

Abstract
Recognition of signaling phospholipids by proteins is a critical requirement for the targeting and initiation of many signaling cascades. Most biophysical methods for measuring protein interactions with signaling phospholipids use purified proteins, which do not take into account the effect of post-translational modifications and other cellular components on these interactions. To potentially circumvent these problems, we have developed a single-molecule fluorescence approach to analyzing lipid-protein interactions in crude cell extracts. As a proof of principle for this assay, we show that a variety of lipid-binding domains (LBDs) can be recruited from cell lysates specifically onto their target phospholipids. With single-molecule analysis in real-time, our assay allows direct determination of binding kinetics for transient lipid-protein interactions, and has revealed unique assembly properties and multiple binding modes of different LBDs. Whereas single-copy LBDs display transient interaction with lipid vesicles, tandem-repeat LBDs, often used as lipid biosensors, tend to form stable interactions that accumulate over time. We have extended the assay to study a cellular protein, Akt, and discovered marked differences in the lipid binding properties of the full-length protein compared to its PH domain. Importantly, we have found that phosphorylation of Akt at T308 and S473 does not affect the lipid binding behaviors of Akt, contrary to the long-standing model of Akt regulation. Overall, this work establishes the single-molecule lipid pulldown assay as a simple and highly sensitive approach to interrogating lipid-protein interactions in a setting that at least partly mimics the cellular environment.
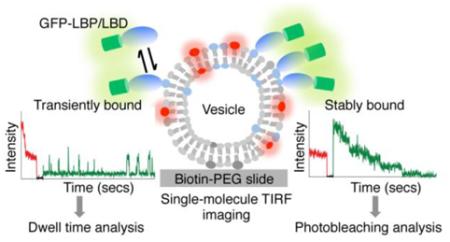
Introduction
Lipids comprise the largest class of biomolecules with a huge diversity in chemical identities.1 In addition to their role as structural components of the biological membrane, they are directly involved in signaling events during cell growth, proliferation, and metabolism.2,3 Among lipids, phospholipids, and predominantly phosphoinositides, interact with proteins directly and regulate the subcellular localization and/or activity of the proteins.4,5 For example, phosphatidylinositol 3-phosphate (PI(3)P) plays a critical role in endocytic and phagocytic trafficking, autophagy, and growth factor signaling.6,7 Phosphatidylinositol 4,5-bisphosphate (PIP2) regulates cell shape, migration, cytokinesis, and membrane trafficking events.8 Phosphatidylinositol 3,4,5-triphosphate (PIP3) is involved in cell proliferation, survival, metabolism, and in diseases such as diabetes and cancer.9 Another important phospholipid is phosphatidic acid (PA), essential for cytoskeletal rearrangement, membrane vesicle trafficking, and growth factor signaling.10
Signaling lipids are typically recognized by specific and structurally conserved lipid-binding domains (LBDs) that are widely present in proteins.11 For example, FYVE domain has a conserved basic amino acid motif (RR/KHHCR) that contributes to a shallow, positively charged binding pocket for PI(3)P. PH domains exhibit a range of lipid selectivity depending on the amino acid residues present and on the cellular context. The PH domain of PLCδ is a specific effector for PIP2,12 while the PH domain of Akt binds preferentially to PIP3.13-15 In the case of PA, no sequence or structure conservation is found among the known PA binding proteins, except for the presence of positively charged amino acids that form electrostatic interactions with the head group of PA.16
In vitro assays such as lipid overlay, centrifugation-based methods, size-exclusion chromatography, surface plasmon resonance (SPR), and isothermal titration calorimetry have traditionally been employed to investigate lipid-protein interactions.17,18 A handful of studies have also probed such interactions with single-molecule resolution,19,20 made possible by the recent development of methods to tether liposomes on surfaces at single-particle densities for imaging.21 A major limitation of the current methods lies in the use of purified recombinant proteins, which can be laborious and technically challenging, and may not recapitulate the post-translational modifications or the native state of the protein. Additionally, most assays typically require the separation of unbound protein or lipid from the lipid-protein complex, which would disrupt equilibrium. Thus, developing a new biophysical method where lipid-protein interaction can be studied in a more physiological setting and in equilibrium is desired.
We previously developed a single-molecule pull-down (SiMPull) assay to study protein complexes directly captured from cell lysates.22 Here we present a novel approach based on the principle of SiMPull to interrogate lipid-protein interactions in crude cell lysates. Proteins are directly pulled down from mammalian cell lysates by surface-immobilized lipid vesicles. We provide evidence of high sensitivity and specificity of our assay for the detection of interactions between several signaling lipids and their respective LBDs, as well as the full-length Akt protein. Our results also provide new insights into the assembly of the LBDs and Akt on lipid vesicles.
