

Abstract
Although dopamine receptor antagonism has long been associated with impairments in motor performance, more recent studies have shown that dopamine D2 receptor (D2R) antagonism, paired with a motor task, not only impairs motor performance concomitant with the pharmacodynamics of the drug, but also impairs future motor performance once antagonism has been relieved. We have termed this phenomenon “aberrant motor learning” and have suggested that it may contribute to motor symptoms in movement disorders such as Parkinson's disease (PD). Here, we show that chronic nicotine (cNIC), but not acute nicotine, treatment mitigates the acquisition of D2R-antagonist-induced aberrant motor learning in mice. Although cNIC mitigates D2R-mediated aberrant motor learning, cNIC has no effect on D1R-mediated motor learning. β2-containing nicotinic receptors in dopamine neurons likely mediate the protective effect of cNIC against aberrant motor learning, because selective deletion of β2 nicotinic subunits in dopamine neurons reduced D2R-mediated aberrant motor learning. Finally, both cNIC treatment and β2 subunit deletion blunted postsynaptic responses to D2R antagonism. These results suggest that a chronic decrease in function or a downregulation of β2-containing nicotinic receptors protects the striatal network against aberrant plasticity and aberrant motor learning induced by motor experience under dopamine deficiency.
SIGNIFICANCE STATEMENT Increasingly, aberrant plasticity and aberrant learning are recognized as contributing to the development and progression of movement disorders. Here, we show that chronic nicotine (cNIC) treatment or specific deletion of β2 nicotinic receptor subunits in dopamine neurons mitigates aberrant motor learning induced by dopamine D2 receptor (D2R) blockade in mice. Moreover, both manipulations also reduced striatal dopamine release and blunt postsynaptic responses to D2R antagonists. These results suggest that chronic downregulation of function and/or receptor expression of β2-containing nicotinic receptors alters presynaptic and postsynaptic striatal signaling to protect against aberrant motor learning. Moreover, these results suggest that cNIC treatment may alleviate motor symptoms and/or delay the deterioration of motor function in movement disorders by blocking aberrant motor learning.
Keywords: aberrant plasticity, Beta2 nAChR, dopamine D2 receptor, dopamine release, inhibitory learning
Introduction
Dopamine plays a critical role in motor performance and motor learning by modulating throughput in the basal ganglia–cortical loop. Recently, we found that dopamine receptor blockade, paired with a motor task, induces experience-dependent, learned motor impairment, which we termed “aberrant motor learning” (Beeler et al., 2010, 2012). In these studies, dopamine blockade paired with rotarod training impaired performance. This impairment persisted even when dopamine signaling was restored and performance only gradually improved with repeated drug-free training sessions. Critically, impairments in rotarod performance were not observed in mice administered dopamine receptor antagonists in their home cage (Beeler et al., 2012). Therefore, we defined aberrant motor learning as an experience-dependent, learned inhibition of movement that inappropriately impedes future drug-free performance of a motor task.
This aberrant learning is dopamine D2 receptor (D2R) dependent and likely arises from abnormal long-term potentiation (LTP) of corticostriatal inputs to indirect pathway neurons under conditions of reduced dopamine signaling (Beeler et al., 2010; Beeler, 2011; Beeler et al., 2012, 2013; Zhuang et al., 2013). Inactivity of D2Rs under low-dopamine conditions or D2R antagonism increases intracellular cAMP levels, promoting LTP in these neurons (Calabresi et al., 2000; Shen et al., 2008; Lerner and Kreitzer, 2011; Augustin et al., 2014). We have suggested that inappropriate D2R-mediated LTP underpins aberrant motor learning and plays a significant role in the development and progression of motor symptoms in Parkinson's disease (PD). Furthermore, we have suggested that the correction of aberrant learning may underlie the long-duration response (LDR) associated with l-DOPA use in PD patients (Beeler et al., 2010; Beeler, 2011; Beeler et al., 2012, 2013; Zhuang et al., 2013). In support of these hypotheses, several studies have reported that manipulations that block or diminish abnormal LTP, or those that restore normal corticostriatal plasticity, are associated with reduced parkinsonian-like symptoms in rodents (Kreitzer and Malenka, 2007; Shen et al., 2008; Peterson et al., 2012; Thiele et al., 2014). In addition, clinical studies report that motor skills progressively worsen in PD patients when practiced in the absence of dopamine replacement therapy (Kang et al., 2012; Anderson et al., 2014). Therefore, we have suggested that targeting aberrant learning mechanisms may delay the deterioration of motor function in PD (Beeler et al., 2010; Beeler, 2011; Beeler et al., 2012; Zhuang et al., 2013).
In the current study, we investigated whether chronic nicotine (cNIC) exposure could mitigate aberrant motor learning in mice. The rationale for these studies stems from the well documented crosstalk between the endogenous acetylcholine and dopamine systems. Endogenous acetylcholine gates striatal dopamine release via presynaptic nicotinic receptors (nAChRs) on dopamine terminals, and nicotine can transiently increase striatal dopamine release (Zhou et al., 2001; Rice and Cragg, 2004; Threlfell et al., 2012; Cachope et al., 2012; Zhang and Sulzer, 2004). However, cNIC exposure attenuates nicotine-elicited dopamine release (Marks et al., 1993; Picciotto et al., 1998) and suppresses electrically stimulated dopamine release even in the absence of nicotine (Exley et al., 2013; Perez et al., 2013; Koranda et al., 2014). Not only does the endogenous acetylcholine system interact directly with the dopamine system, but both systems play a key role in modulating plasticity of cortical inputs to medium spiny synapses (Shen et al., 2007; Kreitzer and Malenka, 2008; Lerner and Kreitzer, 2011; Tozzi et al., 2011). In addition, several studies suggest that smoking and/or nicotinic therapy may modulate motor symptoms in movement disorders such as PD (Moll, 1926; Fagerström et al., 1994; Kelton et al., 2000; Mitsuoka et al., 2002; Villafane et al., 2007), Tourette's syndrome (Sanberg et al., 1997; Silver et al., 2001; Sanberg et al., 2012), and antipsychotic-induced catalepsy (Menza et al., 1991; Quik et al., 2014). Therefore, we hypothesized that cNIC exposure can mitigate aberrant corticostriatal plasticity and aberrant motor learning. To address this hypothesis, we tested the effects of cNIC exposure on the induction of aberrant motor learning by dopamine receptor blockade in mice.
Materials and Methods
Animals.
For all studies, male and female mice aged 8–14 weeks on a C57BL/J background were used. Mice were housed in standard conditions on a 12 h light/dark cycle in a temperature- and humidity-controlled facility and allowed ad libitum access to standard chow and water. Behavioral testing occurred during the light phase. All procedures were in accordance with guidelines of and approved by the Institutional Animal Care and Use Committee at the University of Chicago.
β2KO mice (Picciotto et al., 1995) were crossed with C57BL/J mice (Jackson Laboratories) to generate β2 heterozygote (HET) breeding pairs. β2KO and WT littermates from HET matings were used in global knockout studies. A similar breeding scheme was used to generate the α4KO mice and WT controls.
For conditional deletion of β2 from dopamine neurons, a floxed β2 line was crossed with a DAT-cre transgenic line (Zhuang et al., 2005). The floxed β2 conditional allele was generated previously (Burbridge et al., 2014). In brief, a 2.2 kb CHRNB2 DNA fragment containing exon 5, the largest coding exon (970bp), was floxed with two loxP sites. Targeting constructs also contained a loxP-flanked neocassette that was subsequently removed using EIIa-cre transgenic mice (Jackson Laboratory).
Drugs.
Nicotine, SCH23390 (Sigma-Aldrich), and eticlopride (Sigma-Aldrich) were used in the rotarod studies. Haloperidol (University of Chicago Hospitals) was used for catalepsy studies. All injections were intraperitoneal at 0.01 ml/g body weight and prepared in 0.9% saline.
cNIC treatment.
Mice were treated with nicotine in their drinking water. Once mice were assigned to a nicotine dose, they remained on that specific dose for the duration of behavioral testing. This route of administration allows for minimally invasive nicotine dosing over prolonged periods of time and for intermittent nicotine exposure with numerous small peaks in plasma nicotine levels throughout the day, similar to human smokers. However, it does not allow for precise control of nicotine dosing. Nicotine was dissolved in tap water and no ramping protocol was used. Nicotine intake was monitored daily and treatment did not alter water intake or body weight during the dose–response study (data not shown, H2O consumption: dose main effect: F(4,133) = 0.9639, p = 0.4299; body weight: dose main effect: F(4,20) = 2.224, p = 0.1029) or any subsequent studies using 100 μg/ml (males: t = 1.267, p = 0.2078; females: t = 0.5723, p = 0.5692). Different groups of mice were used for each experiment and cNIC treatment began at least 2 weeks before any experimentation. Nicotine doses were chosen based on previous research showing that these particular nicotine doses administered via the drinking water correlate with nicotine and cotinine blood levels that induce nicotine tolerance and dependence in mice. (Robinson et al., 1996; Matta et al., 2007). Doses are expressed as the free base concentration of nicotine.
Rotarod studies.
The accelerating rotarod test was used to assess motor performance and learning as described previously (Beeler et al., 2010; Beeler et al., 2012). Briefly, a computer-controlled rotarod apparatus (Rotamex-5; Columbus Instruments) with a rat rod (7 cm diameter) was set to accelerate from 4 to 40 rpm over 300 s and time to fall was recorded. Mice received five consecutive trials per session at one session per day with 30 s intertrial intervals (ITIs).
