

Abstract
Study Objectives:
To evaluate the role of orexin-A with respect to cerebrospinal fluid (CSF) Alzheimer disease (AD) biomarkers, and explore its relationship to cognition and sleep characteristics in a group of cognitively normal elderly individuals.
Methods:
Subjects were recruited from multiple community sources for National Institutes of Health supported studies on normal aging, sleep and CSF biomarkers. Sixty-three participants underwent home monitoring for sleep-disordered breathing, clinical, sleep and cognitive evaluations, as well as a lumbar puncture to obtain CSF. Individuals with medical history or with magnetic resonance imaging evidence of disorders that may affect brain structure or function were excluded. Correlation and linear regression analyses were used to assess the relationship between orexin-A and CSF AD-biomarkers controlling for potential sociodemographic and sleep confounders.
Results:
Levels of orexin-A, amyloid beta 42 (Aβ42), phosphorylated-tau (P-Tau), total-tau (T-Tau), Apolipoprotein E4 status, age, years of education, reported total sleep time, number of awakenings, apnea-hypopnea indices (AHI), excessive daytime sleepiness, and a cognitive battery were analyzed. Subjects were 69.59 ± 8.55 years of age, 57.1% were female, and 30.2% were apolipoprotein E4+. Orexin-A was positively correlated with Aβ42, P-Tau, and T-Tau. The associations between orexin-A and the AD-biomarkers were driven mainly by the relationship between orexin-A and P-Tau and were not influenced by other clinical or sleep characteristics that were available.
Conclusions:
Orexin-A is associated with increased P-Tau in normal elderly individuals. Increases in orexin-A and P-Tau might be a consequence of the reduction in the proportion of the deeper, more restorative slow wave sleep and rapid eye movement sleep reported with aging.
Clinical Trial Registration:
Clinicaltrials.gov registration number NCT01962779.
Citation:
Osorio RS, Ducca EL, Wohlleber ME, Tanzi EB, Gumb T, Twumasi A, Tweardy S, Lewis C, Fischer E, Koushyk V, Cuartero-Toledo M, Sheikh MO, Pirraglia E, Zetterberg H, Blennow K, Lu SE, Mosconi L, Glodzik L, Schuetz S, Varga AW, Ayappa I, Rapoport DM, de Leon MJ. Orexin-A is associated with increases in cerebrospinal fluid phosphorylated-tau in cognitively normal elderly subjects. SLEEP 2016;39(6):1253–1260.
Keywords: Alzheimer disease, orexin-A, phosphorylated-tau, sleep
Significance.
Orexin is a key regulator of sleep-wake homeostasis. Deposition of abnormal phosphorylated tau (P-Tau) in neurons and glia is one of the major features of Alzheimer's disease (AD). Our results show a positive association between cerebrospinal fluid (CSF) levels of orexin-A and P-Tau in a group of cognitively normal elderly. Further, this correlation was not influenced by total sleep time, number of awakenings or sleep disordered breathing. Both findings could be explained by the decrease in the proportion of deeper restorative sleep stages that is part of normal aging or, alternatively, by AD pathology causing orexin dysfunction early in the disease process. Understanding the role of orexin dysfunction in older adults might help unfold new preventive therapies for AD.
INTRODUCTION
In humans, a growing body of evidence suggests that synaptic activity is associated with increased production of amyloid beta (Aβ).1–5 The brain maintains its connectivity during light sleep6–8 but reduces its metabolic and electric activity with increasing depth of non-rapid eye movement (NREM) sleep.6–10 This suggests that brain-soluble Aβ levels may fluctuate with a diurnal pattern consistent with higher neuronal activity during wakefulness and decreased neuronal activity during NREM sleep.11 Evidence supporting this Aβ diurnal pattern has been reported in some human studies but not others.12–16 With respect to tau protein, a recent study in transgenic mice showed that chronic sleep disruption resulted in an increased insoluble fraction of tau in the brain,17 but the relationship between the sleep-wake cycle and tau phosphorylation in humans is unknown.
Orexin-A (hypocretin-1), a neuropeptide produced by lateral hypothalamic neurons,18 is involved in the regulation of the sleep-wake cycle by increasing arousal levels19–21 and has been suggested to promote Aβ production and amyloid deposition in transgenic mice.12 The relationship between the orexinergic system and the Alzheimer disease (AD) neurodegenerative process in humans has been analyzed by a variety of cross-sectional studies in different clinical populations showing associations of cerebrospinal fluid (CSF) orexin-A with Aβ42 but also with phosphorylated tau (P-Tau) and total tau (T-Tau). However, the results so far have been inconclusive.22–27 Whether the reported changes in Aβ42, P-Tau or T-Tau are directly related to orexin-A secretion or are secondary to changes in the sleep-wake cycle (as suggested by a recent study in mice)28 is also unknown.
