for total 4-OH-ATX (non-conjugated plus glucuronide conjugate), whereas the concentration-time profiles for the therapeutically active, non-conjugated form are presented within the shaded area. Concentrations of non-conjugated 4-OH-ATX are considerably lower than ATX concentrations at all time points, and do not contribute to any appreciable extent to total therapeutically active drug in any genotype group.
Table 2.
Pharmacokinetic parameters for atomoxetine, total 4-hydroxy atomoxetine, and N-desmethyl atomoxetine
| CYP2D6 Groups | |||||
|---|---|---|---|---|---|
|
| |||||
| Pharmacokinetic Parameter1 | EM2 (n=8) |
EM1 (n=8) |
IM (n=3) |
PM (n=4) |
p |
| Atomoxetine | |||||
| Cmax (µM) | 0.7 ± 0.2A** | 1.0 ± 0.3A,B** | 1.4 ± 0.1B,C** | 2.5 ± 0.3C | <0.0001 |
| Tmax (h) | 1.4 ± 1.2A** | 1.5 ± 0.5A** | 3.3 ± 1.2B | 4.5 ± 1.0B | <0.0001 |
| AUC0-∞ (µM*h) | 3.5 ± 1.9A** | 4.8 ± 1.6A** | 14.1 ± 1.9A** | 49.6 ± 14.3B | <0.0001 |
| AUC0-∞ (µM*h per 0.5 mg/kg) | 4.4 ± 2.7A** | 5.8 ± 1.7A** | 16.3 ± 4.9B** | 50.2 ± 7.3C | <0.0001 |
| T1/2 (h) | 2.9 ± 0.7A** | 3.0 ± 0.2A** | 6.0 ± 1.2A** | 17.1 ± 3.9B | <0.0001 |
| Vd/F (L/kg) | 2.12 ± 0.67A* | 1.50 ± 0.41A | 1.34 ± 0.36A | 0.94 ± 0.14B | 0.0056 |
| CL/F (L/h/kg) | 0.49 ± 0.21A* | 0.32 ± 0.10A,B* | 0.11 ± 0.03B,C | 0.035 ± 0.005C | 0.0002 |
| Urinary Recovery (% Dose) | 0.21 ± 0.13A* | 0.27 ± 0.26A* | 0.85 ± 0.16A* | 2.75 ± 1.64B | 0.0004 |
|
| |||||
| 4-hydroxy atomoxetine | |||||
|
| |||||
| Cmax (µM) | 1.6 ± 0.5A** | 1.9 ± 0.2A,** | 0.7 ± 0.2B | 0.1 ± 0.01B | <0.0001 |
| Tmax (h) | 2.1 ± 0.8A** | 2.0 ± 0.0A** | 3.3 ± 1.2A** | 14.0 ± 4.0B | <0.0001 |
| AUC0-∞ (µM*h) | 9.6 ± 2.2A* | 12.4 ± 1.50B** | 9.7 ± 2.6A,B* | 5.0 ± 0.9C | <0.0001 |
| Urinary Recovery (% Dose) | 85.4 ± 11.7A** | 82.1 ± 5.3A** | 60.4 ± 11.1B* | 27.2 ± 6.0C | <0.0001 |
|
| |||||
| N-desmethyl atomoxetine | |||||
|
| |||||
| Cmax (µM) | 0.05 ± 0.02A** | 0.07 ± 0.03A** | 0.3 ± 0.2A* | 0.7 ± 0.3B | <0.0001 |
| Tmax (h) | 2.3 ± 1.5A** | 2.9 ± 1.6A** | 8.7 ± 3.1A* | 42.0 ± 23.0B | <0.0001 |
| AUC0-∞ (µM*h) | 0.43 ± 0.30A** | 0.70 ± 0.30A** | 9.5 ± 7.9A* | 78.8 ± 51.9B | <0.0001 |
| Urinary Recovery (% Dose) | <0.02** | <0.03A** | <0.4A* | 1.1 ± 0.5B | <0.0001 |
Mean ± SD
Values designated by the same letter are not significantly different by post-hoc analysis using Tukey-Kramer Honestly Significant Difference
p<0.05 vs.CYP2D6 PM
p<0.0001 vs. CYP2D6 PM
Overall, plasma concentrations of NDA were greater in the PM and IM groups as compared with the EM1 and EM2 groups (Figure 1C). Cmax was CYP2D6 genotype-dependent, ranging from 0.7±0.3 μM, 0.3±0.2 μM, 0.07±0.03 μM, and 0.05±0.02 μM in the PM, IM, EM1, and EM2 groups, respectively. Likewise, AUC0-∞ values were correspondingly greater in the PM and IM groups due to considerably prolonged elimination of the metabolite, presumably due to compromised CYP2D6 activity in these two groups. No relationship between CYP2C19 genotype and plasma AUC or urinary recovery of NDA was observed.
Apparent oral clearance (CL/F) of ATX was significantly associated with genotype; in PMs CL/F was 6.0% of that observed in EM2s (0.035±0.005 vs 0.49±0.21 L/h/kg; p=0.005), and was also significantly reduced in IMs (0.11±0.03 L/h/kg; p=0.02), but not the EM1 group (0.32±0.10 L/h/kg). Similarly, ATX half-life was 2.9-fold longer in PMs as compared to IMs, and 5.4- to 5.9-fold longer than in the EM1 and EM2 groups (Table 2). As the actual dose administered ranged from 0.34–0.59 mg/kg, AUC∞ was dose-normalized and varied 29.6-fold across the study cohort, ranging from 4.4±2.7 μM*h in EM2s to 5.8±1.7 μM*h, 16.3±4.9 μM*h and 50.2±7.3 μM*h in EM1s, IMs and PMs, respectively (p<0.0001) (Figure 2; Table 2).