Haloperidol-induced catalepsy.
Mice were injected with haloperidol (0.5 mg/kg or 0.1 mg/kg) 1 h before testing (Wiecki et al., 2009; Wiecki and Frank, 2010). Haloperidol injections were paired in one of two contexts. For Context A, biohazard hood with a metal table surface was used and both the lights and ventilation system were switched on. For Context B, a plastic table with a texture surface located in a separate room was used. In each context, mice were lifted by the tail and placed so that their forepaws rested on an elevated bar and their hindpaws were flat on the table surface. The bar measured 22 cm in length, 7 mm in diameter, and was elevated 4.5 cm above the table. Latency until the mice made an intentional movement was recorded. Mice received three trials per day over several days. Each trial had a maximum of 180 s with a 30 s ITI.
Fast-scan cyclic voltammetry.
Mice were anesthetized using urethane (2.25 g/kg), placed in a stereotaxic frame, and stimulated dopamine release was measured as described previously (Koranda et al., 2014; Fortin et al., 2015). Briefly, a carbon fiber working electrode was lowered into the dorsolateral striatum (Anterior-Posterior: +1.1 mm; Medial-Lateral: +2.0 mm; Dorsal-Ventral: ∼2.9 mm) while a stimulating electrode was lowered into the midbrain (AP: −3.2 mm; LAT: 0.5–0.8 mm; DV: ∼4.5 mm). The carbon fiber electrode was scanned from −400 to +1300 mV and back relative to an Ag/AgCl reference electrode (400 V/s) in a triangular fashion at a rate of 60 Hz for 30 min, after which the scan rate was set to 10 Hz. A dopamine signal was evoked with 24 pulses (60 Hz; 150 μA) and optimized by adjusting the positions of the stimulating and recording electrodes. After optimization, dopamine was evoked using either a single pulse or 5 pulses of stimulation administered at 5, 10, 20, 40 or 60 Hz. A cyclic voltammogram with an oxidation peak occurring at ∼600 mV and a reduction peak occurring at ∼−200 mV was used to identify dopamine as the electrochemically active species. Each stimulation was separated by 120 s. All data were collected using customized software written in LabView (Tar Heel CV). Electrodes were calibrated at the end of each recording session using a flow injection system and exposing the recording electrode to a known concentration of dopamine (1 μm). The average calibration factor was 44.5 ± 3.8 nm/nA.
Fluorescent immunohistochemistry.
Immunostaining was performed as described previously (Ding et al., 2011). Protein phosphatase inhibitors were not included before or after perfusion with fixative. Briefly, after blocking in 0.05 m Tris-buffered saline containing 5% normal serum and 0.3% Triton X-100 for 1 h at room temperature, 40 μm coronal sections were transferred to primary antibody, rabbit anti-phospho-ERK (1:300; Cell Signaling Technology), containing 0.3% Triton X-100 with 3% BSA and incubated at 4°C for 48 h. The secondary antibody, anti-rabbit Cy3 (1:500; Jackson ImmunoResearch), was diluted in 3% BSA and sections were incubated for 1 h at room temperature. Sections were imaged using fluorescent microscopy and analyzed using ImageJ. Analysis of fluorescently immunostained pERK-labeled neurons was based on images taken with a Nikon Eclipse Ti inverted l microscope at 20× from at least two different sections of the dorsal striatum (between bregma, 0.86–1.18 mm) of each individual mouse.
Statistical analysis.
Data are reported as mean ± SEM. Statistical significance was assessed using a Student's t test, one-way ANOVA, or repeated-measures ANOVA using GraphPad Prism software.
Results
To assess aberrant motor learning, we used a variant of the accelerating rotarod test consisting of two training phases: (1) an acquisition phase in which D1Rs and D2Rs are either concurrently or independently blocked while animals run the rotarod and (2) a recovery phase in which animals are reexposed to the rotarod in the absence of dopamine receptor blockade. The two phases of training are separated by a 3 d break during which mice remain undisturbed in their home cages (Fig. 1A). Aberrant motor learning is inferred from a substantial increase in the time required for mice to reach asymptotic performance during the drug-free recovery phase compared with acquisition by naive mice, as described previously (Beeler et al., 2010, 2012). Using pharmacology to block dopamine signaling reversibly allows the dissociation of acute motor effects of dopamine receptor blockade on performance from dopamine receptor-blockade-induced aberrant learning effects, which is not possible with traditional lesion models.
Figure 1.
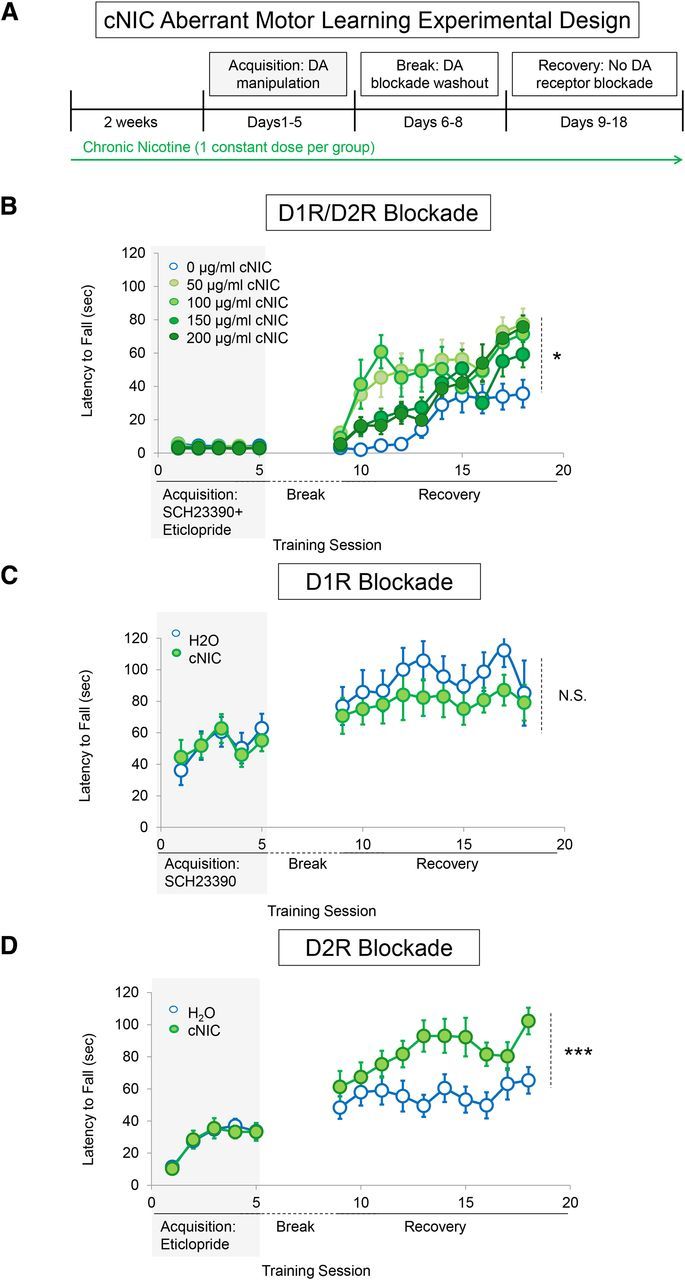
cNIC exposure partially protects against D2R-dependent aberrant motor learning. A, Experimental timeline. B, Latency to fall across subsequent rotarod training sessions with dopamine D1R/D2R antagonist mixture during the acquisition phase and without dopamine blockade during recovery. Each data point represents the average of five trials. n = 5 for each group. C, Average latency to fall across acquisition phase training during D1R antagonism and the recovery phase when dopamine blockade has been relieved. n = 8 for each group. D, Average latency to fall each training day during D2R antagonism during acquisition phase training and recovery when dopamine blockade has been relieved. n = 10 for each group. Shaded rectangles indicate dopamine receptor antagonism. Data are shown as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
cNIC partially protects against aberrant motor learning
To assess possible alterations in aberrant motor learning under cNIC exposure, mice were administered nicotine via the drinking water for 2 weeks before and then throughout behavioral testing (Fig. 1a, arrow represents duration of continuous nicotine treatment at a single dose). This schedule mimics nicotine dosing in human smokers, allowing for repetitive, intermittent exposure (Matta et al., 2007). Mice were assigned to one of five groups, with each group receiving a single concentration of nicotine throughout the experiment [(0, 50, 100, 150, or 200 μg/ml, free base)]. After 2 weeks of nicotine pretreatment, mice were subjected to acquisition phase training on the rotarod under dopamine receptor blockade, which consisted of a mixture injection of D1R (SCH23390, 0.1 mg/kg) and D2R (eticlopride, 0.16 mg/kg) antagonists 30 min before rotarod training. During the 5 d of acquisition phase training, the mixture dramatically impaired rotarod performance with no apparent evidence of learning in either control (water only) or cNIC mice (dose main effect, F(4,80) = 3.02, p = 0.4341; Fig. 1b). After a 72 h break in which the mice remained in their home cage (with cNIC mice still receiving nicotine via the drinking water), the mice were retrained on the rotarod without dopamine receptor blockade (recovery phase). Control mice that did not receive cNIC showed marked slower motor learning during the recovery phase (cf. naive mice; Fig. 2b), indicating that aberrant learning occurred during dopamine blockade (Beeler et al., 2012). Although cNIC mice showed slower motor learning compared with naive mice, cNIC mice performed markedly better than vehicle-treated mice during the same testing period (dose main effect, F(4,180) = 3.337, p = 0.031; Fig. 1b). There was a trend toward a decrease in efficacy of this phenomenon with higher concentrations of nicotine. However, with the highest concentration, 200 μg/ml, mice still performed significantly better than vehicle-treated mice (dose main effect: F(2,72) = 6.847, p = 0.030). Because 100 μg/ml nicotine showed the greatest effect on aberrant learning over the first 5 d of recovery, this concentration was used for the remainder of the studies.