Based on this preliminary evidence, the aim of this study was to investigate the involvement of the orexinergic system in Aβ42, P-Tau and T-Tau concentrations by measuring CSF orexin-A and AD-biomarker levels at cross-section in a group of cognitively normal elderly controlling for sociodemographic, clinical, and sleep characteristics.
METHODS
Subject Recruitment
Sixty-three cognitively normal elderly individuals were recruited at the NYU Center for Brain Health from active National Institutes of Health (NIH)-supported longitudinal studies of normal aging and CSF diagnostic AD-biomarkers that have been ongoing between 1998 and 2015. Subjects had previously been recruited from multiple community sources, including individuals interested in research participation and risk consultation; self-referred individuals with subjective cognitive complaints; spouses, family members, and caregivers of impaired patients participating in other studies; and random sampling using voter registration records from the New York City Manhattan and Brooklyn Borough areas. Individuals with medical conditions or history of significant conditions that may affect the brain structure or function, such as stroke, uncontrolled diabetes mellitus, traumatic brain injury, any neurodegenerative diseases, and active major depression, as well as magnetic resonance imaging evidence of intracranial mass or infarcts, were excluded from the parent studies. Sleep complaints were not part of the inclusion or exclusion criteria of any of the NIH studies that the subjects were recruited from, nor were subjects referred to the study from the NYU Sleep Disorders Center that performed the later sleep analyses.
Clinical and Diagnostic Evaluation
Subjects received a clinical standardized diagnostic assessment that is part of the Uniform Data Set II (UDS II). This battery was incorporated into all US AD Centers in 2005 to standardize data collection across centers and disciplines and includes medical, psychiatric, and neurological evaluations.29 To this battery we added the Hamilton Depression Rating Scale,30 additional home monitoring for sleep disordered breathing (SDB), a detailed sleep interview, and we asked the participants to fill out the Epworth Sleepiness Scale (ESS)31 to measure excessive daytime sleepiness. None of the selected subjects were on active treatment for SDB with continuous positive airway pressure or dental appliances. Eligibility requirements for the present study included having had CSF collected by lumbar puncture (LP) and a diagnostic structural magnetic resonance imaging scan completed prior to the sleep examination (average time interval between the sleep study and the LP was 0.9 ± 1.1 y). Presence of the apolipoprotein E4 (ApoE4) genotype was determined using standard polymerase chain reaction procedures.
Cognitive Evaluation
All subjects received a standard neuropsychological test battery, which has published norm values.32 The tests included to measure declarative memory were subtests of the Guild Memory Scale: verbal paired associates (initial [PRDI], delayed [PRDD] and immediate [PARI]), delayed paragraph recall subtest (PARD) and the Wechsler Memory Scale Revised33: Logical Memory subtests (Logic I and II). A subtest of the Wechsler Intelligence Scale Revised was added to assess working memory (digits backward [WAISDIG-B]).34 The Digit Symbol Substitution Test (DSST)34 was used to evaluate psychomotor speed. Trails A and digits forward (WAISDIG-F)34 were included to evaluate attention and Trails B Test was included to evaluate executive function.35 Category fluency (animals and vegetables) and the Boston Naming Test (BNT)36 were used to evaluate language. The Mini Mental State Examination37 was included as an additional global measure of cognition.