Figure 2.
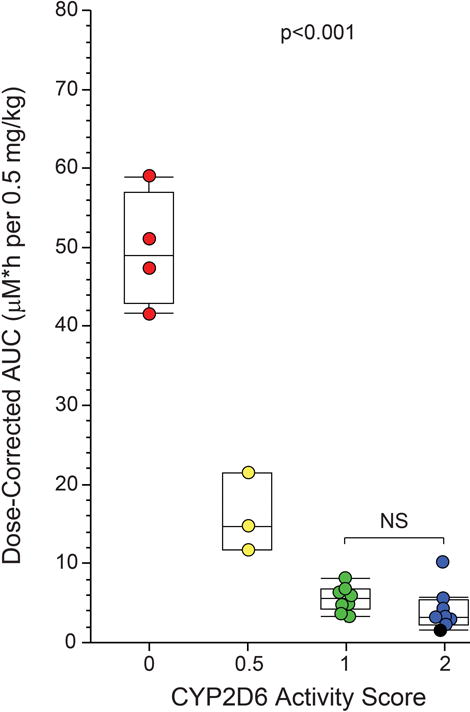
Association between CYP2D6 genotype expressed as activity score and total ATX systemic exposure (AUC0-∞). As a single capsule of the available dosage forms was administered to provide a dose at or near the recommended starting dose of 0.5 mg/kg, the actual dose administered ranged from 0.34–0.59 mg/kg, and AUC0-∞ data were normalized to a 0.5 mg/kg dose. The one participant with a CYP2D6 activity score of 3 (three functional alleles) included in the EM2 group is designated by the black symbol.
Urinary recovery of ATX and metabolites
Complete urine collections were obtained for 19/23 participants; 12-hr collections were complete in all subjects, but 12–24 hr collections were incomplete for two EM1 and two EM2 participants. For those participants with complete urine profiles, 63.5±13.0% of the total dose was recovered in 72 hrs in PMs compared to 70.0±7.4%, 87.1±5.7%, and 90.4±13.2% in 24 hrs in the IM, EM1 and EM2 groups, respectively.
ATX was extensively metabolized with 2.8±1.6% of the administered dose recovered in urine as unchanged drug in PMs, and <1.0% for the other three groups (Table 2). In all groups 4-OH-ATX was the predominant metabolite recovered, primarily as the glucuronide conjugate, and represented 27.2±6.0%, 60.4±11.1%, 82.1±5.3% and 85.4±11.7% of the administered dose in the PM, IM, EM1 and EM2 groups, respectively. Overall, N-demethylation contributed minimally to ATX biotransformation, representing <0.02%, <0.03%, and <0.4% of the administered dose in the EM2, EM1 and IM groups, respectively, and 1.1±0.5% of dose for the PM group. In contrast, 2-methylhydroxylation and subsequent further oxidation of ATX was a quantitatively more important alternative pathway, with summed urinary recovery of the secondary metabolites, 4-OH-carboxy-ATX and OH-carboxy-ATX, as the corresponding glucuronides and representing 4.3±2.6%, 4.9±1.8%, 8.5±5.0% and 32.4±7.7% of the administered dose for the EM2, EM1, IM and PM groups, respectively (Figure 3).
Figure 3.
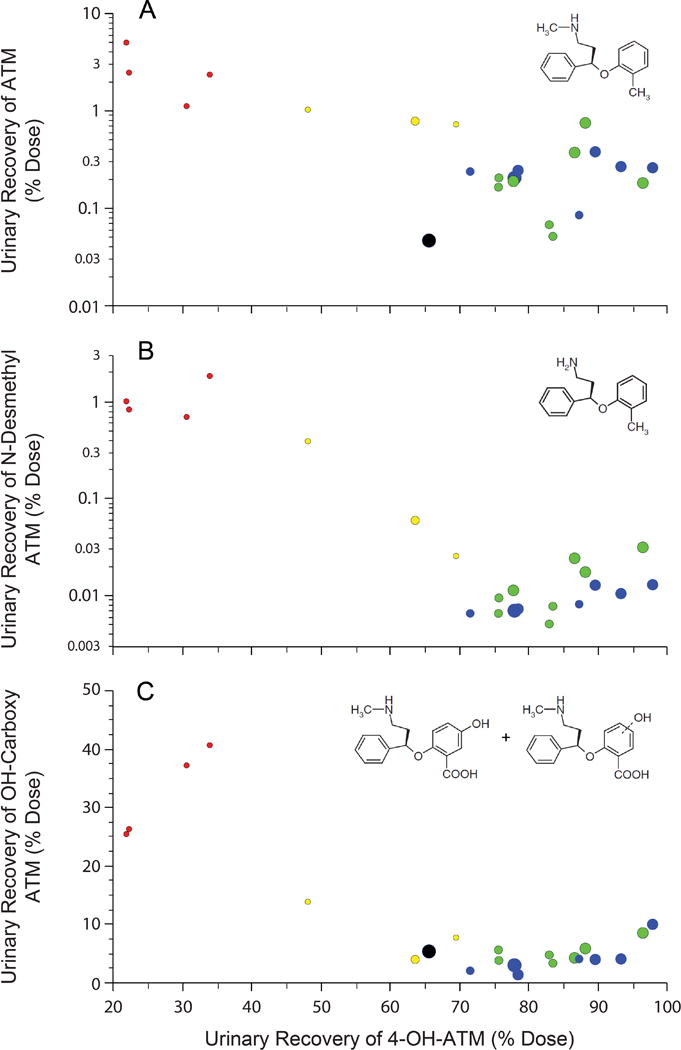
Urinary recovery of ATX and metabolites. The percentage of the administered dose recovered in 24 h (IM, EM1, EM2) or 72 h (PM) urine collections as ATX (panel A), NDA (panel B) and carboxy metabolites, products of subsequent biotransformation of initial 2-methylhydroxylation (panel C), is presented as a function of the percentage of the administered dose recovered as 4-OH-ATX. Recovery exceeded 100% of the dose in two subjects (~105%). 4-OH-ATX was the most abundant single metabolite recovered in all CYP2D6 genotype groups, but the hydroxycarboxy products of initial 2-methylhydroxylation constituted a considerably larger proportion of overall ATX biotransformation in the PM group. Individual data points are color-coded by genotype (PM, red; IM, yellow; EM1, green; EM2, blue), and the size of the circles represent the apparent oral clearance relative to the largest apparent oral clearance in an individual with three functional CYP2D6 alleles (black symbol); NDA was not detected in the urine of this participant. The extremely small size of the PM data points relative to the reference demonstrates that the increased contribution of 2-hydroxylation in PMs does not “compensate” for the absence of CYP2D6 activity.