Material and methods
Lipids, Antibodies, Plasmids, and Other Reagents
These are all described in detail in the Supporting Information section.
Cell Culture, Transfection, and Cell Lysis
HEK293 cells were cultured in DMEM containing 10% (vol/vol) FBS at 37 °C with 5% (vol/vol) CO2. Transfection was performed using PolyFect following the manufacturer’s recommendations (Qiagen), and cells were mechanically lysed in detergent-free buffer. See Supporting Information for further details. [Move next line here.] The concentrations of GFP-fused LBDs in cell lysates were determined by single-molecule pulldown, with purified recombinant GFP as a reference. All GFP-LBDs were present at 2-5 μM in cell lysates in our study. Lysates were then diluted 10 to 100-fold with dilution buffer (20 mM Tris·HCl pH 8.0, 150 mM NaCl) before loading into the imaging chamber.
Vesicle Preparation
Lipid vesicles were prepared by probe sonication with 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindodicarbocyanine percholorate (DiD) as a label. Detailed procedures are described in the Supporting Information section.
Single-Molecule Imaging and Quantification
A prism-type total internal reflection fluorescence (TIRF) microscope was used to acquire single-molecule data.22 Quartz slides were passivated with methoxy polyethylene glycol (PEG, MW 5000) doped with 2-5% biotinylated PEG (Laysan Bio Inc). Immobilized DiD-labeled vesicles were excited at 640 nm and GFP-tagged LBDs were excited at 488 nm. A 665LP from Semrock for DiD, and HQ 535/30 from Chroma Technology for GFP were used to detect emission signals. All experiments were performed at room temperature (22–25°C). The number of GFP or DiD fluorescence spots was counted per imaging area of 4,500 μm2. The emission signal from each spot was recorded for 16 frames (100 ms each) and the intensity average calculated. Standard deviation from mean was calculated from 10 or more different imaging areas. Cell lysate was present in the flow chamber during data acquisition. Single-molecule co-localization and assembly plot analysis are described in Supporting Information.
Results
Detection of Lipid-Protein Interaction in Crude Mammalian Cell Lysates
To establish the single-molecule binding assay, we picked three lipid-LBD interaction pairs known in the literature: PI(3)P and Hrs-FYVE domain,23 PIP2 and PLCδ-PH domain,12 PA and Spo20-PA-binding domain (PABD).24 The design principle of the assay is depicted in Figure 1a. Lipids were presented on small unilamellar vesicles, each containing the scaffolding lipid phosphatidylcholine (PC), biotin-phosphatidylethanol-amine (PE), the lipophilic dye DiD, and a specific signaling phospholipid. The lipid vesicles were immobilized on biotin-PEG-passivated microscope slides aided by neutravidin at single-vesicle resolution and visualized by TIRF microscopy. The number of DiDs incorporated into each vesicle was determined by photobleaching analysis of DiD fluorescence to peak at 6-7 (Figure S1). The LBDs were expressed in HEK293 cells as GFP-fusions of tandem repeats (i.e., GFP-2xFYVE, GFP-2xPLC-PH, and GFP-2xPABD), which are commonly used as lipid sensors.25 Detergent-free cell lysates expressing the LBDs were added to slide chambers containing various lipid vesicles, followed by TIRF microscope visualization (Figure 1a). As shown in Figure 1b & 1c, 2xFYVE bound specifically to PI(3)P as indicated by the appearance of GFP fluorescence spots, and no binding to PC, PA, PS, PIP2, or PIP3 was detected above background. Similarly, 2xPABD was pulled down by PA vesicles (Figure 1d and Figure S2a) and 2xPLC-PH by PIP2 vesicles (Figure 1e and Figure S2b), without any cross-interaction with other vesicles.
Figure 1. Detection of lipid-protein interactions in fresh cell lysates.

(a) Schematic depiction of the single-molecule lipid-protein interaction assay. (b) Single-molecule TIRF images of various vesicles (DiD, top row) and 2xFYVE (GFP, bottom row). (c-e) Quantification of 2xFYVE (c), 2xPABD (d), and 2xPLC-PH (e) pull-down by various lipid vesicles. Background fluorescence spots in the GFP channel obtained by adding lysates in the absence of lipid vesicles were subtracted from all samples. Error bars: SD of data from 10 or more imaging areas. (f) Single-molecule TIRF images showing the overlay of 2xFYVE and PI(3)P vesicles from the same region (top row) and different regions (bottom row). (g) Colocalization of 2xLBDs (GFP) with corresponding lipid vesicles (DiD) from the same region or different regions is shown as percentage of GFP spots that overlapped with DiD spots. Error bars: SD of data from 3 independent experiments. Scale bars: 10μm.