CSF Analysis
LPs were performed using a 25-gauge needle guided by fluoroscopy between 11:00 and 13:00 (median time of the LP 12:39; interquartile range [IQR] 01:48 h) to be consistent with respect to circadian variation of Aβ peptides, as previously published studies indicate that there is a diurnal fluctuation of CSF Aβ42 (with lower levels during the late morning hours).12,14 However, there is a marked decrease of Aβ42 circadian amplitude with increasing age,38 so it is unlikely that the results were affected considerably by the small variability in the time of the LPs in our sample. The average ‘lag time’ between midsleep (reported bedtime + reported total sleep time [TST]/2) and the LP start time was of 10.02 ± 01:35 h. All CSF samples were kept on ice until centrifuged for 10 min at 1,500 g, at 4°C. Samples were aliquoted to 0.25 mL polypropylene tubes and stored at −80°C until assayed. CSF P-Tau 181 (pg/mL), T-Tau (pg/mL), and Aβ42 (pg/mL) were analyzed in a blind manner in batch mode using enzyme-linked immunosorbent assay (Fujirebo, Ghent, Belgium).39 CSF orexin-A (pg/mL) was measured using an in-house radioimmunoassay.25
Sleep Evaluation
A full sleep evaluation was performed on all subjects. This included a sleep interview, detailed snoring history, and self-administration of the ESS.31 Home monitoring of SDB was completed using either an ARES” Unicorder (Watermark, Boca Raton, FL)40 or an Embletta” MPR (Natus Medical Inc, San Carlos, CA)41 systems during a 2-night period. Both systems record flow from a nasal cannula and oximetry via finger or forehead. The variables calculated and included in this study were: (1) the apnea/hypopnea index with 4% desaturation (AHI4%), defined as the sum of all apneas (> 90% reduction in airflow for > 10 sec) and all hypopneas (> 30% reduction in airflow for 10 sec) associated with > 4% oxygen (O2) desaturation divided by the total time where both flow and oximetry signals were valid; (2) the AHIall, which was defined as the sum of all apneas and all hypopneas (hypopneas 4% + events with visible reduction in airflow amplitude and presence of inspiratory flattening ending in breaths with normalization of airflow as a surrogate for arousal42) identified divided by the total time where there was a valid flow signal irrespective of O 2 saturation; and, (3) mean saturation of oxygen (SpO2) saturation during the night. Although the systems used different techniques of oximetry measurement, we have previously shown that SDB metrics between these two measurements are highly correlated.40 Both systems and AHI indices have been compared with the recommended definitions of AHI based on full in-laboratory nocturnal polysomnography that included electroencephalographic measures of sleep and show good comparability.40,41 Reported total sleep time (TST) duration was assessed using one question: “During the past month, how many hours of sleep did you usually get each night, what is your best estimate?” Although reported TST by a single question has shown to have a large individual variability, in our global dataset of elderly subjects (n = 143), reported TST shows a correlation of 0.45 (P < 0.05) with actigraphy which is consistent with the literature43 (data not published). Additionally, in contrast to previous reports of overestimation by subjective measure, our global data show little systematic bias of reported TST (−0.1 ± 1.1 h). Awakenings from sleep were assessed using a second question: “During the past month, how many times do you wake up each night, on average?”
Statistical Analysis
Logarithm transformation was applied to normalize right skewed variables prior to analysis (these included: P-Tau, T-Tau, AHI4%, AHIall, TST, number of awakenings, and mean SpO2 saturation). Regression-based z-scores corrected for age, sex, race, and education derived from our normative sample32 were used to assess cognitive measures. Correlation analyses were used to assess for the relations between continuous dependent (Aβ42, P-Tau, and T-Tau) and explanatory variables (orexin-A) to obtain correlation coefficients. Finally, stepwise multivariable linear regression models were examined for Aβ42, lnP-Tau, and lnT-Tau to determine which characteristics independently predicted their CSF levels. We used a stepwise selection process with variables significant at the P < 0.05 entry and exit criteria to determine the final multivariate models. The following variables were included in the models: orexin-A, P-Tau (for Aβ42 analyses), Aβ42 (for P-Tau analyses), age, ApoE4 status, years of education, AHI4%, AHIall, TST, and number of awakenings. Analyses were done with SPSS 20.0 (Chicago, IL, USA).
RESULTS
Participant Characteristics
Demographic, sleep and cognitive characteristics of all subjects (n = 63) are shown in Table 1. Subjects were 69.59 ± 8.55 y of age, 57.1% were female, and 30.2% were ApoE4+. All participants had at least 12 y of education (mean value: 16.51 ± 2.27); a Mini Mental State Examination score > 26 (mean value: 29.29 ± 0.96); a Clinical Dementia Rating score of 0; and who received a diagnosis of cognitively normal and not actively depressed by the study clinician. Of all subjects, four were being treated with fluoxetine (one with fluoxetine and lorazepam), one was taking bupropion, one was taking lorazepam, one was taking trazodone, two were taking diazepam, two were taking gabapentin, and one was taking nonprescription sleep medication (melatonin).
Table 1.
demographic, cognitive and sleep characteristics for all subjects.