Adverse Events
Adverse events considered to be related to the study drug were reported in 10 of the 23 participants, with drowsiness being the most commonly reported adverse event (8/23). One PM participant reported palpitations with concurrent tachycardia. Additionally, one EM1 participant complained of a self-described “hot flash,” one EM1 participant complained of feeling light-headed, and one IM participant complained of dizziness, nausea, and headache. The average maximum increase from baseline for pulse was similar across the four groups (30.3±14.6 bpm in PMs, 24.0±6.57 bpm in IMs, 28.0±14.7 bpm in EM1s, and 28.0±14.9 bpm in EM2s). One PM, one EM1, and one EM2 experienced an increase in systolic blood pressure of greater than 20 mm Hg from baseline.
Discussion
ATX is one of a few non-stimulant alternatives currently used to treat ADHD, and is considered to have a more attractive safety profile relative to stimulants, at least with respect to risk of behavioral changes and appetite suppression. Nevertheless, use of ATX has declined between 2004 and 2010 since the initial spike in prescribing rates following FDA approval in 200212 despite an increase in ADHD prevalence.13 A retrospective analysis of published clinical trials of atomoxetine in children by Newcorn et al reported that 40%of patients were considered to be non-responders to the drug as defined by a <25% decrease from baseline ADHD Rating Score after nine weeks of treatment.14 As will discussed in more detail below, the results of our study suggest one possible factor contributing to this phenomenon.
Given that ATX is predominantly metabolized by CYP2D6, variability in the dose-exposure relationship could give rise to variability in response, ranging from increased risk of treatment-limiting toxicity at one extreme to a perceived lack of efficacy at the other. The product label acknowledges a 10-fold difference in AUC between CYP2D6 poor and extensive metabolizers as reported by Sauer et al.2 Studies in adult Chinese6 and Japanese7 participants reported approximately two-fold higher systemic exposure in subjects with the CYP2D6*10/*10 genotype (reduced function alleles corresponding to our EM1 group) compared to subjects with CYP2D6*1/*1 or *1/*10 genotypes. Although the difference in dose-corrected AUC0-∞ between the EM1 and EM2 groups in our pediatric population was less pronounced (1.3-fold) than that observed in Asian adults, the 11.4-fold difference in dose-corrected AUC0-∞ between the PM and EM2 groups in our study is comparable to the 9-fold lower clearance between PM and EM children reported by others.15 An important finding from our study is the dose-exposure data for the IM participants who were compound heterozygous for a nonfunctional and a reduced function allele. Systemic exposure for this group was intermediate between the PM and EM1 groups, indicating that simple classification of patients into PM and non-PM (EM) groups is insufficient for genotype-based dosing strategies.
Decisions regarding the practical value of genotype test information prior to initiation of treatment are driven by an understanding of the likely consequences of genetic variation, and generally focus on safety/toxicity. Although many adverse drug reactions are reported to be approximately 2-fold higher more frequent in PMs compared to EMs,9 a series of dose titration studies conducted over several weeks by investigators blinded with respect to participants’ CYP2D6 genotype status led to the conclusion that pre-emptive genotyping was unnecessary as physicians were able to appropriately titrate patient doses without regard to CYP2D6 genotype.16 Conversely, based on a retrospective analysis involving several patients with reduced CYP2D6 activity and either a delayed response or adverse effects, another group has advocated that knowledge of an individual’s CYP2D6 genotype and/or phenotype may be clinically useful when initiating ATX treatment.8 Regardless, available data also suggest that the increased frequency of adverse effects associated with PM status is accompanied by a better clinical response,4 implying that an exposure-response relationship may exist. As CYP2D6 genotyping is not routinely conducted, our data clearly demonstrate that ATX levels at the 6-hour mark correlate extremely well (r=0.986) with dose-corrected AUC (data not shown), implying that a single point assessment of ATX concentration could act as a surrogate for ATX exposure and provide clinicians with an accurate estimate of the patient’s ATX exposure.