To further confirm that the LBDs were indeed pulled down by vesicles and not non-specifically by the PEG surface, we superimposed the DiD images of vesicles with the corresponding GFP images of the LBDs, and found that ~50-60% of GFP spots colocalized with vesicles (Figure 1f, g and Figure S2c, d). Colocalization was not complete most likely due to unlabeled vesicles, inactive chromophores, and surface impurities. The results of superimposing DiD and GFP images from two non-overlapping regions indicated that only 6% colocalization occurred by chance (Figure 1f, g). In summary, our assay can detect lipid-protein interactions from whole cell lysates with exquisite specificity at single-molecule resolution.
Distinct Assembly Properties of Tandem LBDs
Next, we examined the assembly and binding dynamics of each LBD to its target phospholipid vesicles using single-molecule fluorescence imaging. We recorded movies (frame rate 10 Hz), where first DiD was briefly excited followed by longer GFP excitation (Figure S3b). This strategy allowed us to selectively record the time course of GFP fluorescence signal colocalized with vesicle, and to eliminate GFP signal arising from surface impurities and LBDs bound to unlabeled vesicles. The movies were acquired in the presence of cell lysate, allowing the capture of transient binding events if there were any.
We detected two distinct behaviors of GFP-LBDs in the single-vesicle traces – stable and transient binding. Stable binding was defined by GFP fluorescence intensity remaining unchanged until the GFP photobleached causing an abrupt decrease in intensity. The average phobleaching lifetime of GFP under our assay conditions was 8.6 secs (Figure S3a). A transient binding event was observed as repeated cycles of abrupt increase in GFP fluorescence intensity (binding) followed by abrupt decrease (dissociation) on a single vesicle, with dwell times less than GFP lifetime. To determine the number of stable LBDs bound per vesicle (NGFP) we counted the number of photobleaching steps of GFP upon GFP excitation. Because the noise in fluorescence traces of GFP made it difficult to clearly distinguish single photobleaching steps,26 we used Chung-Kennedy filtration algorithm to average out noise from the raw GFP intensity profiles (Figure S3b-d).27 This enabled us to confidently determine the number of photobleaching steps even when more than four GFP molecules were bound.
For 2xFYVE binding to PI(3)P, we observed an increase in GFP spot intensity over the time that lysates were incubated in the slide chamber (Figure S4a), likely due to the accumulation of LBDs on the immobilized vesicles. Single-molecule traces from different incubation times (Figure 2a) revealed two distinct behaviors of 2xFYVE – stable and transient binding. The number of stably bound molecules increased overtime, while transient binding events occurred similarly through the incubation time. The dissociation rate koff determined from the average lifetime of the transiently bound state was 0.44 ± 0.03 sec−1 (Figure S4d). Figure 2b shows the time evolution of the fraction of vesicles displaying different NGFP values (NGFP = 0, 1, …, 5). For example, the fraction of vesicles with NGFP = 1 (gray line) increased in the first 35 mins and then decreased afterwards as more LBDs bound. NGFP = 0 depicts the time evolution of unbound vesicles, and the data indicates that by ~30 mins more than half of the PI(3)P vesicles were occupied by 2xFYVE. The bound vesicles shifted to higher NGFP (up to 5) over time.
Figure 2. Assembly properties of tandem repeat LBDs on lipid vesicles.

(a) GFP photobleaching traces of 1, 2, and 4 copies of 2xFYVE pulled down by PI(3)P vesicles, from 3 incubation times. (b) Distribution of 2xFYVE stably bound on PI(3)P vesicles at various NGFP over incubation time. (c, d) Similar to (a, b), for binding of 2xPABD to PA vesicles. (e, f) Similar to (a, b), for binding of 2xPLC-PH to PIP2 vesicles. Scale bars: 10μm.