CSF AD Biomarker Levels and Orexin-A
A summary of CSF levels of Aβ42, P-Tau, T-tau, and orexin-A is presented in Table 2. There was no significant correlation between Aβ42 and time of LP or lag time. Across all subjects, significant correlations were found between all three CSF biomarkers and orexin-A (Aβ42: r = 0.40, P < 0.01; lnP-Tau: r = 0.52, P < 0.01; and lnT-Tau: r = 0.42, P < 0.01) (Figure 1). Similarly, all CSF biomarkers were positively associated with lnP-Tau (Aβ42: r = 0.46, P < 0.01; lnT-Tau: r = 0.87, P < 0.01) and with Aβ42 (lnT-Tau: r = 0.35, P < 0.01) (Figure 2).
Table 2.
Summary of CSF levels of Aβ42, P-Tau, T-Tau, and Orexin-A.

Figure 1.

Scatterplots of CSF orexin-A, Aβ42, and the natural log of CSF P-Tau and T-Tau for all subjects are shown. Significant correlations were found between all CSF biomarkers and orexin-A levels. Aβ42, amyloid beta 42; CSF, cerebrospinal fluid; P-Tau, phosphorylated tau; T-Tau, total tau.
Figure 2.

Scatterplots of natural log of CSF P-Tau and the natural log of CSF T-Tau and Aβ42 for all subjects are shown. Both CSF biomarkers were found to have significant positive associations with P-Tau. Aβ42, amyloid beta 42; CSF, cerebrospinal fluid; P-Tau, phosphorylated tau; T-Tau, total tau.
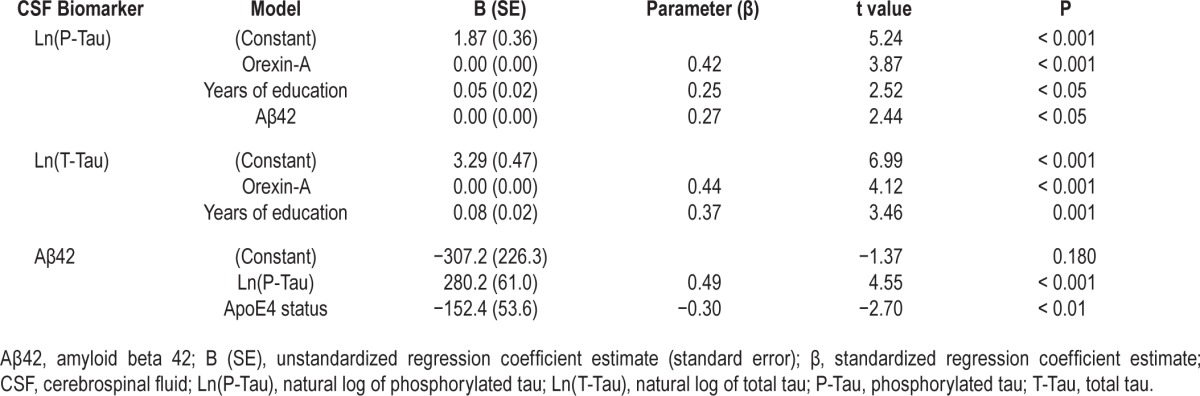
Stepwise linear regression models using Aβ42, lnP-Tau and lnT-Tau as the dependent variables revealed that the positive associations shown previously were driven by the correlations between orexin-A and tau proteins as only P-Tau and T-Tau were associated with orexin-A in the final regression models: (1) LnP-Tau: β = 0.42, partial correlation coefficient = 0.45 (P < 0.01), suggesting that lnP-Tau increased by 0.17 (0.42 times the standard deviation [SD] of lnP-tau of 0.41), equivalent to an approximately 1.19-fold increase in P-Tau, for every SD increase in orexin-A (SD = 156.70 pg/mL) after adjusting for years of education and CSF Aβ42. (2) LnT-Tau: β = 0.44, partial correlation coefficient = 0.47 (P < 0.01), suggesting that lnT-Tau increased by 0.22 (0.44 times the SD of lnT-tau of 0.5), equivalent to approximately 1.25-fold increase in T-Tau, for every SD increase in orexin-A after adjusting for years of education (full models are shown in Table 3). The best-fit model for CSF Aβ42 did not include orexin-A but P-Tau instead: β = 0.49, partial correlation coefficient = 0.51 (P < 0.01), suggesting that Aβ42 increased by 112.7 pg/mL (0.49 times SD of 229.91 pg/mL) for every SD increase in lnP-Tau (SD = 0.41) after adjusting for ApoE4 (full models are shown in Table 3). Sensitivity analyses, removing lnP-Tau and Aβ42 from the linear regression models, showed that the correlation between lnP-Tau and orexin-A remained significant (β = 0.54, partial correlation coefficient = 0.55, P < 0.01) after adjusting for years of education; the correlation between orexin-A and Aβ42 changed and became significant once again (β = 0.53, partial correlation coefficient = 0.50, P < 0.01) after adjusting for sex and ApoE4 status, suggesting that P-tau could mediate some of the observed effects between orexin-A and Aβ42.