Traditionally, pharmacogenetic testing is considered to be most relevant for narrow therapeutic index drugs. In recent commentaries, de Leon has presented a compelling argument for the importance of pharmacogenetic variability for drugs with broad therapeutic indices as well.10, 11 Specifically, dose escalation studies in a population with a wide range of CYP2D6 activity may lead to excessive exposure for PMs that, in turn, results in a capping of the maximum dose with the potential unintended consequence of inadequate exposure in extensive or ultrarapid metabolizers. Thus, the average dose in a population with such wide variation in CYP2D6 activity becomes increasingly inappropriate at the extreme ends of the treatment population. This phenomenon appears to be the case for ATX. Michelson et al,4 using an inhibitory Emax model, estimated that the maximum improvement in ADHD symptoms corresponded to a peak ATX concentration (60–90 min post dose) of 800 ng/ml (~3.1 μM), and this concentration threshold has been incorporated into some study designs.17 In one Eli Lilly and Company study, B4Z-MC-LYCL,18 the majority (256/294; 87%) of patients did not achieve the 800 ng/ml threshold after 6 weeks of an ATX dose of 1.2 mg/kg/d. In this analysis, only 39% of subjects with ATX concentrations <800 ng/ml had adequate symptom reduction compared to 55% of subjects with concentrations >800 ng/ml. The 1.2 mg/kg/d dose used in the study represents the recommended target dose in the current product label, and 88% of ATX concentration measurements obtained after 6 weeks of treatment exceeded 800 ng/ml in PMs whereas only 9% of values reached this threshold for EMs. Thus, it is likely that a significant number of children may not achieve adequate drug exposure using the current dosing guidelines, a consequence of the extensive variability in the dose-exposure relationship, even using weight-based dosing. However, the response model presented by Michelson et al implies that near maximal responses may occur at concentrations exceeding 400 ng/ml4, and 39% of subjects in study B4Z-MC-LYCL 18 achieved a therapeutic response at peak concentrations <800 ng/ml. Thus the issue of what the right exposure is for a given patient remains unresolved and is an important additional area to be considered in future studies. When the participant-specific pharmacokinetic parameters observed in the present study were used to simulate a once daily dosing regimen at the maximum recommended dose of 1.4 mg/kg/day (maximum of 100 mg/day), 0/8 of the EM2s, 2/8 of the EM1s, 2/3 of the IMs, and all four of the PMs achieved values greater than 800 ng/ml or 3.1 μM (Figure 4). On average, PMs reached a peak concentration of 2,360 ng/ml (9.1 μM) with approximately 82% of the 24-hr dosing interval exceeding the target threshold.
Figure 4.
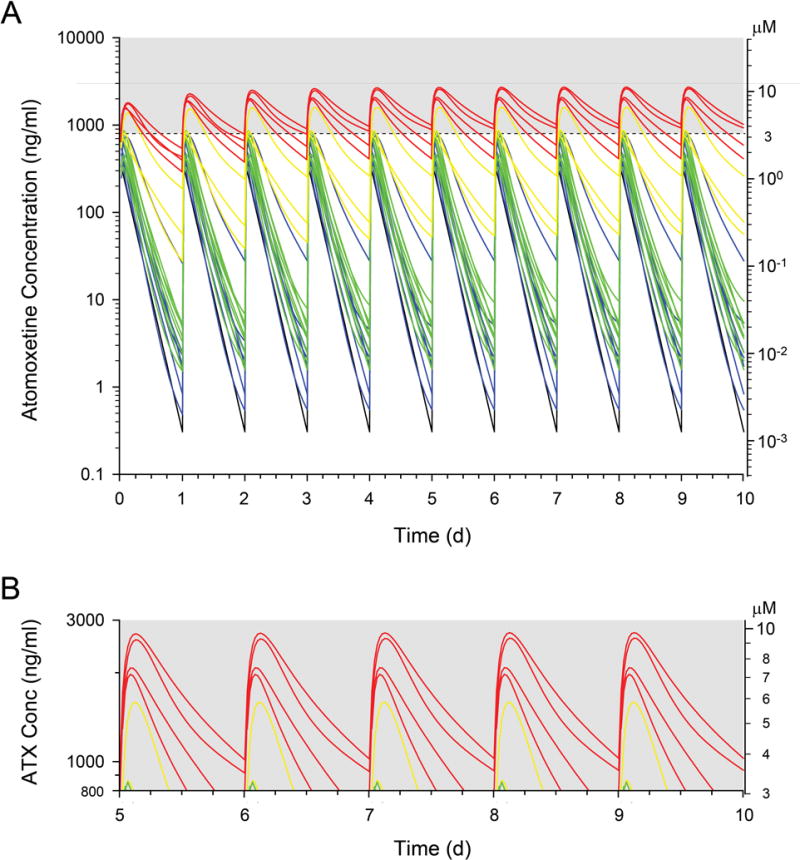
Simulated atomoxetine plasma concentration-time profiles for each study participant over the first 10 days of treatment with the current maximum FDA-recommended dose of 1.4 mg/kg up to 100 mg/d. A. The dashed line represents the threshold for therapeutic benefit proposed by Michelson et al.4 Plasma concentration profiles are color-coded for the genotype-stratified groups: PM (red), IM (yellow), EM1 (green) and EM2 (blue). The one participant with a CYP2D6 activity score of 3 (three functional alleles) included in the EM2 group is represented by the black profile. B. The gray shaded area in panel A is expanded to demonstrate that at steady state, only five study participants would exceed the proposed 800 ng/ml (~3.1 μM) threshold for any appreciable period of time under current FDA-approved dosing guidelines. Ordinate axes are provided for interconversion of ng/ml and μM units.