Next, in our measurements for 2xPABD binding to PA vesicles, we again observed an increase in the intensity of GFP spots with incubation time (Figure S4b). Single-vesicle traces indicated that 2xPABD assembled stably onto the PA vesicles and did not show any transient binding events (Figure 2c). Consistent with the increasing GFP intensity, we observed that NGFP increased from zero to up to eight (Figure 2d) such that the fraction of vesicles with NGFP = 1 (gray line) reached the maximum by 5 min and decreased thereafter as more 2xPABD bound to the vesicle. For the third LBD-lipid pair, 2xPLC-PH binding to PIP2 vesicles, the GFP intensity remained largely unchanged with the incubation time of the lysate (Figure S4c), and we observed both stable and transient populations in the single-vesicle traces (Figure 2e). In agreement with the GFP intensity, NGFP for 2xPLC-PH did not change markedly with the incubation time (Figure 2f). Most vesicles had one (50%) or two (25%) copies of 2xPLC-PH bound starting from the earliest observation time point (1 min). We calculated the dissociation rate, koff, of the transient species to be 0.52 ± 0.04 sec−1 (Figure S4e).
For these tandem LBDs, the GFP fusions were mostly monomeric as analyzed by SiMPull using an anti-GFP antibody (Figure S5a-c), ruling out vesicles-independent oligomerization of the GFP-LBDs. In summary, we observed differences among the lipid-LBD pairs in NGFP over time and binding dynamics (summarized in Table 1). The number of stably bound molecules per vesicle (NGFP) for 2xFYVE and 2xPABD, but not 2xPLC-PH, increased over time. Additionally, both transient and stable interactions with the target lipids were observed for 2xFYVE and 2xPLC-PH, but only stable binding was found for 2xPABD. Taken together, our results clearly indicate that these lipid-protein interactions are governed by distinct assembly mechanisms, which may arise from different binding affinities and structural variations between the LBDs and lipids.
Table 1.
Summary of LBD-lipid interactions
| # of repeats | FYVE [PI(3)P] |
PABD [PA] |
PLCδ-PH [PIP2] |
Akt-PH [PIP3] |
Akt [PIP3] |
|---|---|---|---|---|---|
|
1x
(Single) |
Binding not detect- ed |
Stable binding (rapid equilibrium) Transient binding (koff = 0.71 s−1) |
Transient binding only (koff = 2.33 s−1) |
Stable binding (slow accumulation) Transient binding (koff = 0.22 s−1) |
Stable binding only (rapid equilibrium) |
|
2x
(Tandem) |
Stable binding (slow accumulation) Transient binding (koff = 0.44 s−1) |
Stable binding only (slow accumula- tion) |
Stable binding (rapid equilibrium) Transient binding (koff = 0.21 s−1) |
Not determined | Not determined |
Transient interactions between lipids and single-copy LBDs
Having established the assay using tandem LBDs commonly used as lipid biosensors, we wished to investigate the lipid binding behaviors of single-copy LBDs because they represent the configuration found in most cellular proteins. Cell lysates expressing 1xPABD, 1xPLC-PH, or 1xFYVE were prepared and subjected to analyses as described above. Specific pull-down of 1xPABD and 1xPH by PA and PIP2 vesicles, respectively, was observed (Figure 3a, b and Figure S6a, b). About 55-60% of GFP spots colocalized with the vesicles (Figure 3c and Figure S6c, d). We did not detect any pull-down of 1xFYVE by PI(3)P, consistent with the requirement of FYVE dimerization for PI(3)P recognition.23,28
Figure 3.

Assembly properties of single-copy LBDs on lipid vesicles. (a,b) Quantification of 1xPABD (a) and 1xPH (b) pull-down by various lipid vesicles. Error bars: SD of data from 10 or more imaging areas. (c) Colocalization of 1xLBDs (GFP) with corresponding lipid vesicles (DiD) from the same region or different regions is shown as percentage of GFP spots that overlapped with DiD spots. Error bars: SD of data from 3 independent experiments. (d) GFP photobleaching traces of 1, 2, and 3 copies of 1xPABD bound to PA vesicles. (e) Distribution of 1xPABD stably bound on PA vesicles at various NGFP over incubation time. (f) GFP photobleaching traces of 1xPH bound to PIP2 vesicles at two different laser intensities (20 mW and 5 mW).