Table 3.
Stepwise multivariate linear regression models of P-Tau, T-Tau, and Aβ42.
Sleep Characteristics and Orexin-A
Sleep characteristics: objective (AHI4%, AHIall, and mean SpO2 during the night) and subjective (reported TST, reported number of awakening and ESS) are shown in Table 1. Among the 63 participants, 22 subjects were considered free of SDB (AHI4% < 5/h), 26 had mild SDB (AHI4% 5–14.99/h), and 15 had moderate to severe SDB (AHI4% > 15/h). Thirteen subjects were classified as short sleepers (≤ 6 h per night), 34 were normal sleepers (6 to 8 h per night) and 16 were long sleepers (≥ 8 h per night). Only eight subjects complained of excessive daytime sleepiness (ESS > 10). Using analysis of variance and simple bivariate correlations, orexin-A levels were negatively correlated with body mass index (r = −0.43, P < 0.01) and were higher in females (F(1,61) = 11.83, P < 0.01). Orexin-A was not significantly correlated with any other available sleep characteristics.
DISCUSSION
This study was unique in systematically testing the relationship between orexin-A and CSF AD-biomarkers in a group of cognitively normal elderly individuals with available objective and subjective ambulatory measurements of sleep. CSF orexin-A levels were positively correlated with Aβ42, P-Tau, and T-Tau but the associations were not independent, suggesting that they could have been mediated by the observed relationship between orexin-A and tau. Further, they were not influenced by reported TST, number of awakenings or SDB indices.
Previous publications are conflicting with reports of positive or no associations between orexin-A and CSF biomarkers of risk for AD.22–27,44 From these, three studies have similarly found associations between CSF orexin-A and tau but not with Aβ42. The first study analyzed a group of 48 AD patients and 28 inpatient controls without dementia admitted for suspected subarachnoid hemorrhage or chronic polyneuropathy.27 CSF orexin-A levels were correlated with T-Tau but only in the AD group. The second study included two different groups of AD patients and compared them with two groups of depressed patients in full remission.44 When the whole group was analyzed, orexin-A was related to increases in P-Tau, T-Tau, and to a lesser extent Aβ40, but not Aβ42. The third study analyzed a group of 26 AD patients, 18 patients with Lewy body dementia and 24 non-demented controls.22 Orexin-A was linked to T-Tau in female non-demented controls whereas associations between orexin-A and Aβ42 were absent in all groups regardless of sex. The variability of results from these studies is hard to interpret due to the small sample sizes, heterogeneity of comparison groups, presence of psychotropic medication (which could have influenced orexin-A levels or neuronal activity, whereas less than 20% of subjects in our sample were receiving psychotropic medication), lack of objective sleep assessments, wide age ranges, and the possibility that the orexinergic system is affected by the neurodegenerative process itself, as shown by a postmortem study that reported a 40% decreased cell number and 14% lower CSF orexin-A levels in patients with advanced AD.45 Together, these results suggest that the relationship between CSF levels of orexin-A and AD biomarkers may vary depending on age, clinical diagnoses, medication, dementia severity, hypothalamic damage, and presence or absence of senile plaques.