From a biostatistical viewpoint, assessment of the effect of an independent variable, such as genotype, on a dependent variable like AUC involves the comparison of the mean AUC between genotypes and satisfies scientific and regulatory purposes. These data are summarized in publications and occasionally in product labels. However, data presented in this manner are not conducive to individualization or “precision medicine” as clinicians, parents, and patients are less concerned about differences in the dose-exposure relationship on average between subgroups within a population than they are about what dose of the medication is most likely to produce the desired therapeutic response in the individual patient. From this perspective, a key finding of our study is the nearly 30-fold range of AUC values observed following a single 0.5 mg/kg dose. Most relevant, however, is the observation that if, in clinical practice, a pediatrician is likely to administer the single available dosage strength that most closely approximates 0.5 mg/kg as we did in this study, the range of AUC values varies 51.3-fold between the PM with the most impaired clearance and the UM with the highest clearance. Given that the product label indicates that CYP2D6 genotyping is not necessary, this information is unlikely to be available when a prescription is written, and the pediatric prescriber has no way of knowing where within this 50-fold range of drug exposure a given patient will be following the initial nominal 0.5 mg/kg dose.
Knowledge of CYP2D6 genotype alone may, however, not allow precise prediction of ATX Cmax or AUC following a given dose. Although dose-corrected AUC0-∞ was the least variable in the PM group (1.4-fold), the absolute difference in AUC0-∞ in the PM group was 17.5 μM*h, more than three-times the range of AUC0-∞ values for the EM1 and EM2 groups (~5.1 μM*h). As 4-hydroxylation remains a primary route of ATX elimination by PMs, sources of variability in non-CYP2D6-mediated 4-hydroxylation will need to be identified. Examination of urinary metabolite data in our study revealed that almost half of the metabolic clearance of ATX in PMs involves initial benzylic hydroxylation to form 2-OH-ATX, which subsequently undergoes further biotransformation to hydroxycarboxy metabolites. It should be noted that increased 2-hydroxylation does not “compensate” for the absence of CYP2D6 activity as apparent oral clearance of ATX in PMs remains dramatically reduced compared to EMs. Nevertheless, this pathway does account for a greater proportion of biotransformation in individuals with reduced CYP2D6 function, and therefore the enzymes involved will also need to be characterized for inclusion in individualized dosing algorithms.
Within the IM, EM1, and EM2 groups, interindividual variation in dose-normalized AUC0-∞ varied 1.8-, 2.5- and 5.1-fold, respectively, and can be primarily attributable to variability in CYP2D6 activity. Wang et al identified an enhancer SNP about 100 kb downstream of the CYP2D6 gene locus affecting mRNA expression levels,19, 20 but genotyping for the enhancer SNP rs5758550 did not account for the within group variability observed in our study (not shown), albeit given a limited sample size. Although the genotype-stratified pharmacokinetic design we employed efficiently estimated the magnitude of the dose-exposure relationship in a population, the number of participants in each CYP2D6 activity score group was insufficient to capture the range of exposures within each group. Thus, larger studies will be necessary to identify additional factors contributing to inter-individual variability in CYP2D6 activity within groups with nominally the same CYP2D6 genotypes.
Our data have important implications for individualization of drug therapy. In situations where a drug is highly dependent upon a polymorphically expressed gene product for its elimination and activity of the pathway is highly variable in the population, the “average dose” may be associated with clinically significant consequences at both extremes of the activity distribution. In the case of atomoxetine, the currently approved dose of atomoxetine appears to represent a compromise intended to minimize the risk of toxicity in PMs. An unintended consequence of this compromise is that current dosing guidelines are likely to result in inadequate exposures for a considerable proportion of the intended population, supporting the concern raised by de Leon.11 However, it is important to note that these conclusions should be confirmed through additional studies that further refine the exposure-response relationship of atomoxetine. For example, given the extensive variability in atomoxetine systemic exposure even with weight-based dosing, individualized dosing to achieve a target exposure will be necessary to investigate the role of genetic variation in the drug target and downstream effector pathways to optimize clinical effectiveness and minimize adverse events.
Methods
Subjects
Children and adolescents (n=24) between 6–17 years of age were eligible for enrollment. Participants were selected based upon a diagnosis of ADHD and previous participation in a longitudinal phenotyping study wherein CYP2D6 genotype was determined. Participants were excluded if they were currently taking ATX, known CYP2D6 inhibitors, had any known serious structural cardiac abnormalities, any flu like symptoms within 14 days, diagnosis of inflammatory bowel disease or Crohn’s disease, or those with any hepatic and/or renal insufficiency. Participants were allowed to continue their daily medications. All participants provided written assent, while their parent provided written permission. The protocol and permission/assent documents were approved by the Institutional Review Board at Children’s Mercy Kansas City.
Study Design
The study cohorts for this open label, CYP2D6 genotype-stratified, single-dose pharmacokinetic study of ATX consisted of participants with 1) a CYP2D6 activity score of 2 or more (n=8) defined as extensive metabolizers with two or more functional alleles (EM2;5 one participant had three functional alleles), 2) a CYP2D6 activity score of 1 (extensive metabolizers with one functional and one nonfunctional allele, or two reduced function alleles; EM1, n=8), 3) a CYP2D6 activity score of 0.5 (intermediate metabolizers with one reduced function allele and one nonfunctional allele; IM, n=3), and 4) a CYP2D6 activity score of 0 (poor metabolizer with 2 nonfunctional alleles; PM n=4). Notably, three of the four participants in the PM group were siblings.