Figure 2.

cNIC, but not aNIC, blocks the initial acquisition of aberrant motor learning. A, Top, Behavioral timeline showing cNIC regimen according to training phase. Bottom, Average latency to fall from rotarod in mice differentially treated with cNIC during dopamine blockade or during recovery after aberrant motor learning has been induced. n = 8 for each group. B, Top, Behavioral timeline showing cNIC regimen according to training phase. Bottom, cNIC effects on average latency to fall from the rotarod in dopamine antagonist naive mice. C, Top, Experimental timeline. Bottom, Effects of aNIC injections either during mixture administration or during recovery when dopamine blockade is no longer present. Arrows represent acute intraperitoneal injections of nicotine (0.5 mg/kg). n = 8 for each group. Shaded rectangles indicate dopamine receptor antagonism. Data are shown as means ± SEM. n = 8. *p < 0.05.
cNIC protects against D2R-mediated, but not D1R-mediated, aberrant motor learning
To test independently whether cNIC alters motor learning in the direct versus indirect pathway, D1R (SCH23390) and D2R (eticlopride) antagonists were administered separately to mice that were treated with cNIC or vehicle. cNIC treatment had no effect on rotarod performance during acquisition phase training when mice were administered either the D1R antagonist (nicotine treatment main effect, F(1,56) = 0.0007, p = 0.9796; Fig. 1c) or D2R antagonist alone (nicotine treatment main effect, F(1,92) = 0.0326, p = 0.8583; Fig. 1d). All groups were equally impaired compared with drug-naive mice (D1R main effect, F(2,80) = 19.27, p < 0.0001; D2R main effect, F(2,116) = 66.95, p < 0.0001, cf. Figs. 1c,d, 2b). During the recovery phase, there was no significant difference between cNIC- and vehicle-treated mice trained previously with a D1R antagonist (nicotine treatment main effect, F(1,126) = 2.319, p = 0.1501; Fig. 1c) and the performance of both groups increased to levels of drug-naive mice (drug main effect, F(2,80) = 0.8998, p = 0.4225). In contrast, cNIC mice treated previously with a D2R antagonist during acquisition phase training demonstrated significantly better rotarod performance during the recovery phase compared with controls (nicotine treatment main effect: F(1,207) = 6.607, p = 0.0171; Fig. 1d). These observations are consistent with our earlier studies showing that D1R antagonism dose-dependently impairs motor performance concomitant with the pharmacodynamics of the drug, but does not affect future drug-free performance, whereas D2R antagonism both impairs rotarod performance acutely and impedes future performance in the absence of antagonism (Beeler et al., 2010, 2012). The observation that cNIC diminishes the persistent performance impairment associated with D2R blockade suggests that cNIC mitigates aberrant learning mediated by the indirect pathway.
cNIC acts during acquisition, but not recovery, to reduce aberrant motor learning
To determine whether cNIC protected against acquisition of aberrant learning under dopamine blockade condition or facilitated recovery after aberrant learning, two groups of mice were treated with cNIC at different points in the experiment (Fig. 2a). cNIC was initiated in Group 1 at 2 weeks before acquisition phase training and mice were maintained on this regimen for the remainder of the experiment (cNIC-acquisition). Group 2 was subjected to acquisition phase rotarod training under dopamine blockade in the absence of cNIC. After the 5 d of acquisition phase training, cNIC treatment was initiated and maintained for the remainder of the experiment (cNIC-recovery). Because our previous research has shown that the acquisition of aberrant learning is experience dependent and does not dissipate with prolonged intervals in the home cage (Beeler et al., 2010, 2012) and because we wanted to expose the second group to nicotine for 2 weeks (i.e., chronic), all mice had a 14 d rather than a 72 h break between the acquisition and recovery phases.
As shown above, cNIC had no effect on rotarod performance during acquisition phase training when mice were trained with the D1R/D2R antagonist mixture. During the recovery phase, performance was significantly better in the cNIC-acquisition mice (treatment main effect, F(1,108) = 7.381, p = 0.0187; Fig. 2a). However, cNIC-recovery mice that began cNIC treatment after acquisition phase training but before the recovery phase showed no performance improvement compared with mixture-treated control mice (treatment main effect, F(1,108) = 0.1127, p = 0.7380; Fig. 2a). These results suggest that cNIC blocks the acquisition of aberrant learning, but cannot rescue performance once aberrant learning has been established.
To control for direct nicotine effects on motor learning and performance independent of protection against dopamine blockade, we used a corresponding nicotine group that began cNIC treatment either before acquisition phase training or during the 14 d break and received saline instead of the D1R/D2R antagonist mixture. cNIC did not alter initial acquisition of the rotarod task (treatment main effect, F(1,48) = 2.151, p = 0.1682; Fig. 2b, cNIC-acquisition vs H2O; treatment main effect, F(1,108) = 0.2187, p = 0.6484; Fig. 2b, cNIC-recovery vs H2O). This suggests that cNIC exposure does not enhance or hamper motor performance when dopamine signaling is intact.
Acute nicotine does not prevent the acquisition of aberrant learning and worsens recovery
We next tested whether acute nicotine (aNIC) could protect against aberrant motor learning. Although the nicotine half-life in mice is ∼6–7 min (Matta et al., 2007), the physiological effects of a single nicotine injection (e.g., increase in dopamine release and altered locomotor activity) can persist for at least 1 h (Picciotto et al., 1998, 2008). To assess the effects of aNIC on aberrant motor learning, WT mice were coadministered nicotine (0.5 mg/kg, i.p.) with the D1R/D2R antagonist mixture during each day of acquisition phase training (aNIC-acquisition). Seventy-two hours later, the mice were retested on the rotarod under drug-free conditions (no nicotine or dopamine blockade) during the recovery phase. Mice coadministered aNIC showed no difference in rotarod performance during mixture treatment (treatment main effect, F(1,32) = 1.377, p = 0.2744; Fig. 2c), nor was there a significant difference in performance during the drug-free recovery phase (treatment main effect, F(1,126) = 0.4300, p = 0.5226).
To assess whether aNIC could facilitate performance during the recovery phase, another group of mice was first trained on the rotarod under dopamine receptor blockade but with no nicotine. During recovery, this group was injected with 0.5 mg/kg nicotine 30 min before each session (aNIC-recovery). aNIC administered during the recovery phase worsened performance compared with control mice (treatment main effect, F(1,126) = 3.620, p = 0.0779; Fig. 2c). Together, these results suggest that aNIC cannot mitigate aberrant learning, but can impair recovery. These results suggest that neuroadaptations that accompany long-term nicotine exposure rather than the direct actions of nicotine at nAChRs may underlie nicotine's protective effect against aberrant motor learning.
Deletion of the β2 nAChR subunit protects against D2R -mediated aberrant motor learning
cNIC exposure is well known to upregulate high-affinity β2 nicotinic-binding sites. To determine whether β2-containing nAChRs mediate cNIC's ability to diminish aberrant motor learning, we trained mice lacking the β2 subunit (β2KO) on the rotarod as described above. Deletion of β2 subunits did not affect rotarod performance in saline-injected mice (genotype main effect, F(1,52) = 1.909, p = 0.1904; Fig. 3a). As seen with cNIC, β2 deletion had no effect on rotarod performance during acquisition phase training when mice were trained with the D1R/D2R antagonist mixture (genotype main effect, F(1,56) = 1.241, p = 0.2841; Fig. 3b). However, deletion of β2 increased performance during the recovery phase (genotype main effect, F(1,126) = 11.83, p = 0.0040; Fig. 3c).
Figure 3.

Conventional deletion of β2 nAChR subunits protects against D2R-mediated aberrant motor learning. A, Average latency to fall in β2KO mice treated with saline. n = 8. B, Average latency to fall with D1R/D2R mixture on board during acquisition and during drug-free recovery. n = 8 for each group. C, Average latency to fall in the presence of a D1R antagonist and during drug-free recovery stage. D, Average latency to fall in the presence of a D2R antagonist and during the drug-free recovery phase. n = 8 for each group. E, Effects of α4 nAChR subunit deletion on aberrant motor learning. n = 8 for each group. Shaded rectangles indicate dopamine receptor antagonism. Data are shown as means ± SEM. n = 8. **p < 0.01.
β2 deletion did not rescue rotarod performance during acquisition phase training when mice were independently trained with either a D1Rantagonist (genotype main effect, F(1,56) = 0.0014, p = 0.9702; Fig. 3c) or D2R antagonist (genotype main effect, F(1,56) = 4.163, p > 0.05; Fig. 3d). Drug-free recovery was similar in β2KO and WT mice previously administered a D1R antagonist during acquisition phase training (genotype main effect, F(1,126) = 0.1609, p = 0.6944; Fig. 3c). In contrast, β2 deletion improved performance during the recovery phase in mice previously administered a D2R antagonist during acquisition phase training (genotype main effect, F(1,126) = 22.02, p = 0.003; Fig. 3d).