The orexinergic system is a key regulator of sleep onset, transitions between vigilance states, and energy expenditure.46 It shows a wake-on and rapid eye movement (REM)-off pattern of firing47 that helps maintain sleep-wake homeostasis.48 REM deprivation has been shown to differentially increase orexin-A levels in discrete brain areas in rats in what could potentially comprise a positive feed forward cycle between REM sleep disruption and CSF orexin-A level increases.48 Normal aging is accompanied by a decrease in the ability to initiate and maintain sleep, sleep consolidation, and the proportion of deeper, more restorative slow wave and REM sleep stages.49 This suggests that increases in orexin-A could be a biomarker of the reduced sleep depth observed as part of the aging process. Further, the coexistence of narcolepsy and AD49 (which demonstrates that AD pathology can also develop in the absence of orexin) and recent studies from Roh et al.28 showing that focal overexpression of orexin does not alter the amount of Aβ pathology in transgenic mice support the hypothesis that it is the effect of orexin (or aging) on the sleep-wake cycle and not the expression of the neuropeptide itself that modulates Aβ pathology. Relatedly, sleep deprivation has been associated with GSK3β activation50 and better sleep consolidation has been shown to attenuate the effect of ApoE4 genotype on incident AD and development of neurofibrillary tangles postmortem,51 in what could comprise a second positive feed forward cycle between sleep disruption and increases in CSF P-Tau. Thus, one model that accounts for the observed positive correlation between P-Tau and orexin-A is that age-related increases in orexin-A promote wakefulness and sleep fragmentation, which then promote accumulation of P-Tau. However, we could not confirm this hypothesis because we were not able to show associations between orexin-A levels and our available ambulatory measures of sleep fragmentation (AHIall and reported number of awakenings). Dual orexin receptor antagonists, which have recently been approved for the treatment of insomnia,51 should therefore be investigated in elderly patients with elevated levels of CSF orexin-A and P-Tau as a possible preventive measure for development of AD pathology. It remains to be determined whether such an intervention would only benefit those with insomnia symptoms or objective measurements of poor sleep quality, as they may prove ineffective in normal sleepers.
An alternative model that explains the positive correlation between CSF P-Tau and orexin-A would be that AD pathology causes overexpression of CSF orexin-A. In cell cultures, Davies et al.52 have reported that application of Aβ42 induces both amyloid plaque formation and tau phosphorylation mimicking an AD milieu that results in downregulation of orexin receptors (OxRs). Although levels of orexin-A were not specifically evaluated, these results suggest a plausible homeo-static increase in orexin-A levels secondary to OxR reduction induced by AD neuropathology. In humans, current consensus is that the AD pathological process begins decades before clinical symptoms occur.53 This long ‘preclinical’ phase of AD might first become detectable in middle age as deposits of P-tau in the transentorhinal cortex and in subcortical nuclei such as the locus coeruleus (LC).54,55 The LC is a cluster of norepinephrine (NE) neurons that project throughout the neuraxis and provide the sole source of NE to the neocortex.56 LC-NE neurons express Ox1R57,58 and have two modes of firing: tonic, which is highest during wakefulness, decreases during NREM and is silent during REM sleep20,59,60; and phasic, which occurs in response to sensory stimuli.59,61 Projection neurons are remarkably sturdy and a large number of them containing pretangle material can survive until an individual dies.62 However, several studies have clearly shown that loss of LC-NE neurons63–78 occurs later in the disease process and exacerbates AD pathogenesis and disease progression.79,80 These findings suggest that overexpression of orexin could result from an imbalance between the LC-NE and orexinergic systems after the norepinephrine pathways are damaged by neurofibrillary tangles.
The clinical relevance of the observed positive correlation between Aβ42 and tau protein levels is unknown as most studies indicate that changes in P-Tau are usually downstream to decreases in CSF Aβ42. However, recent studies have shown that secreted tau may cause neuronal hyperactivity and increase Aβ production, suggesting another mechanism of feed forward regulation.81 To our knowledge, only two studies have found similar incidental cross-sectional evidence of a positive association between CSF tau and Aβ42. The first was performed in a group of familial AD mutation carriers82 and the second in cognitively normal elderly patients with normal CSF Aβ42 levels.83 Because amyloid precursor protein and tau are highly expressed proteins in neurons, their increased levels in CSF could reflect overall synaptic function83 and/or the effects of impaired sleep.84
Limitations of this study include the cross-sectional nature of the findings; the time interval between the sleep studies and the LP; the absence of nocturnal polysomnography recordings with measurements of sleep architecture and REM duration that could confirm our explanatory hypothesis; and the use of reported TST instead of objective measurements of sleep duration. The results of this study should therefore be validated in independent cohorts with in-laboratory encephalographic measures of sleep architecture, young comparison groups, and LPs performed shortly after or before the sleep studies.