Prior to dosing, participants underwent a full physical exam, an electrocardiogram, urinalysis, vital signs, a complete blood count, serum chemistries, electrolytes, glucose, and creatinine, all of which were interpreted and approved by a physician. Participants were dosed according to a naturalistic protocol to mimic the clinical setting such that each participant <70 kg received a single capsule of the available dosage forms to provide a dose at or near the recommended starting dose of 0.5 mg/kg; participants greater than 70 kg were given a maximum of 40 mg. Participants completed the study with an overnight stay within the pediatric clinical research unit. Plasma samples were obtained at predose, 0.5, 1, 2, 4, 6, 8, 12, 16, 20, and 24 hours after dosing. Heart rate (CARESCAPE™ V100 Vital Signs Monitor, GE Healthcare) and blood pressure (DINAMAP™ ProCare Monitor, GE Healthcare) were measured at each time point. For participants with a CYP2D6 activity score of 0 (i.e. PM), 48- and 72-hour plasma samples were also collected due to the extended half-life of ATX and its metabolites. Urine was collected throughout the pharmacokinetic sampling period.
Analytical method
Concentrations of ATX and metabolites in plasma and urine were determined by an UHPLC-tandem mass spectrometry method developed in-house that was validated according to the FDA guidelines for a bioanalytical assay (van Haandel L, Brown JT, Gibson KT, Leeder JS, manuscript in preparation). Briefly, samples were extracted using solid phase extraction (SPE) in a 96 well plate format (OASIS hydrophilic lipophilic balance μElution plates (Waters Corp., Milford, MA)). Analytes were separated on a Waters Acquity UPLC C18 reversed phase column (BEH 1.7μm, 2.1 x100mm) using gradient elution and mobile phases consisting of 30:70 MeOH: ammonium formate buffer, pH 3.5 (A) and 90:10 MeOH: ammonium formate buffer, pH 3.5 (B). The elution profile started with 30% mobile phase B and was linearly ramped up to 70% over 1.2 minutes, followed by an isocratic hold for 1.0 minute.
ATX was quantitated against a standard reference solution (Sigma-Aldrich), whereas NDA and 4-OH-ATX were quantitated against standards obtained from Toronto Research Chemicals. Isotope labeled (deuterated) standards of ATX, 4-OH-ATX and NDA (Toronto Research Chemicals), were utilized as internal standards. The secondary metabolites 4-OH-carboxy-ATX and OH-carboxy-ATX were determined semiquantitatively, utilizing the 4-OH-ATX calibration curves and internal standard, due to lack of a commercial source of standards for these metabolites. For both plasma and urine, Total 4-OH-ATX concentration was determined following deconjugation (18 h, 37°C with β-glucuronidase from Helix pomatis (>100,000 units/mL; Sigma Aldrich Chemical Co.) whereas free 4-OH-ATX was measured in the absence of the deconjugation step. 4-OH-ATX glucuronide metabolite was calculated as the difference between total and free 4-OH-ATX.
Pharmacokinetic Analysis
Pharmacokinetic analyses were conducted using Kinetica™ version 5.0 (Thermo Scientific, Philadelphia, PA). Pharmacokinetic parameters for each participant were estimated using non-compartmental analyses. The area under the plasma concentration versus time curve during the sampling period (AUC0-t) was calculated using the mixed log-linear rule and extrapolated to infinity (AUC0-∞). Apparent oral clearance was calculated by dividing the weight-based dose by AUC0-∞. 4-OH-ATX and NDA pharmacokinetics were evaluated similarly to ATX. For the purposes of simulation, ATX concentration versus time data was curve fit in Kinetica™ using a peeling algorithm to generate initial polyexponential parameter estimates. Model-dependent pharmacokinetic parameters were calculated from final polyexponential parameter estimates. Each participant’s parameter estimates were then used to simulate steady-state concentrations by the principle of superposition using Kinetica™ at the maximum recommended dose of 1.4 mg/kg, not to exceed 100 mg.
CYP2D6 and CYP2C19 Genotyping
CYP2D6 genotyping was carried out according to procedures described in further detail elsewhere.5, 21–24 Briefly, a 6.6 kb long-range PCR (XL-PCR) fragment was amplified with CYP2D6-specific primers and the product verified by agarose gel electrophoresis prior to 2000-fold dilution with 10 mM Tris pH 8 and used as a template for TaqMan (*2–*4, *6, *7, *9–*12, *15, *17, *29, *35, *36, *40–*42; Thermo Fisher Scientific, formerly Life Technologies, Foster City, CA, USA) or PCR-RFLP-based (*31 and *45/46) genotyping assays. Each assay included study samples as well as positive and negative controls. For TaqMan assays, 6 μl reactions were conducted in 96-well plates using the TaqMan Genotyping Master Mix or the KAPA Probe qPCR Master Mix (KAPA Biosystems, Wilmington, MA). Cycling was performed on the Applied Biosystems 7900 Real Time PCR System according to manufacturer’s specifications; data were analyzed with the SDS2.4 software. The CYP2D6*5 gene deletion, the presence of gene duplications/multiplications and CYP2D6*13-like CYP2D7/2D6 hybrid genes were assayed by XL-PCR. Copy number variation was assessed by quantitative multiplex PCR interrogating four different gene regions as previously described.25 The exon 9 conversion defining CYP2D6*36 was genotyped from genomic DNA using a duplex PCR assay,21 and CYP2D6*3123 and *45/4626 were genotyped by PCR-RFLP. To characterize gene duplication events, an 8.6 kb long XL-PCR product encompassing the duplicated gene copy was generated,25 and the amplified fragment genotyped for the presence of sequence variations as described above. Alleles are defined according to the Human Cytochrome P450 (CYP2D6) Allele Nomenclature Database at www.cypalleles.ki.se/cyp2d6.htm, and converted to activity scores according to Gaedigk et al.5
All subjects were genotyped for CYP2C19*2, *3, *4 and *17 using genomic DNA and commercially available TaqMan genotyping assays in the presence of positive and negative controls. Six μl reactions were carried out in 96-well plates using the KAPA Probe qPCR Master Mix. Cycling and data analysis was performed as described above for CYP2D6 genotyping.