Next we compared the assembly behavior of single-copy LBDs with their tandem repeat counterparts. Whereas 2xPABD displayed only stable binding to PA (Figure 2c), transient binding events with a koff of 0.71 ± 0.04 sec−1 were detected for 1xPABD in addition to stable binding events (Figure 3d and Figure S6e). Unlike 2xPABD, which accumulated up to 8 copies of the protein per vesicle over time (Figure 2d), the distribution of NGFP for 1xPABD did not change markedly over the course of 60 min and stayed below 5 copies (Figure 3e). 1xPLC-PH also shifted to all transient binding (Figure 3f) with a koff of 2.33 ± 0.3 sec−1 (Figure S6f), from the combination of transient and stable binding detected for 2xPLC-PH (Figure 2e). We measured the duration of transient binding of 1xPLC-PH to PIP2 at two different laser intensities and obtained the same average dwell times, hence the same koff values (Figure 3f), confirming that laser-induced photobleaching or photoblinking of GFP emission did not contribute to the koff determination. We also confirmed that these single-copy LBDs do not exhibit self-oligomerization in the absence of vesicles by performing anti-GFP pull-down and photobleaching analysis (Figure S7a, b).
Overall, our results indicate that some single-copy LBDs bind only transiently to the lipid targets, while tandem LBDs bind lipid vesicles more stably and at higher copy numbers per vesicle than their single-copy counterparts. Thus, the additional copy of LBD appears to stabilize the protein-lipid interaction and, in the case of FYVE domain, is necessary for detectable lipid binding.
PIP3 binding by Akt and its PH domain
Upon validating the versatility and specificity of our assay using LBDs, we next explored its applicability to intact proteins containing an LBD. We chose Akt, a protein kinase critically involved in many cellular processes,29 as a model system. Akt contains a PH domain that binds to PIP3 with nanomolar affinity.13,14 We expressed GFP-tagged full-length Akt in HEK293 cells and observed its specific pull-down by immobilized PIP3 vesicles (Figure 4a and Figure S8a), with 47% of GFP spots colocalizing with the vesicles (Figure 4c and Figure S8b). This interaction was highly specific, as no binding to PC, PA, PI(3)P or PIP2 was detected. Hence, the assay is both sensitive and specific for a full-length protein in a whole cell lysate environment.
Figure 4. Characterization of Akt and Akt-PH binding to PIP3 lipid vesicles.

(a,b) Quantification of Akt pull-down by various lipid vesicles. Error bars: SD of data from 10 or more imaging areas. (c) Colocalization of Akt and Akt-PH (GFP) with PIP3 vesicles (DiD) from the same region or different regions is shown as percentage of GFP spots that overlapped with DiD spots. (d) Binding of Akt and Akt-PH to PIP3 vesicles over incubation time is shown as fraction of bound vesicles. (e) GFP photobleaching traces of 1 and 2 copies of Akt bound to PIP3. (f) Distribution of Akt stably bound on PIP3 vesicles at various NGFP over incubation time. (g) GFP photobleaching traces of 1, 3, and 4 copies of Akt-PH bound to PIP3. (h) Distribution of Akt-PH stably bound on PIP3 vesicles at various NGFP over incubation time.
GFP-tagged Akt-PH domain expressed in HEK293 cells was also specifically pulled down by PIP3 vesicles (Figure 4b and Figure S8c) with 53% colocalization (Figure 4c and Figure S8d). The comparison between Akt-PH and full-length Akt revealed remarkable differences in their assembly behaviors on vesicles. While Akt-PH association with vesicles occurred very rapidly with 90% of the vesicles bound within two minutes of lysate addition, only half of the vesicles were occupied by Akt after 30 min incubation (Figure 4d). Additionally, Akt displayed only stable binding to the vesicles (Figure 4e), and the majority of bound vesicles had one copy of Akt (Figure 4f). In contrast, Akt-PH showed both stable and transient binding to the vesicles (Figure 4g) with NGFP increasing over time (Figure 4h). The transient interactions were determined to have a koff of 0.22 ± 0.01 sec−1 (Figure S8e). The difference cannot be due to different expression levels because we confirmed by anti-GFP pull-down that Akt and Akt-PH were present at similar levels in the cell lysate (Figure S8f). Also, photobleaching analysis of the GFP pull-down indicated that Akt was mostly monomeric in the cell lysates (Figure S8g).