DISCLOSURE STATEMENT
This was not an industry supported study. Supported by grants from: NIH/ NIA/NHLBI R01HL118624, R01HL111724, R21AG049348, R01AG035137, R01AG032554, R01AG022374, R01AG13616 and R01AG1210; Foundation for Research in Sleep Disorders and CTSI UL1TR000038. Dr. Blennow has served on Advisory Boards for Pfizer, Roche and Innogenetics, Belgium, IBL International, Novartis, Eli Lilly, and Sanofi; has served on speakers bureaus for Fujirebio Europe and Lundbeck; and has performed research supported by the Swedish Research Council, grant #14002. Drs. Blennow and Zetterberg are co-founders of Brain Biomarker Solutions in Gothenburg AB, a GU-holding based company at University of Gothenburg. Dr. Ayappa has received research support from Fisher & Paykel Healthcare and holds multiple US and foreign patents covering techniques and analysis algorithms for the diagnosis of OSAHS and techniques for administering CPAP - several of which have been licensed to Fisher & Paykel Healthcare. Dr. Rapoport has received research support from Fisher & Paykel Healthcare and has participated in speaking and consulting engagements for Fisher & Paykel Healthcare and holds multiple US and foreign patents covering techniques and analysis algorithms for the diagnosis of OSAHS and techniques for administering CPAP – several of which have been licensed to Fisher & Paykel Healthcare and Sefam Medical. Dr. Glodzik received grant/research support from the NIH. Mony de Leon serves on the external advisory board of Roche Pharmaceuticals and holds patents issued through NYU related to the image analysis of PET and MRI scans. The other authors have indicated no financial conflicts of interest. Study procedures were performed at the NYU Center for Brain Health and the NYU Sleep Disorders Center. Cerebrospinal Fluid samples were analyzed at the Clinical Neurochemistry Laboratory, Institute of Neuroscience and Physiology, the Sahlgrenska Academy at the University of Gothenburg. Additional support is acknowledged from the philanthropy of Mr. James B. Kuhn. None of the funding sources played a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
REFERENCES
- 1.Buckner RL, Snyder AZ, Shannon BJ, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–17. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid-b deposition. Nature Neuroscience. 2011;14:750–6. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 4.Gouras GK, Relkin NR, Sweeney D, Munoz DG, Mackenzie IR, Gandy S. Increased apolipoprotein E epsilon 4 in epilepsy with senile plaques. Ann Neurol. 1997;41:402–4. doi: 10.1002/ana.410410317. [DOI] [PubMed] [Google Scholar]
- 5.Mackenzie IR, Miller LA. Senile plaques in temporal lobe epilepsy. Acta Neuropathol. 1994;87:504–10. doi: 10.1007/BF00294177. [DOI] [PubMed] [Google Scholar]
- 6.Horovitz SG, Braun AR, Carr WS, et al. Decoupling of the brain's default mode network during deep sleep. Proc Natl Acad Sci U S A. 2009;106:11376–81. doi: 10.1073/pnas.0901435106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larson-Prior LJ, Power JD, Vincent JL, et al. Modulation of the brain's functional network architecture in the transition from wake to sleep. Prog Brain Res. 2011;193:277–94. doi: 10.1016/B978-0-444-53839-0.00018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spoormaker VI, Schroter MS, Gleiser PM, et al. Development of a large-scale functional brain network during human non-rapid eye movement sleep. J Neurosci. 2010;30:11379–87. doi: 10.1523/JNEUROSCI.2015-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samann PG, Wehrle R, Hoehn D, et al. Development of the brain's default mode network from wakefulness to slow wave sleep. Cereb Cortex. 2011;21:2082–93. doi: 10.1093/cercor/bhq295. [DOI] [PubMed] [Google Scholar]
- 10.Wu CW, Liu PY, Tsai PJ, et al. Variations in connectivity in the sensorimotor and default-mode networks during the first nocturnal sleep cycle. Brain Connect. 2012;2:177–90. doi: 10.1089/brain.2012.0075. [DOI] [PubMed] [Google Scholar]
- 11.Lucey BP, Bateman RJ. Amyloid-beta diurnal pattern: possible role of sleep in Alzheimer's disease pathogenesis. Neurobiol Aging. 2014;35(Suppl 2):S29–34. doi: 10.1016/j.neurobiolaging.2014.03.035. [DOI] [PubMed] [Google Scholar]
- 12.Kang JE, Lim MM, Bateman RJ, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Llano DA, Ellis T, et al. Effect of human cerebrospinal fluid sampling frequency on amyloid-beta levels. Alzheimers Dement. 2012;8:295–303. doi: 10.1016/j.jalz.2011.05.900. [DOI] [PubMed] [Google Scholar]