Safety
Adverse events were recorded on the case report form, and were defined as any undesirable sign, symptom, or medical condition occurring after the informed consent/assent form was signed.
Statistical methods
The sample size for the PM, EM1 and EM2 groups was calculated based on observed 2-fold differences in ATX AUC between EM1 (CYP2D6*10/*10) and EM2 adults;6 groups of n=8 for EM1 and EM2 provided 86% power to detect a 2-fold difference in AUC between groups with a one-sided alpha value of 0.05. Two PM subjects were sufficient to detect a 10-fold difference under the same conditions.
ATX pharmacokinetic data for each CYP2D6 activity score group were examined using standard descriptive statistics. Analysis of variance was utilized for comparisons of pharmacokinetic parameters between the four different CYP2D6 activity score groups, and post-hoc analyses were conducted using Tukey-Kramer Honestly Significant Difference Analyses using SPSS version 20.0 (SPSS, Chicago, IL) and JMP 11.0 (SAS Institute Inc., Cary, NC) with the significance limit set at α=0.05.
Study Highlights
What is the current knowledge on the topic?
Atomoxetine’s metabolic profile is greatly impacted by CYP2D6 activity. Pharmacokinetic studies in adults show 10-fold differences in AUC values between individuals who are CYP2D6 extensive versus poor metabolizers.
What question did this study address?
The magnitude of effect that the number of functional CYP2D6 alleles has on the dose-exposure profile of atomoxetine and metabolites in children with ADHD.
What this study adds to our knowledge?
Dosing children at the recommended 0.5 mg/kg starting dose, without consideration of CYP2D6 genotype or predicted phenotype, results in a 30-fold range in exposure as measured by dose-corrected AUC0-∞. Simulated steady-state exposure profiles at the maximum recommended dose reveal that the majority of children are unlikely to attain adequate atomoxetine exposure.
How this might change clinical pharmacology and therapeutics?
The data from this study can be used to inform a systems pharmacology-based dosing algorithm to reduce the wide range of exposures to atomoxetine experienced with current clinical dosing recommendations and ultimately lead to new, more rational individualized dosing strategies to improve safety and efficacy of the drug in children with ADHD.
Acknowledgments
Support was provided by grants from the National Institutes of Health’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) grant 1R01 HD058556 (Leeder, Lin, co-PIs). J.T.B was supported by grant T32 HD069038 “Postdoctoral Training in Pediatric Clinical Pharmacology” from NICHD (Kearns, PD). The authors would like to gratefully acknowledge Krista J. Wright for her assistance in participant recruitment and clinical trial execution.
Footnotes
Conflict of Interest: The authors declare no conflicts of interest.
Author Contributions: J.T.B, S.M.A., L.V.H., A.G., and J.S.L. wrote the article; J.T.B, S.M.A., Y.S.L., and J.S.L. designed the research; J.T.B. and S.M.A. performed the research; J.T.B, S.M.A., L.V.H. and J.S.L. analyzed the data.
References
- 1.Ring BJ, Gillespie JS, Eckstein JA, Wrighton SA. Identification of the human cytochromes P450 responsible for atomoxetine metabolism. Drug Metab Dispos. 2002;30:319–23. doi: 10.1124/dmd.30.3.319. [DOI] [PubMed] [Google Scholar]
- 2.Sauer JM, et al. Disposition and metabolic fate of atomoxetine hydrochloride: the role of CYP2D6 in human disposition and metabolism. Drug Metab Dispos. 2003;31:98–107. doi: 10.1124/dmd.31.1.98. [DOI] [PubMed] [Google Scholar]
- 3.Witcher JW, et al. Atomoxetine pharmacokinetics in children and adolescents with attention deficit hyperactivity disorder. J Child Adolesc Psychopharmacol. 2003;13:53–63. doi: 10.1089/104454603321666199. [DOI] [PubMed] [Google Scholar]
- 4.Michelson D, Read HA, Ruff DD, Witcher J, Zhang S, McCracken J. CYP2D6 and clinical response to atomoxetine in children and adolescents with ADHD. J Am Acad Child Adolesc Psychiatry. 2007;46:242–51. doi: 10.1097/01.chi.0000246056.83791.b6. [DOI] [PubMed] [Google Scholar]
- 5.Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–42. doi: 10.1038/sj.clpt.6100406. [DOI] [PubMed] [Google Scholar]
- 6.Cui YM, et al. Atomoxetine pharmacokinetics in healthy Chinese subjects and effect of the CYP2D6*10 allele. Br J Clin Pharmacol. 2007;64:445–9. doi: 10.1111/j.1365-2125.2007.02912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui A, et al. Pharmacokinetics, safety, and tolerability of atomoxetine and effect of CYP2D6*10/*10 genotype in healthy Japanese men. J Clin Pharmacol. 2012;52:388–403. doi: 10.1177/0091270011398657. [DOI] [PubMed] [Google Scholar]
- 8.ter Laak MA, Temmink AH, Koeken A, van ’t Veer NE, van Hattum PR, Cobbaert CM. Recognition of impaired atomoxetine metabolism because of low CYP2D6 activity. Pediatr Neurol. 2010;43:159–62. doi: 10.1016/j.pediatrneurol.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Strattera [Product Label] http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021411s046lbl.pdf. Accessed June 29, 2015.