Lipid binding independent of Akt phosphorylation
A long-standing model of Akt activation involves membrane recruitment of Akt30 and subsequent phosphorylation of its A loop (T308) and hydrophobic motif (S473), both sites critical for full activation of Akt.31 Upon activation, Akt detaches from the membrane and translocates into the cytoplasm and the nucleus to exert its functions.32-34 T308-phosphorylated catalytic domain has been found to interact with the PH domain, which is believed to be critical for the release of Akt from the plasma membrane.32 An extrapolation of this model is that active/phosphorylated Akt has a lower affinity for PIP3 than inactive Akt or PH domain alone. Our observations above indeed indicated more binding of Akt-PH domain to PIP3 vesicles than full-length Akt. However, those experiments were performed with lysates prepared from asynchronously growing cells, in which Akt activation status was most likely heterogeneous. To probe a possible correlation between Akt activity and its lipid binding properties, we compared inactive Akt from serum-starved cells with active Akt from serum-stimulated cells for their binding to PIP3. As shown in Figure S9, robust activation of Akt by serum was confirmed by western analysis of pS473 (Figure S9a) as well as TIRF imaging of pS473 pulldown (Figure S9b), but a similar number of GFP-Akt was pulled down by the vesicles with and without serum stimulation (Figure S9c).
However, it is formally possible that only a small fraction of Akt was activated by serum stimulation and inactive Akt dominated the outcome of the assays above. To unequivocally examine a potential effect of phosphorylation (and thus activation) of Akt on its lipid binding properties, we created GFP fusions of phosphomimetic and nonphosphorylatable mutants of Akt: T308D/S473D, T308A/S473A, T308A/S473D, and T308D/S473A. Alanine substitution at Thr308 and Ser473 has been reported to inactivate Akt, whereas aspartic acid substitution at these two sites confers constitutive activity.31 Interestingly, all the mutants bound PIP3 to a similar degree (Figure 5a, b), and they displayed patterns of NGFP comparable to that of WT-Akt over incubation time (Figure 5c). These results suggest that, contrary to the long-held belief, phosphorylation of T308 and S473 does not appear to impact Akt binding to PIP3.
Figure 5. Phosphorylation of T308 and S473 does not affect Akt binding to PIP3.

(a) Quantification of pull-down of various Akt proteins by PIP3 vesicles. DD: T308D/S473D; AA: T308A/S473A; AD: T308A/S473D; DA: T308D/S473A. Error bars: SD of data from 10 or more imaging areas. (b) Colocalization of Akt mutants (GFP) with PIP3 vesicles (DiD) from the same region or different regions. Error bars: SD of data from 3 independent experiments. (c) Distribution of Akt mutants stably bound on PIP3 vesicles at various NGFP over incubation time.
Discussion
We have developed a single-molecule approach to studying lipid-protein interactions, which allows detection of specific binding, acquisition of kinetic parameters and determination of assembly properties in crude cell extracts, simultaneously. We first demonstrated the versatility of our assay to detect specific binding of several LBDs to their lipid targets, followed by application to a full-length protein containing an LBD. A distinct advantage of our approach is its applicability to proteins in freshly prepared whole cell lysates without purification, simplifying experimental procedures and also potentially preserving the native state of the protein as well as recapitulating post-translational modifications. The assay requires only 50 μL of cell lysate, and could potentially be performed with as few as 10 cells, as previously demonstrated for SiMPull.22 This assay could be readily extended to using liposomes of complex compositions or even endogenous vesicular membranes to offer a closer mimic of physiological conditions. The assay could also be coupled with microfluidic devices, and applied to high-throughput screens.
The use of a relatively large fluorescent protein tag in our current assays could potentially introduce artifacts. It is feasible to ameliorate this issue by using small genetically encoded fluorescent tags.35 Another inherent limitation of the assay is that it would not be possible to definitively determine if a protein-lipid interaction is direct, due to the presence of other cellular proteins. Furthermore, expressed LBDs could be pre-occupied by cellular lipids, or masked by their protein binding partners, or competed by endogenous proteins for binding vesicles. At the same time, however, the effects of endogenous proteins on an LBD-lipid interaction can potentially be probed by experimentally manipulating the cell lysates, which will be an interesting direction for future investigations.
In our assays each binding and unbinding event of an LBD to a single immobilized vesicle can be monitored. This single-vesicle resolution has led to the revelation of heterogeneity in the number of LBDs bound per vesicle. Since the brightness of DiD as an indicator of vesicle size did not correlate with copy number of bound LBD (data not shown), the observed heterogeneity is most likely an intrinsic property of the lipid-LBD binding and not a reflection of the vesicle size heterogeneity. Distinct assembly properties of three well-known LBDs – PH, FYVE and PABD – in single-copy as well as tandem-repeat form have been deciphered in this study. As summarized in Table 1, although these LBDs assemble on their target lipids at different copy numbers and dissociation rates, there is a common trend of more stable binding by tandem-repeat LBDs than their single-copy counterparts. In the case of the FYVE domain this stabilizing effect allows detection of 2xFYVE binding to PI(3)P, albeit transient, while 1xFYVE does not display any binding. This is consistent with previous observations that two tandem copies of the Hrs-FYVE domain, or dimerization by a chemical dimerizer, was necessary for the targeting of Hrs-FYVE to endosomal membranes containing PI(3)P.23,28 Our findings extend the stabilizing effect of LBD dimerization to the binding of PIP2 and PA to their target proteins, which have not been reported thus far, and suggest that multivalent interactions may be a common mechanism for stabilized protein binding to lipids.