We also tested whether deletion of α4 nicotinic subunits (α4KO) could mitigate aberrant motor learning. There was no effect of α4 deletion during either acquisition phase training (genotype main effect, F(1,56) = 3.207, p = 0.0950; Fig. 3e) or the recovery phase (genotype main effect, F(1,126) = 2.2253, p = 0.1556; Fig. 3e). This result suggests that α6(non-α4)β2* nAChRs mediate protection against aberrant motor learning. However, several studies have suggested that α6 nicotinic subunits may be incorporated into functional β2* nAChRs in place of α4 subunits and vice versa (Champtiaux et al., 2003; Marubio et al., 2003). Therefore, the lack of effect on aberrant motor learning in α4KO mice may be indicative of α6 subunit substitution. More studies are warranted to determine the specific subpopulation of β2-containing nAChRs that mediate protection against aberrant motor learning.
In short, β2 deletion mitigates D2R blockade-induced aberrant motor learning, recapitulating the pattern observed with cNIC. Because β2 deletion can protect against aberrant motor learning, these results suggest that a functional downregulation of β2*nAChR expression and/or activity may protect against the acquisition of aberrant motor learning.
cNIC and β2 deletion both reduce the sensitization of haloperidol-induced catalepsy
In humans, chronic D2R antagonist treatment (e.g., haloperidol treatment in schizophrenia) can lead to parkinsonism, including rigidity of movement and increasingly severe cataleptic events (Arnt, 2008; Gao et al., 2008). Previous research has suggested that inappropriate learning mechanisms (i.e., LTP) in the D2R indirect pathway underlie the sensitization of haloperidol-induced catalepsy (Centonze et al., 2004; Wiecki et al., 2009; Wiecki and Frank, 2010). Therefore, to determine whether cNIC's protective effect against aberrant motor learning was specific to the rotarod task or if it generalized to other D2R-mediated motor learning tasks, we investigated whether cNIC exposure could mitigate the sensitization of haloperidol-induced catalepsy. Mice treated with cNIC (100 μg/ml) or vehicle for 2 weeks before and throughout were injected with 0.5 mg/kg haloperidol 1 h before catalepsy testing. The forepaws of the mice were placed on an elevated bar and the latency to first overt movement was recorded. Both control and cNIC mice showed sensitization of haloperidol-induced catalepsy that was context dependent—that is mice showed increasing freezing time when injected with haloperidol in Context A (see Materials and Methods) over several days. However, when animals were given the same haloperidol dose in a different context (Context B, see Materials and Methods), there was no evidence of catalepsy consistent with prior studies showing that contextual stimuli become associated with learned catalepsy (Centonze et al., 2004; Wiecki et al., 2009; Wiecki and Frank, 2010). Although both control and cNIC mice showed sensitization of haloperidol-induced catalepsy, there was less sensitization in cNIC mice compared with control mice (treatment main effect, F(1,84) = 12.59, p < 0.0001; Fig. 4a). We also tested the effect of aNIC on the sensitization of haloperidol-induced catalepsy. In this cohort of mice, haloperidol was injected 1 h before testing and a second injection containing nicotine (0.5 mg/kg, i.p.) was administered 5 min before testing. Although cNIC reduced the expression of haloperidol-induced catalepsy, aNIC enhanced this behavior compared with control mice (treatment main effect, F(1,91) = 29.34, p = 0.0001; Fig. 4b). These results mirror those from the rotarod studies in which cNIC mitigated D2R-mediated aberrant motor learning, whereas aNIC had no effect or worsened performance.
Figure 4.

cNIC and β2 subunit deletion reduces the sensitization of haloperidol-induced catalepsy. Mice were tested for sensitization of catalepsy in response to repeated injections of haloperidol (0.5 mg/kg) in either Context A or Context B. A, Experimental design. B, Haloperidol-induced cataleptic response in control mice (CON), cNIC- treated mice (cNIC), and mice receiving aNIC injections just before behavioral testing (aNIC). C, Haloperidol-induced response in mice lacking the β2 nAChR (β2KO) subunit and WT littermates. Control, n = 8; cNIC, n = 8, aNIC, n = 5; WT, n = 8; β2KO, n = 8. Data are shown as means ± SEM. ***p < 0.01, ***p < 0.001.
We also tested the effect of β2 deletion on the sensitization of haloperidol-induced catalepsy. Similar to cNIC, deletion of the β2 nAChR subunit reduced the expression of catalepsy (genotype main effect, F(1,98) = 12.07, p = 0.0037; Fig. 4c). Together, results from the rotarod and catalepsy studies suggest that cNIC, likely through downregulation of β2-containing nAChR function or expression, provides protection against maladaptive D2R antagonist-induced impairments in motor learning
Selective β2 nAChR subunit deletion in dopamine cells is sufficient to prevent aberrant learning
The principal role of striatal β2*nAChRs is the regulation of dopamine release (Zhou et al., 2001; Rice and Cragg, 2004; Salminen et al., 2004; Zhang and Sulzer, 2004; Cachope et al., 2012; Threlfell et al., 2012; Koranda et al., 2014) However, β2*nAChRs are also expressed in GABAergic interneurons and may be expressed on other cell types such as cholinergic interneurons. To rule out a possible contribution to aberrant learning of β2 deletion in other cells, we specifically deleted β2 nAChR subunits in dopamine neurons by crossing a floxed β2 line (fβ2; Burbridge et al., 2014) with transgenic mice expressing Cre recombinase under control of the dopamine transporter (DAT) promoter (Zhuang et al., 2005). Mice lacking the β2 subunit in dopamine neurons were trained on the rotarod as described above. During acquisition phase training under D1R/D2R antagonist mixture, there was no difference in performance of mice lacking the β2 subunit in dopamine neurons (f/f; DAT-cre) compared with controls (WT mice, f/f mice, and DAT-cre mice) (genotype main effect, F(3,196) = 1.715, p = 0.1761; Fig. 5a). During the recovery phase, deletion of dopamine neuron-specific β2 subunits improved motor performance compared with control groups (genotype main effect, F(3,441) = 4.413, p = 0.0080; Fig. 5a). These results suggest that deletion of β2 subunits in dopamine neurons is sufficient to mitigate aberrant motor learning.
Figure 5.
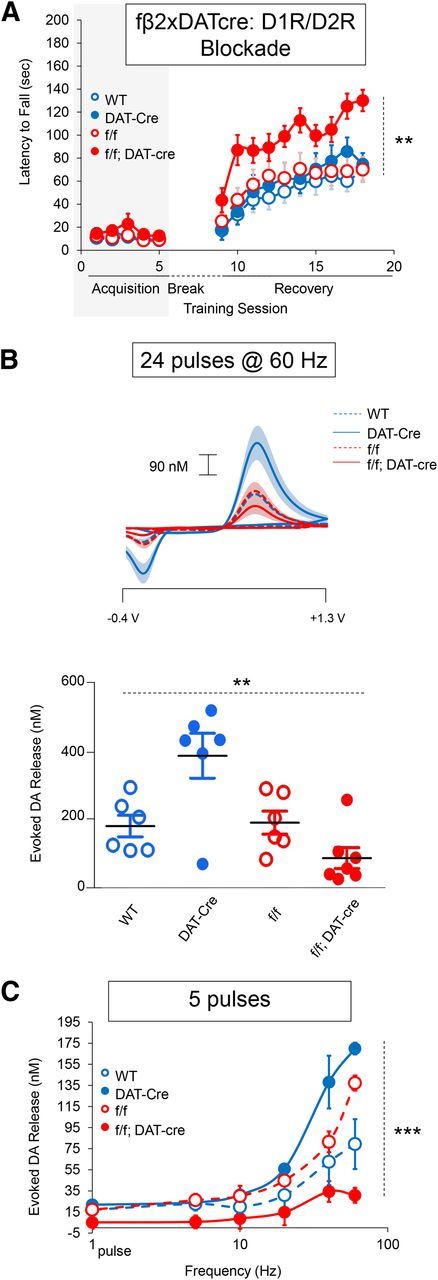
Specific deletion of β2 nAChR subunits in dopamine neurons is sufficient to mitigate aberrant motor learning and diminish stimulated dopamine release. A, Average latency to fall from rotarod. Shaded rectangle indicates dopamine receptor antagonism. n = 10–12. B, Stimulated dopamine release after supraphysiological midbrain stimulation. Top, Average current–voltage plots. Bottom, Peak dopamine release. Each point represents an individual mouse. Horizontal black bar represents group average. n = 6–8. C, Frequency dependence of stimulated dopamine release after either a single pulse of stimulation or five pulses of stimulation administered at various frequencies. n = 3. Data are shown as means ± SEM. **p < 0.01, ***p < 0.001.