- 10.de Leon J. The crucial role of the therapeutic window in understanding the clinical relevance of the poor versus the ultrarapid metabolizer phenotypes in subjects taking drugs metabolized by CYP2D6 or CYP2C19. J Clin Psychopharmacol. 2007;27:241–5. doi: 10.1097/JCP.0b013e318058244d. [DOI] [PubMed] [Google Scholar]
- 11.de Leon J. Translating pharmacogenetics to clinical practice: Do cytochrome P450 2D6 ultrarapid metabolizers need higher atomoxetine doses? J Am Acad Child Adolesc Psychiatry. 2015;54:532–4. doi: 10.1016/j.jaac.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 12.Chai G, Governale L, McMahon AW, Trinidad JP, Staffa J, Murphy D. Trends of outpatient prescription drug utilization in US children, 2002–2010. Pediatrics. 2012;130:23–31. doi: 10.1542/peds.2011-2879. [DOI] [PubMed] [Google Scholar]
- 13.Boyle CA, et al. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics. 2011;127:1034–42. doi: 10.1542/peds.2010-2989. [DOI] [PubMed] [Google Scholar]
- 14.Newcorn JH, Sutton VK, Weiss MD, Sumner CR. Clinical responses to atomoxetine in Attention-Deficit/Hyperactivity Disorder: The Integrated Data Exploratory Analysis (IDEA) study. J Am Acad Child Adolesc Psychiatry. 2009;48:511–8. doi: 10.1097/CHI.0b013e31819c55b2. [DOI] [PubMed] [Google Scholar]
- 15.Witcher J, Kurtz D, Heathman M, Sauer JM, Smith BP. Population pharmacokinetic analysis of atomoxetine in pediatric patients [abstract] Clin Pharmacol Ther. 2004;75:46. [Google Scholar]
- 16.Trzepacz PT, Williams DW, Feldman PD, Wrishko RE, Witcher JW, Buitelaar JK. CYP2D6 metabolizer status and atomoxetine dosing in children and adolescents with ADHD. Eur Neuropsychopharmacol. 2008;18:79–86. doi: 10.1016/j.euroneuro.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 17.Hazell P, et al. Relationship between atomoxetine plasma concentration, treatment response and tolerability in attention-deficit/hyperactivity disorder and comorbid oppositional defiant disorder. ADHD Attn Def Hyp Disord. 2009;1:201–10. doi: 10.1007/s12402-009-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eli Lilly & Company. http://www.lillytrials.com/results/Strattera.pdf. Accessed June 29, 2015.
- 19.Wang D, Papp DC, Sun X. Functional characterization of CYP2D6 enhancer polymorphisms. Hum Mol Genet. 2015;24:1556–62. doi: 10.1093/hmg/ddu566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D, Poi MJ, Sun X, Gaedigk A, Leeder JS, Sadee W. Common CYP2D6 polymorphisms affecting alternative splicing and transcription: long-range haplotypes with two regulatory variants modulate CYP2D6 activity. Hum Mol Genet. 2014;23:268–78. doi: 10.1093/hmg/ddt417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaedigk A, Bradford LD, Alander SW, Leeder JS. CYP2D6*36 gene arrangements within the CYP2D6 locus: association of CYP2D6*36 with poor metabolizer status. Drug Metab Dispos. 2006;34:563–9. doi: 10.1124/dmd.105.008292. [DOI] [PubMed] [Google Scholar]
- 22.Gaedigk A, Fuhr U, Johnson C, Berard LA, Bradford D, Leeder JS. CYP2D7-2D6 hybrid tandems: identification of novel CYP2D6 duplication arrangements and implications for phenotype prediction. Pharmacogenomics. 2010;11:43–53. doi: 10.2217/pgs.09.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaedigk A, et al. Identification of novel CYP2D7-2D6 hybrids: Non-functional and functional Variants. Front Pharmacol. 2010;1:121. doi: 10.3389/fphar.2010.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaedigk A, et al. Cytochrome P4502D6 (CYP2D6) gene locus heterogeneity: characterization of gene duplication events. Clin Pharmacol Ther. 2007;81:242–51. doi: 10.1038/sj.clpt.6100033. [DOI] [PubMed] [Google Scholar]
- 25.Gaedigk A, Twist GP, Leeder JS. CYP2D6, SULT1A1 and UGT2B17 copy number variation: quantitative detection by multiplex PCR. Pharmacogenomics. 2012;13:91–111. doi: 10.2217/pgs.11.135. [DOI] [PubMed] [Google Scholar]
- 26.Gaedigk A, et al. Identification and characterization of novel sequence variations in the cytochrome P4502D6 (CYP2D6) gene in African Americans. Pharmacogenomics J. 2005;5:173–82. doi: 10.1038/sj.tpj.6500305. [DOI] [PMC free article] [PubMed] [Google Scholar]