Notably, the dissociation rate (koff) we have determined for 1xPLCδ-PH and PIP2 (2.33 sec−1) is ~300-fold higher than that reported using SPR (0.0079 sec−1).36 A potentially major contributing factor to this difference is the density of surface-immobilized vesicles. While our assay is performed at a very low density to allow single-vesicle resolution, the SPR method typically requires much higher density of vesicles on the surface (~500-fold).37 It is known that higher surface densities utilized in SPR result in underestimation of dissociation rate constants due to re-binding events.38 In addition, a difference in the environment – whole cell lysates versus purified proteins – may have also contributed to the discrepancy. It is also important to point out that binding of purified Hrs-FYVE to PI(3)P vesicles has been observed using SPR,23,39 whereas we did not detect any binding with the single-molecule assay. This further suggests that our assay conditions may be closer to those in a cell, in which Hrs-FYVE is incapable of detecting cellular PI(3)P.23
It is interesting to note the difference between the two PH domains, from PLCδ and Akt, in their binding to PIP2 and PIP3, respectively. The dissociation rate of PLCδ-PH from PIP2 (2.33 sec−1) is 10 times higher than that of Akt-PH from PIP3 (0.22 sec−1). This may reflect the biophysical nature of these lipid-protein interactions, and may also underline the biological functions of these two signaling proteins.
The comparison between Akt and its PH domain has revealed striking differences in their lipid binding dynamics. At similar expression levels, Akt-PH binds PIP3 vesicles faster than Akt, but Akt binding is more stable, extending its residence time on the membrane. Interestingly, this stable binding by full-length Akt is not accompanied by binding of multiple copies of the protein on vesicles as seen with the PH domain alone. It seems that the additional domains of Akt both stabilize the lipid binding and prevent the protein from accumulating to high copy numbers on a single vesicle. This would imply that Akt is unlikely to be recruited to the membrane at high densities in a cell, further highlighting the importance of residence time of Akt that does get recruited to the membrane to allow activation.
A long-held model of Akt activation suggests that phosphorylation on T308 and/or S473 impacts the association of Akt with PIP3-containing membrane. To our surprise, we have found that activation of Akt by serum stimulation does not affect the lipid binding properties of Akt. Furthermore, mutating both T308 and S473 to either abolish phosphorylation or mimic phosphorylation has no effect on Akt binding to PIP3 vesicles. These results challenge the notion of T308/S473 phosphorylation modulating Akt-lipid interaction, and suggest the existence of regulatory mechanisms involving other post-translational modifications or regulatory proteins.
Supplementary Material
ACKNOWLEDGMENT
We thank Benjamin Leslie, and members of the Ha and Chen labs for helpful discussions. This work was supported by grants from the National Institute of Health (AG042332 to JC and TH, GM089771 to JC), and by the National Science Foundation (PHY 1430124). T.H. is an investigator with the Howard Hughes Medical Institute
Footnotes
Supporting Information
These material is available free of charge via the internet at http://pub.acs.org.
Experimental details including materials, methods, additional figures, and references. (PDF)
Author contributions: E.A., V.A., A.J., T.H., and J.C. designed research; E.A., V.A., and A.J. performed experiments; E.A., and V.A. analyzed data; E.A., V.A., T.H., and J.C. wrote the paper.