Dopamine neuron-specific β2 nAChR deletion diminishes stimulated dopamine release
To confirm β2 deletion in dopamine neurons, we measured evoked dopamine release in vivo using fast-scan cyclic voltammetry in anesthetized mice lacking the β2 subunit specifically in dopamine neurons. Our previous study found that constitutive β2KO mice showed diminished stimulated dopamine release in the striatum at nearly every stimulation intensity tested, with the greatest differences at high frequency and pulse number (Koranda et al., 2014). Based on this, we used strong stimulation parameters to test the impact of β2 deletion in dopamine neurons on evoked striatal dopamine release (24 pulses at 60 Hz). As observed in the β2KO animals, evoked dopamine release was significantly decreased in dorsal striatum when β2 was selectively eliminated from dopamine neurons compared with control groups (genotype main effect, F(3,24) = 9.314, p = 0.0004; Fig. 5b). We also noted that significantly more dopamine was released by DAT-Cre mice. This is not surprising because the DAT-Cre line loses one copy of DAT (Zhuang et al., 2005), which will reduce dopamine reuptake rate. Nevertheless, the combination of DAT-Cre and dopamine neuron deletion of β2 still reduced dopamine release and, behaviorally, this is the only group protected from aberrant motor learning (Fig. 5a). In a subset of mice, we stimulated dopamine release by administering 5 stimulation pulses at frequencies ranging from 5 to 60 Hz. Specific deletion of β2 subunits in dopamine neurons decreased dopamine release at all measured frequencies (genotype main effect, F(3,50) = 16.38, p = 0.0009; Fig. 5c), which is consistent with previous results using β2KO mice and cNIC treatment (Zhang et al., 2009; Exley et al., 2013; Perez et al., 2013; Koranda et al., 2014; Perez et al., 2015) and confirms the significant contribution of β2*nAChRs in dopamine neurons. Given the key role of dopamine in corticostriatal plasticity, these results suggest that inhibiting β2 nAChR function in dopamine neurons can modify a key aspect of motor learning.
cNIC blunts D2R antagonist-induced phosphorylation of ERK
The above voltammetry results, combined with previous studies, suggests that cNIC and β2 nAChR deletion reduce dopamine release (Exley et al., 2013; Perez et al., 2013; Koranda et al., 2014; Perez et al., 2015). Therefore, we next investigated whether these mice develop changes in postsynaptic signaling in the striatopallidal pathway. Although several studies have implicated the importance of AC5 and cAMP signaling in corticostriatal plasticity and striatal learning (Kheirbek et al., 2009; Augustin et al., 2014), a previous study indicated that β2 nAChR subunit deletion did not alter either AC5 or cAMP activity (Picciotto et al., 1998). Extracellular signal-regulated kinase (ERK) regulates the intrinsic excitability of medium spiny neurons (Adams et al., 2000; Schrader et al., 2006; Johnston and Narayanan, 2008), has been implicated in sensorimotor learning (Bureau et al., 2010; Shiflett et al., 2010; Shiflett and Balleine, 2011), and contributes to corticostriatal LTP (Calabresi et al., 2000; Mazzucchelli et al., 2002; Flajolet et al., 2008; Shiflett and Balleine, 2011). Although aNIC reportedly induces phosphorylation of ERK (pERK) through activation of α7 (Dajas-Bailador et al., 2004; Dickinson et al., 2008) and α4β2* nAChRs (Nakayama et al., 2001) and cNIC can either increase or decrease pERK dependent upon the brain region examined (Brunzell et al., 2003), there are no reports examining the effects of D2R antagonist-induced pERK expression (Welsh et al., 1998; Wang et al., 2005; Bertran-Gonzalez et al., 2008) after cNIC treatment or β2 deletion. Therefore, we investigated whether cNIC treatment alters pERK expression in response to repeated D2R antagonism paired with rotarod training. Mice were injected with 0.16 mg/kg eticlopride 20 min before rotarod training for 5 consecutive days. Immediately after rotarod training on day 5, mice were perfused and brains removed and prepared for fluorescent immunohistochemistry (Fig. 6a). cNIC and β2 subunit deletion significantly decreased pERK expression to 44.3 ± 6.3% and 26.3 ± 4.8%, respectively, compared with WT and nicotine-naive counterparts (treatment main effect, F(2.25) = 9.246, p = 0.0010; Fig. 6c). Decreased D2R antagonist-induced pERK in cNIC and β2KO mice suggests that corticostriatal synapses in the indirect pathway in these animals will show less plasticity under conditions of suppressed dopamine signaling after either dopamine antagonist treatment or dopamine neuron loss. Furthermore, these results suggest that cNIC, acting via β2-containing nAChRs, chronically decreases striatal dopamine release, leading to alterations in postsynaptic signaling in the indirect pathway. Such changes may protect the animal from aberrant motor learning.
Figure 6.

cNIC and β2 nAChR subunit deletion reduce D2R antagonist-induced phosphorylation of ERK. A, Behavioral timeline. B, Representative images showing pERK fluorescence using 20× magnification. C, Left, Quantification of fluorescent cell count. Individual points represent cell count from an individual mouse. Black horizontal bar represents mean cell count. Right, Percentage change in pERK cell count relative to WT average. Data are shown as means ± SEM. n = 9–10. ***p < 0.001.
Discussion
Dopamine has been implicated in the acquisition of new motor skills and maintenance of established motor skills (Yin et al., 2009; Jin and Costa, 2010; Beeler et al., 2012). Although low dopamine conditions lead to acute motor impairments, we demonstrated previously that dopamine blockade paired with a motor task induces a D2R- and experience-dependent learned motor impairment in mice that likely arises from inappropriate LTP in the indirect pathway (Beeler et al., 2010, 2012). We suggested this aberrant motor learning (i.e., plasticity) contributes to degradation of motor function in PD. In support of this aberrant motor hypothesis, motor skills in PD patients worsen progressively if practiced in the absence of dopamine replacement therapy (Kang et al., 2012; Anderson et al., 2014). Here, we expand upon our earlier work and show that nicotine administered chronically, but not acutely, diminishes D2R-dependent aberrant motor learning in mice. Importantly, when aberrant motor learning was established before nicotine treatment, cNIC could not rescue motor performance.
Mechanistically, our data suggest that cNIC's protection against aberrant motor learning is mediated by changes in presynaptic dopamine release. Specific deletion of β2 nAChR subunits in dopamine neurons both protected against aberrant motor learning and reduced stimulated striatal dopamine release in vivo. These results suggest that chronic decreases in function or downregulation of a subpopulation of β2-containing nAChRs in dopamine neurons mediate protection against aberrant motor learning. Although several studies suggest that cNIC upregulates α4β2 nAChR-binding sites (Marks et al., 1985; McCallum et al., 2006a; Marks et al., 2011), others suggest that cNIC downregulates α6(non-α4)β2*nAChRs (McCallum et al., 2006a; Walsh et al., 2008; Exley et al., 2013; Perez et al., 2013) and α6(non-α4)β2*nAChRs are more sensitive than α4β2*nAChRs to regulation by cNIC (Marks et al., 2014). Here, we found that α4 deletion could not mitigate aberrant motor learning, suggesting that α6(non-α4)β2*nAChRs play a key role in protection against aberrant motor learning. Regardless of the specific subpopulation of β2-containing nAChRS involved, the current results strongly suggest that cNIC protects against aberrant motor learning by chronically reducing striatal dopamine release.
Postsynaptically, a cNIC-induced reduction of striatal dopamine release may lead to signaling alterations that ultimately protect the striatal network from aberrant plasticity when dopamine levels are lowered. We found that cNIC treatment or β2 nAChR subunit deletion reduced striatal levels of ERK phosphorylation induced by the combination of D2R antagonism and rotarod training. Although we did not examine differential D2R-induced pERK expression in striatopallidal neurons versus acetylcholine interneurons, these results suggest that cNIC exposure alters corticostriatal plasticity. Because increased pERK induces corticostriatal LTP (Mazzucchelli et al., 2002; Flajolet et al., 2008) and we have hypothesized that LTP at striatopallidal synapses induced under dopamine loss or blockade mediates aberrant learning (Beeler et al., 2010; Beeler, 2011; Beeler et al., 2012, 2013; Zhuang et al., 2013), it is possible that cNIC mitigates aberrant motor learning by reducing the propensity for induction of corticostriatal LTP at striatopallidal synapses.
Increasingly aberrant plasticity is recognized as contributing to motor symptoms in PD patients (Kang et al., 2012; Anderson et al., 2014). Moreover, several studies have reported that manipulations that block or diminish abnormal LTP or those that restore normal corticostriatal plasticity are also associated with reduced parkinsonian-like symptoms in rodents (Kreitzer and Malenka, 2007; Shen et al., 2008; Peterson et al., 2012; Thiele et al., 2014). Interestingly, Quik et al. (2006) showed that cNIC protected corticostriatal LTD induction in MPTP-treated nonhuman primates. This latter finding is intriguing given the well documented protective effect of smoking against PD risk (Chen et al., 2010; van der Mark et al., 2014). Together, these data suggest a link between PD protection and stronger corticostriatal LTD or a shift in plasticity away from LTP. Future investigations into plasticity mechanisms underlying aberrant motor learning may provide novel PD treatments independent of dopamine replacement.
Along with its protective effect against PD risk, several clinical studies have investigated the efficacy of nicotine and nicotinic agents to ameliorate PD symptoms. Although some studies report modest improvements in motor symptoms (Moll, 1926; Ishikawa and Miyatake, 1993; Fagerström et al., 1994; Kelton et al., 2000; Mitsuoka et al., 2002), others report no change (Clemens et al., 1995; Vieregge et al., 2001; Lemay et al., 2004; Parkinson's Study Group, 2006) or a worsening of motor symptoms (Ebersbach et al., 1999). Discrepancies in study design likely contribute to the conflicting results, including route of drug administration, drug dose, length of administration, smoking history, whether nicotinic therapy was coadministered with dopamine replacement therapy, and whether testing occurred on or off of dopamine replacement therapy.
Our findings suggest an alternative strategy for the clinical use of nicotinic agents in PD. Our data show that cNIC did not alter rotarod performance with intact dopamine signaling, suggesting that nicotinic therapy may not modify motor responses during peaks of dopamine replacement therapy. During medication troughs, when motor performance deteriorates, however, nicotine may lessen these symptoms by protecting against aberrant motor learning. We suggested previously that reduction of aberrant motor learning may underlie the clinically important LDR to l-DOPA (Beeler et al., 2010; Beeler, 2011; Beeler et al., 2012; Zhuang et al., 2013). Therefore, cNIC-induced reduction of aberrant motor learning may act synergistically with l-DOPA and augment the LDR, potentially allowing lower doses of l-DOPA. Because nicotinic receptors mediating reduced aberrant learning are located on dopamine neuron terminals that are largely lost in advanced PD, the effective therapeutic window would likely be during earlier stages of the disease to slow functional deterioration. Future studies are warranted to determine whether cNIC can augment the LDR and preserve motor skills in PD patients.
In addition to providing a therapeutic effect in PD patients, our results suggest that cNIC treatment may also have beneficial effects in patients prescribed D2R antagonists. Most commonly prescribed to treat psychosis, chronic D2R antagonism can lead to parkinsonism, including rigidity of movement and increasingly severe cataleptic events (Arnt, 2008; Gao et al., 2008). This dysfunction likely arises from inappropriate corticostriatal LTP in the indirect pathway (Centonze et al., 2004; Wiecki et al., 2009; Wiecki and Frank, 2010). In the current study, we found that cNIC and β2 deletion, but not aNIC, reduced the sensitization of haloperidol-induced catalepsy, similar to the effect of cNIC on aberrant motor learning in the rotarod task. Given that haloperidol treatment reportedly increases pERK expression in striatopallidal neurons (Bertran-Gonzalez et al., 2008) and because we found that cNIC treatment and β2 deletion attenuated D2R antagonist-induced increases in pERK, cNIC-induced reduction in the sensitization of haloperidol-induced catalepsy likely reflects decreased corticostriatal LTP at striatopallidal synapses. The results of the present study suggest a novel therapeutic strategy that consists of maintenance of normal synaptic plasticity and prevention or reversal of aberrant plasticity for a variety of neurological disorders.
It is important to point out that the present studies do not address mechanisms of dopamine neuron degeneration or its protection. Although we suggest that protection against aberrant motor learning may contribute to the inverse correlation between PD incidence and smoking, our data do not preclude possible protective effects of nicotine against dopamine cell death. For example, nicotine can diminish oxidative stress and neuroinflammation (Gahring et al., 2003; Parain et al., 2003; Fujii et al., 2007; Park et al., 2007; Hosur and Loring, 2011), inhibit endoplasmic reticulum stress responses (Kuryatov et al., 2005; Sallette et al., 2005; Srinivasan et al., 2012, 2014), and stimulate prosurvival signaling factors (Kawamata and Shimohama, 2011). However, the extent to which these observations extend to whole animals and PD patients has not been demonstrated.
Alternatively, several studies suggest that nicotine-induced alterations in synaptic transmission might protect against dopamine cell death. After dopamine denervation, surviving dopamine terminals release more dopamine than sham controls (Bergstrom and Garris, 2003; McCallum et al., 2006b; Perez et al., 2008). Dopamine itself can be toxic through the production of free radicals (Chen et al., 2008; Segura-Aguilar et al., 2014). Therefore, a compensatory upregulation of dopamine release during dopamine neuron degeneration could contribute to cell death. By desensitizing presynaptic nAChRs on surviving dopamine terminals, cNIC treatment may protect against cell death by reducing stimulated dopamine release (Exley et al., 2013; Perez et al., 2013; Koranda et al., 2014). Furthermore, a study by Montine et al. (2010) found that lifetime cigarette smoking correlates with reduced Lewy-related pathology (LRP). This suggests that smoking may mitigate LRP-associated disruptions in neuronal signaling, which often precede frank dopamine neuron death. Therefore, because multiple studies show that cNIC exposure can both protect against dopamine cell death and mitigate disruptions in dopamine signaling, we propose that the reduced PD risk associated with smoking may reflect a convergence of neuroprotection against dopamine cell loss and protection against aberrant plasticity that arises from distorted dopamine signaling.
Finally, although the present study focuses on the role of cNIC in protection against aberrant motor learning and plasticity, it is important to note that an earlier study showed that the xanthine theophylline, a nonselective adenosine receptor antagonist similar to caffeine, reduces D2R antagonist-induced corticostriatal LTP at striatopallidal synapses and mitigates aberrant motor learning in mice (Beeler et al., 2012), similar to our observations here with cNIC. Heavy caffeine consumption, like smoking, reduces PD incidence (Qi and Li, 2014; van der Mark et al., 2014). Therefore, it is an interesting observation that two lifestyle factors, caffeine consumption and smoking, that are associated with decreased PD risk also protect against the acquisition of aberrant motor learning and plasticity. It is intriguing to speculate that the prevention of aberrant plasticity is an important deterrent to the progression of motor symptoms in movement disorders such as PD.
Footnotes
This work was supported by the National Institutes of Health (Grant DA25875 to J.A.B., National Institute of Neurological Disorders and Stroke Grant R21NS083383 to X.Z., and Grant 5R01MH099114 to A.C.). We thank Mitch Roitman and Jackson Cone for providing the voltammetry equipment and Susie Turkson for providing excellent technical assistance.
The authors declare no competing financial interests.
References
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- Anderson ED, Horak FB, Lasarev MR, Nutt JG. Performance of a motor task learned on levodopa deteriorates when subsequently practiced off. Mov Disord. 2014;29:54–60. doi: 10.1002/mds.25702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnt J. Pharmacological differentiation of classical and novel antipsychotics. Int Clin Psychopharmacol. 2008;13:S7–S14. doi: 10.1097/00004850-199803003-00002. [DOI] [PubMed] [Google Scholar]
- Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34:6692–6699. doi: 10.1523/JNEUROSCI.3906-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA. Preservation of function in Parkinson's disease: what's learning got to do with it? Brain Res. 2011;1423:96–113. doi: 10.1016/j.brainres.2011.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Ding Y, Koranda J, Murakami M, Kang UJ, Zhuang X. Dopamine-dependent motor learning: insight into levodopa's long-duration response. Ann Neurol. 2010;67:639–647. doi: 10.1002/ana.21947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Frank MJ, McDaid J, Alexander E, Turkson S, Bernardez Sarria MS, Bernandez MS, McGehee DS, Zhuang X. A role for dopamine-mediated learning in the pathophysiology and treatment of Parkinson's disease. Cell Rep. 2012;2:1747–1761. doi: 10.1016/j.celrep.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Petzinger G, Jakowec MW. The enemy within: propagation of aberrant corticostriatal learning to cortical function in Parkinson's disease. Front Neurol. 2013;4:134. doi: 10.3389/fneur.2013.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergstrom BP, Garris PA. ‘Passive stabilization’ of striatal extracellular dopamine across the lesion spectrum encompassing the presymptomatic phase of Parkinson's disease: a voltammetric study in the 6-OHDA-lesioned rat. J Neurochem. 2003;87:1224–1236. doi: 10.1046/j.1471-4159.2003.02104.x. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunzell DH, Russell DS, Picciotto MR. In vivo nicotine treatment regulates mesocorticolimbic CREB and ERK signaling in C57BL/6J mice. J Neurochem. 2003;84:1431–1441. doi: 10.1046/j.1471-4159.2003.01640.x. [DOI] [PubMed] [Google Scholar]