REFERENCES
- (1).van Meer G, de Kroon AI. J Cell Sci. 2011;124:5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- (2).De Matteis MA, Godi A. Nat Cell Biol. 2004;6:487–492. doi: 10.1038/ncb0604-487. [DOI] [PubMed] [Google Scholar]
- (3).Hannun YA, Obeid LM. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- (4).Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV. Nature. 2014;510:172–175. doi: 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lemmon MA. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- (6).Burman C, Ktistakis NT. FEBS Lett. 2010;584:1302–1312. doi: 10.1016/j.febslet.2010.01.011. [DOI] [PubMed] [Google Scholar]
- (7).Yoon MS, Du G, Backer JM, Frohman MA, Chen J. J Cell Biol. 2011;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Di Paolo G, De Camilli P. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- (9).Cantley LC. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- (10).Jenkins GM, Frohman MA. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).DiNitto JP, Cronin TC, Lambright DG. Sci STKE. 2003;2003:re16. doi: 10.1126/stke.2132003re16. [DOI] [PubMed] [Google Scholar]
- (12).Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J. Proc Natl Acad Sci U S A. 1995;92:10472–10476. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Biochem J. 1996;315 ( Pt 3):709–713. doi: 10.1042/bj3150709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. The Journal of biological chemistry. 1997;272:8474–8481. doi: 10.1074/jbc.272.13.8474. [DOI] [PubMed] [Google Scholar]
- (15).Rowland MM, Gong D, Bostic HE, Lucas N, Cho W, Best MD. Chem Phys Lipids. 2012;165:207–215. doi: 10.1016/j.chemphyslip.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Stace CL, Ktistakis NT. Biochim Biophys Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- (17).Narayan K, Lemmon MA. Methods. 2006;39:122–133. doi: 10.1016/j.ymeth.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rusten TE, Stenmark H. Nat Methods. 2006;3:251–258. doi: 10.1038/nmeth867. [DOI] [PubMed] [Google Scholar]
- (19).Knight JD, Falke JJ. Biophys J. 2009;96:566–582. doi: 10.1016/j.bpj.2008.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rozovsky S, Forstner MB, Sondermann H, Groves JT. J Phys Chem B. 2012;116:5122–5131. doi: 10.1021/jp210045r. [DOI] [PubMed] [Google Scholar]
- (21).Christensen AL, Lohr C, Christensen SM, Stamou D. Lab on a chip. 2013;13:3613–3625. doi: 10.1039/c3lc50492a. [DOI] [PubMed] [Google Scholar]
- (22).Jain A, Liu R, Ramani B, Arauz E, Ishitsuka Y, Ragunathan K, Park J, Chen J, Xiang YK, Ha T. Nature. 2011;473:484–488. doi: 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nakanishi H, de los Santos P, Neiman AM. Mol Biol Cell. 2004;15:1802–1815. doi: 10.1091/mbc.E03-11-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Balla T, Varnai P. Sci STKE. 2002;2002:pl3. doi: 10.1126/stke.2002.125.pl3. [DOI] [PubMed] [Google Scholar]
- (26).Dickson RM, Cubitt AB, Tsien RY, Moerner WE. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- (27).Chung SH, Kennedy RA. J Neurosci Methods. 1991;40:71–86. doi: 10.1016/0165-0270(91)90118-j. [DOI] [PubMed] [Google Scholar]
- (28).Hayakawa A, Hayes SJ, Lawe DC, Sudharshan E, Tuft R, Fogarty K, Lambright D, Corvera S. The Journal of biological chemistry. 2004;279:5958–5966. doi: 10.1074/jbc.M310503200. [DOI] [PubMed] [Google Scholar]
- (29).Manning BD, Cantley LC. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kohn AD, Takeuchi F, Roth RA. The Journal of biological chemistry. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- (31).Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Embo j. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- (32).Ananthanarayanan B, Fosbrink M, Rahdar M, Zhang J. The Journal of biological chemistry. 2007;282:36634–36641. doi: 10.1074/jbc.M706227200. [DOI] [PubMed] [Google Scholar]
- (33).Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. The Journal of biological chemistry. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- (34).Calleja V, Alcor D, Laguerre M, Park J, Vojnovic B, Hemmings BA, Downward J, Parker PJ, Larijani B. PLoS Biol. 2007;5:e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Shi X, Jung Y, Lin LJ, Liu C, Wu C, Cann IK, Ha T. Nat Methods. 2012;9:499–503. doi: 10.1038/nmeth.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ferguson CG, James RD, Bigman CS, Shepard DA, Abdiche Y, Katsamba PS, Myszka DG, Prestwich GD. Bioconjugate chemistry. 2005;16:1475–1483. doi: 10.1021/bc050197q. [DOI] [PubMed] [Google Scholar]
- (37).Cooper MA, Hansson A, Lofas S, Williams DH. Analytical biochemistry. 2000;277:196–205. doi: 10.1006/abio.1999.4389. [DOI] [PubMed] [Google Scholar]
- (38).Myszka DG. Current opinion in biotechnology. 1997;8:50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- (39).Blatner NR, Stahelin RV, Diraviyam K, Hawkins PT, Hong W, Murray D, Cho W. The Journal of biological chemistry. 2004;279:53818–53827. doi: 10.1074/jbc.M408408200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.