- Burbridge TJ, Xu HP, Ackman JB, Ge X, Zhang Y, Ye MJ, Zhou ZJ, Xu J, Contractor A, Crair MC. Visual circuit development requires patterned activity mediated by retinal acetylcholine receptors. Neuron. 2014;84:1049–1064. doi: 10.1016/j.neuron.2014.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau G, Carrier M, Lebel M, Cyr M. Intrastriatal inhibition of extracellular signal-regulated kinases impaired the consolidation phase of motor skill learning. Neurobiol Learn Mem. 2010;94:107–115. doi: 10.1016/j.nlm.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, Lovinger DM, Cheer JF. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41. doi: 10.1016/j.celrep.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Picconi B, Bernardi G, Chergui K, Svenningsson P, Fienberg AA, Greengard P. Dopamine and cAMP-regulated phosprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. J Neurosci. 2000;20:8443–8451. doi: 10.1523/JNEUROSCI.20-22-08443.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Borrelli E, Calabresi P. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–8222. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Léna C, Clementi F, Moretti M, Rossi FM, Le Novère N, McIntosh JM, Gardier AM, Changeux JP. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–7829. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Huang X, Guo X, Mailman RB, Park Y, Kamel F, Umbach DM, Xu Q, Hollenbeck A, Schatzkin A, Blair A. Smoking duration, intensity, and risk of Parkinson disease. Neurology. 2010;74:878–884. doi: 10.1212/WNL.0b013e3181d55f38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W, Hastings TG, Kang UJ, Zhuang X. Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci. 2008;28:425–433. doi: 10.1523/JNEUROSCI.3602-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens P, Baron JA, Coffey D, Reeves A. The short-term effect of nicotine chewing gum in patients with Parkinson's disease. Psychopharmacology (Berl) 1995;117:253–256. doi: 10.1007/BF02245195. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador FA, Soliakov L, Wonnacott S. Nicotine activates the extracellular signal-regulated kinase 1/2 via the alpha7 nicotinic acetylcholine receptor and protein kinase A, in SH-SY5Y cells and hippocampal neurones. J Neurochem. 2002;80:520–530. doi: 10.1046/j.0022-3042.2001.00725.x. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic α2- and β2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Ding Y, Won L, Britt JP, Lim SA, McGehee DS, Kang UJ. Enhanced striatal cholinergic neuronal activity mediates L-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci U S A. 2011;108:840–845. doi: 10.1073/pnas.1006511108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersbach G, Stöck M, Müller J, Wenning G, Wissel J, Poewe W. Worsening of motor performance in patients with Parkinson's disease following transdermal nicotine administration. Mov Disord. 1999;14:1011–1013. doi: 10.1002/1531-8257(199911)14:6%3C1011::AID-MDS1016%3E3.0.CO%3B2-F. [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Franklin M, Bermudez I, Cragg SJ. Striatal dopamine transmission is reduced after chronic nicotine with a decrease in α6-nicotinic receptor control in nucleus accumbens. Eur J Neurosci. 2013;38:3036–3043. doi: 10.1111/ejn.12298. [DOI] [PubMed] [Google Scholar]
- Fagerström KO, Pomerleau O, Giordani B, Stelson F. Nicotine may relieve symptoms of Parkinson's disease. Psychopharmacology (Berl) 1994;116:117–119. doi: 10.1007/BF02244882. [DOI] [PubMed] [Google Scholar]
- Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, Wallach I, Nairn AC, Surmeier DJ, Greengard P. FGF acts as a co-transmitter through adenosine A(2A) receptor to regulate synaptic plasticity. Nat Neurosci. 2008;11:1402–1409. doi: 10.1038/nn.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin SM, Cone JJ, Ng-Evans S, McCutcheon JE, Roitman MF. Sampling phasic dopamine signaling with fast-scan cyclic voltammetry in awake, behaving rats. Curr Protoc Neurosci. 2015;70:7.25.1–7.25.20. doi: 10.1002/0471142301.ns0725s70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii YX, Fujigaya H, Moriwaki Y, Misawa H, Kasahara T, Grando SA, Kawashima K. Enhanced serum antigen-specific IgG1 and proinflammatory cytokine production in nicotinic acetylcholine receptor alpha7 subunit gene knockout mice. J Neuroimmunol. 2007;189:69–74. doi: 10.1016/j.jneuroim.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Gahring LC, Meyer EL, Rogers SW. Nicotine-induced neuroprotection against N-methyl-D-aspartic acid or beta-amyloid peptide occur through independent mechanisms distinguished by pro-inflammatory cytokines. J Neurochem. 2003;87:1125–1136. doi: 10.1046/j.1471-4159.2003.02074.x. [DOI] [PubMed] [Google Scholar]
- Gao KM, Kemp DE, Ganocy SJ, Gajwani P, Xia G, Calabrese JR. Antipsychotic-induced extrapyramidal side effects in bipolar disorder and schizophrenia–a systematic review. J Clin Psychopharmacol. 2008;28:203–209. doi: 10.1097/JCP.0b013e318166c4d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanagasi HA, Lees A, Johnson JO, Singleton A, Emre M. Smoking-responsive juvenile-onset Parkinsonism. Mov Disord. 2007;22:115–119. doi: 10.1002/mds.21177. [DOI] [PubMed] [Google Scholar]
- Hosur V, Loring RH. Alpha4beta2 nicotinic receptors partially mediate anti-inflammatory effects through Janus inase 20signal transducer and activator of transcription 3 but not calcium or cAMP signaling. Mol Pharmacol. 2011;79:167–174. doi: 10.1124/mol.110.066381. [DOI] [PubMed] [Google Scholar]
- Huang LZ, Parameswaran N, Bordia T, Michael McIntosh J, Quick M. Nicotine is neuroprotective when administered before but not after nigrostriatal damage in rats and monkeys. J Neurochem. 2009;109:826–837. doi: 10.1111/j.1471-4159.2009.06011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Miyatake T. Effects of smoking in patients with early-onset Parkinson's disease. J Neurol Sci. 1993;117:28–32. doi: 10.1016/0022-510X(93)90150-W. [DOI] [PubMed] [Google Scholar]
- Jin X, Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature. 2010;466:457–462. doi: 10.1038/nature09263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Narayanan R. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 2008;31:309–316. doi: 10.1016/j.tins.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Kang UJ, Auinger P, Parkinson Study Group ELLDOPA Investigators Activity enhances dopaminergic long-duration response in Parkinson disease. Neurology. 2012;78:1146–1149. doi: 10.1212/WNL.0b013e31824f8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata J, Shimohama S. Stimulating nicotinic receptors trigger multiple pathways attenuating cytotoxicity in models of Alzheimer's and Parkinson's diseases. J Alzheimers Dis. 2011;24:95–109. doi: 10.3233/JAD-2011-110173. [DOI] [PubMed] [Google Scholar]
- Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. The effects of nicotine on Parkinson's disease. Brain Cogn. 2000;43:274–282. [PubMed] [Google Scholar]
- Kheirbek MA, Britt JP, Beeler JA, Ishikawa Y, McGehee DS, Zhuang X. Adenylyl cyclase type 5 contributes to corticostriatal plasticity and striatum-dependent learning. J Neurosci. 2009;29:12115–12124. doi: 10.1523/JNEUROSCI.3343-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koranda JL, Cone JJ, McGehee DS, Roitman MF, Beeler JA, Zhuang X. Nicotinic receptors regulate the dynamic range of dopamine release in vivo. J Neurophysiol. 2014;111:103–111. doi: 10.1152/jn.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Luo J, Cooper J, Lindstrom J. Nicotine acts as a pharmacological chaperone to up-regulate human alpha4beta2 acetylcholine receptors. Mol Pharmacol. 2005;68:1839–1851. doi: 10.1124/mol.105.012419. [DOI] [PubMed] [Google Scholar]
- Lemay S, Chouinard S, Blanchet P, Masson H, Soland V, Beuter A, Bédard MA. Lack of efficacy of anicotine transdermal treatment on motor and cognitive deficits in Parkinson's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:31–39. doi: 10.1016/S0278-5846(03)00172-6. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. Neuromodulatory control of striatal plasticity and behavior. Curr Opin Neurobiol. 2011;21:322–327. doi: 10.1016/j.conb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 1985;235:619–628. [PubMed] [Google Scholar]
- Marks MJ, Grady SR, Collins AC. Downregulation of nicotinic receptor function after chronic nicotine infusion. J Pharmacol Exp Ther. 1993;266:1268–1276. [PubMed] [Google Scholar]
- Marks MJ, McClure-Begley TD, Whiteaker P, Salminen O, Brown RW, Cooper J, Collins AC, Lindstrom JM. Increased nicotinic acetylcholine receptor protein underlies chronic nicotine-induced upregulation of nicotinic agonist binding sites in mouse brain. J Pharmacol Exp Ther. 2011;337:187–200. doi: 10.1124/jpet.110.178236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Grady SR, Salminen O, Paley MA, Wageman CR, McIntosh JM, Whiteaker P. α6β2*-subtype nicotinic acetylcholine receptors are more sensitive than α4β2*-subtype receptors to regulation by chronic nicotine administration. J Neurochem. 2014;130:185–198. doi: 10.1111/jnc.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, McIntosh JM, Rossi F, Champtiaux N, Zoli M, Changeux JP. Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci. 2003;17:1329–1337. doi: 10.1046/j.1460-9568.2003.02564.x. [DOI] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pagès G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouysségur J, Brambilla R. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron. 2002;34:807–820. doi: 10.1016/S0896-6273(02)00716-X. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Parameswaran N, Bordia T, Fan H, McIntosh JM, Quik M. Differential regulation of mesolimbic alpha 3/alpha 6 beta 2 and alpha 4 beta 2 nicotinic acetylcholine receptor sites and function after long-term nicotine to monkeys. J Pharmacol Exp Ther. 2006a;318:381–388. doi: 10.1124/jpet.106.104414. [DOI] [PubMed] [Google Scholar]
- McCallum SE, Parameswaran N, Perez XA, Bao S, McIntosh JM, Grady SR, Quik M. Compensation in pre-synaptic dopaminergic function following nigrostriatal damage in primates. J Neurochem. 2006b;96:960–972. doi: 10.1111/j.1471-4159.2005.03610.x. [DOI] [PubMed] [Google Scholar]
- Menza MA, Grossman N, Van Horn M, Cody R, Forman N. Smoking and movement disorders in psychiatric patients. Biol Psychiatry. 1991;30:109–115. doi: 10.1016/0006-3223(91)90163-G. [DOI] [PubMed] [Google Scholar]
- Mitsuoka T, Kaseda Y, Yamashita H, Kohriyama T, Kawakami H, Nakamura S, Yamamura Y. Effects of nicotine chewing gum on UPDRS score and P300 in early-onset parkinsonism. Hiroshima J Med Sci. 2002;51:33–39. [PubMed] [Google Scholar]
- Moll H. The treatment of post-encephalitic parkinsonism by nicotine. Br Med J. 1926;1:1079–1081. doi: 10.1136/bmj.1.3416.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J Neurochem. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- Parain K, Hapdey C, Rousselet E, Marchand V, Dumery B, Hirsch EC. Cigarette smoke and nicotine protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Parkinsonian toxin. Brain Res. 2003;984:224–232. doi: 10.1016/S0006-8993(03)03195-0. [DOI] [PubMed] [Google Scholar]
- Park HJ, Lee PH, Ahn YW, Choi YJ, Lee G, Lee DY, Chung ES, Jin BK. Neuroprotective effect of nicotine on dopaminergic neurons by anti-inflammatory action. Eur J Neurosci. 2007;26:79–89. doi: 10.1111/j.1460-9568.2007.05636.x. [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. Randomized placebo-controlled study of the nicotinic agonist SIB-1508Y in Parkinson disease. Neurology. 2006;66:408–410. doi: 10.1212/01.wnl.0000196466.99381.5c. [DOI] [PubMed] [Google Scholar]
- Perez XA, Parameswaran N, Huang LZ, O'Leary KT, Quik M. Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J Neurochem. 2008;105:1861–1872. doi: 10.1111/j.1471-4159.2008.05268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez XA, McIntosh JM, Quik M. Long-term nicotine treatment down-regulates α6β2* nicotinic receptor expression and function in nucleus accumbens. J Neurochem. 2013;127:762–771. doi: 10.1111/jnc.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez XA, Khroyan TV, McIntosh JM, Quik M. Varenicline enhances dopamine release facilitation more than nicotine after long-term nicotine treatment and withdrawal. Pharmacol Res Perspect. 2015;3:e00105. doi: 10.1002/prp2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson JD, Goldberg JA, Surmeier DJ. Adenosine A2a receptor antagonists attenuate striatal adaptations following dopamine depletion. Neurobiol Dis. 2012;45:409–416. doi: 10.1016/j.nbd.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Léna C, Bessis A, Lallemand Y, Le Novère N, Vincent P, Pich EM, Brûlet P, Changeux JP. Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature. 1995;374:65–67. doi: 10.1038/374065a0. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Addy NA, Mineur YS, Brunzell DH. It is not “either/or”: Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors releated to nicotine addiction and mood. Prog Neurobiol. 2008;84:329–342. doi: 10.1016/j.pneurobio.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Li S. Dose-response meta-analysis on coffee, tea and caffeine consumption with risk of Parkinson's disease. Geriatr Gerontol Int. 2014;14:430–439. doi: 10.1111/ggi.12123. [DOI] [PubMed] [Google Scholar]
- Quik M, Chen L, Parameswaran N, Xie X, Langston JW, McCallum SE. Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J Neurosci. 2006;26:4681–4689. doi: 10.1523/JNEUROSCI.0215-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, Zhang D, Perez XA, Bordia T. Role for the nicotinic cholinergic system in movement disorders; therapeutic implications. Pharmacol Ther. 2014;144:50–59. doi: 10.1016/j.pharmthera.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice ME, Cragg SJ. Nicotine amplifies reward-related dopamine signals in striatum. Nat Neurosci. 2004;7:583–584. doi: 10.1038/nn1244. [DOI] [PubMed] [Google Scholar]
- Robinson SF, Marks MJ, Collins AC. Inbred mouse strains vary in oral self-selection of nicotine. Psychopharmacology (Berl) 1996;124:332–339. doi: 10.1007/BF02247438. [DOI] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Silver AA, Shytle RD, Philipp MK, Cahill DW, Fogelson HM, McConville BJ. Nicotine for the treatment of Tourette's syndrome. Pharmacol Ther. 1997;74:21–25. doi: 10.1016/S0163-7258(96)00199-4. [DOI] [PubMed] [Google Scholar]
- Sanberg PR, Vindrola-Padros C, Shytle RD. Translating laboratory discovery to the clinic: from nicotine and mecamylamine to Tourette's, depression, and beyond. Physiol Behav. 2012;107:801–808. doi: 10.1016/j.physbeh.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol. 2006;290:C852–C861. doi: 10.1152/ajpcell.00358.2005. [DOI] [PubMed] [Google Scholar]
- Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA. Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem. 2014;129:898–915. doi: 10.1111/jnc.12686. [DOI] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett MW, Balleine BW. Molecular substrates of action control in cortico-striatal circuits. Prog Neurobiol. 2011;95:1–13. doi: 10.1016/j.pneurobio.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett MW, Brown RA, Balleine BW. Acquisition and performance of goal-directed instrumental actions depends on ERK signaling in distinct regions of dorsal striatum in rats. J Neurosci. 2010;30:2951–2959. doi: 10.1523/JNEUROSCI.1778-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver AA, Shytle RD, Philipp MK, Wilkinson BJ, McConville B, Sanberg PR. Transdermal nicotine and haloperidol in Tourette's disorder: a double-blind placebo-controlled study. J Clin Psychiatry. 2001;62:707–714. doi: 10.4088/JCP.v62n0908. [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Richards CI, Xiao C, Rhee D, Pantoja R, Dougherty DA, Miwa JM, Lester HA. Pharmacological chaperoning of nicotinic acetylcholine receptors reduces the endoplasmic reticulum stress response. Mol Pharmacol. 2012;81:759–769. doi: 10.1124/mol.112.077792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Henderson BJ, Lester HA, Richards CI. Pharmacological chaperoning of nAChRs: a therapeutic target for Parkinson's disease. Pharmacol Res. 2014;83:20–29. doi: 10.1016/j.phrs.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele SL, Chen B, Lo C, Gertler TS, Warre R, Surmeier JD, Brotchie JM, Nash JE. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol Dis. 2014;71:334–344. doi: 10.1016/j.nbd.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampà C, Fusco FR, Picconi B, Calabresi P. The distinct role of medium spiny neurons and cholinergic interneurons in the D2/A2 receptor interaction in the striatum: Implications for Parkinson's disease. J Neurosci. 2011;31:1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuang D, Larson EB, Li G, Shofer JB, Montine KS, Thompson ML, Sonnen JA, Crane PK, Leverenz JB, Montine TJ. Association between lifetime cigarette smoking and Lewy body accumulation. Brain Pathol. 2010;20:412–418. doi: 10.1111/j.1750-3639.2009.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallejo YF, Buisson B, Bertrand D, Green WN. Chronic nicotine upregulates nicotinic receptors by a novel mechanism. J Neurosci. 2005;25:5563–5572. doi: 10.1523/JNEUROSCI.5240-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Mark M, Nijssen PC, Vlaanderen J, Huss A, Mulleners WM, Sas AM, van Laar T, Kromhout H, Vermeulen R. A case-control study of the protective effect of alcohol, coffee and cigarette consumption on Parkinson disease risk: time-since-cessation modifies the effect of tobacco smoking. PLoS One. 2014;9:e95297. doi: 10.1371/journal.pone.0095297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieregge A, Sieberer M, Jacobs H, Hagenah JM, Vieregge P. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology. 2001;57:1032–1035. doi: 10.1212/WNL.57.6.1032. [DOI] [PubMed] [Google Scholar]
- Villafane G, Cesaro P, Rialland A, Baloul S, Azimi S, Bourdet C, Le Houezec J, Macquin-Mavier I, Maison P. Chronic high dose transdermal nicotine in Parkinson's disease: and open trial. Eur J Neurol. 2007;14:1313–1316. doi: 10.1111/j.1468-1331.2007.01949.x. [DOI] [PubMed] [Google Scholar]
- Walsh H, Govind AP, Mastro R, Hoda JC, Bertrand D, Vallejo Y, Green WN. Up-regulation of nicotinic receptors by nicotine varies with receptor subtype. J Biol Chem. 2008;283:6022–6032. doi: 10.1074/jbc.M703432200. [DOI] [PubMed] [Google Scholar]
- Wang, Buck DC, Yang R, Macey TA, Neve KA. Dopamine D2 receptor stimulation of mitogen-activated protein kinases mediated by cell type-dependent transactivation of receptor tyrosine kinases. J Neurochem. 2005;93:899–909. doi: 10.1111/j.1471-4159.2005.03055.x. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Hall DA, Warnes A, Strange PG, Proud CG. Activation of microtubule-associated protein kinase (Erk) and p70 S6 kinase by D2 dopamine receptors. J Neurochem. 1998;70:2139–2146. doi: 10.1046/j.1471-4159.1998.70052139.x. [DOI] [PubMed] [Google Scholar]
- Wiecki TV, Frank MJ. Neurocomputational models of motor and cognitive deficits in Parkinson's disease. Prog Brain Res. 2010;183:275–297. doi: 10.1016/S0079-6123(10)83014-6. [DOI] [PubMed] [Google Scholar]
- Wiecki TV, Riedinger K, Ameln-Mayerhofer von A, Schmidt WJ, Frank MJ. A neurocomputational account of catalepsy sensitization induced by D2 receptor blockade in rats: context dependency, extinction, and renewal. Psychopharmacology (Berl) 2009;204:265–277. doi: 10.1007/s00213-008-1457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, Costa RM. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Sulzer D. Frequency-dependent modulation of dopamine release by nicotine. Nat Neurosci. 2004;7:581–582. doi: 10.1038/nn1243. [DOI] [PubMed] [Google Scholar]
- Zhang L, Doyon WM, Clark JJ, Phillips PE, Dani JA. Controls of tonic and phasic dopamine transmission in the dorsal and ventral striatum. Mol Pharmacol. 2009;76:396–404. doi: 10.1124/mol.109.056317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R. Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods. 2005;143:27–32. doi: 10.1016/j.jneumeth.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Zhuang X, Mazzoni P, Kang UJ. The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat Rev Neurol. 2013;9:248–256. doi: 10.1038/nrneurol.2013.57. [DOI] [PubMed] [Google Scholar]