- 14.Ooms S, Overeem S, Besse K, Rikkert MO, Verbeek M, Claassen JA. Effect of 1 night of total sleep deprivation on cerebrospinal fluid beta-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 2014;71:971–7. doi: 10.1001/jamaneurol.2014.1173. [DOI] [PubMed] [Google Scholar]
- 15.Slats D, Claassen JA, Verbeek MM, Overeem S. Reciprocal interactions between sleep, circadian rhythms and Alzheimer's disease: focus on the role of hypocretin and melatonin. Ageing Res Rev. 2013;12:188–200. doi: 10.1016/j.arr.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 16.Xie L, Kang H, Xu Q, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Di MA, Joshi YB, Pratico D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer's disease with plaques and tangles. Neurobiol Aging. 2014;35:1813–20. doi: 10.1016/j.neurobiolaging.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 18.de Lecea L, Kilduff TS, Peyron C, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A. 1998;95:322–7. doi: 10.1073/pnas.95.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–63. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 20.Hagan JJ, Leslie RA, Patel S, et al. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci U S A. 1999;96:10911–6. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kantor S, Mochizuki T, Janisiewicz AM, Clark E, Nishino S, Scammell TE. Orexin neurons are necessary for the circadian control of REM sleep. Sleep. 2009;32:1127–34. doi: 10.1093/sleep/32.9.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wennstrom M, Londos E, Minthon L, Nielsen HM. Altered CSF orexin and alpha-synuclein levels in dementia patients. J Alzheimers Dis. 2012;29:125–32. doi: 10.3233/JAD-2012-111655. [DOI] [PubMed] [Google Scholar]
- 23.Slats D, Claassen JA, Lammers GJ, Melis RJ, Verbeek MM, Overeem S. Association between hypocretin-1 and amyloid-beta42 cerebrospinal fluid levels in Alzheimer's disease and healthy controls. Curr Alzheimer Res. 2012;9:1119–25. doi: 10.2174/156720512804142840. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt FM, Kratzsch J, Gertz HJ, et al. Cerebrospinal fluid melanin-concentrating hormone (MCH) and hypocretin-1 (HCRT-1, orexin-A) in Alzheimer's disease. PLoS One. 2013;8:e63136. doi: 10.1371/journal.pone.0063136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portelius E, Soininen H, Andreasson U, et al. Exploring Alzheimer molecular pathology in Down's syndrome cerebrospinal fluid. Neurodegener Dis. 2014;14:98–106. doi: 10.1159/000358800. [DOI] [PubMed] [Google Scholar]
- 26.Dauvilliers YA, Lehmann S, Jaussent I, Gabelle A. Hypocretin and brain beta-amyloid peptide interactions in cognitive disorders and narcolepsy. Front Aging Neurosci. 2014;6:119. doi: 10.3389/fnagi.2014.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liguori C, Romigi A, Nuccetelli M, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 2014;71:1498–505. doi: 10.1001/jamaneurol.2014.2510. [DOI] [PubMed] [Google Scholar]
- 28.Roh JH, Jiang H, Finn MB, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J Exp Med. 2014;211:2487–96. doi: 10.1084/jem.20141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 30.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14:540–5. doi: 10.1093/sleep/14.6.540. [DOI] [PubMed] [Google Scholar]
- 32.De Santi S, Pirraglia E, Barr WB, et al. Robust and conventional neuropsychological norms: diagnosis and prediction of age-related cognitive decline. Neuropsychology. 2008;22:469–84. doi: 10.1037/0894-4105.22.4.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wechsler D. New York, NY: Harcourt Brace Jovanovich; 1981. Wechsler Adult Intelligence Scale--Revised. [Google Scholar]
- 34.Wechsler D. New York, NY: Psychological Corporation; 1955. Wechsler Adult Intelligence Scale. [Google Scholar]
- 35.Army. Washington, DC: War Department, Adjutant General's Office; 1944. Army Individual Test battery Manual of Directions and Scoring. [Google Scholar]
- 36.Kaplan E, Goodglass H, Weintraub S. Philadelphia, PA: Lea and Febiger; 1983. Boston Naming Test. [Google Scholar]
- 37.Folstein MF, Folstein SE, McHugh PR. Mini-mental state: a practical method for grading the cognitive state of patients for the clinician. J Psychiat Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Potter R, Sigurdson W, et al. Effects of age and amyloid deposition on abeta dynamics in the human central nervous system. Arch Neurol. 2012;69:51–8. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–34. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- 40.Ayappa I, Norman RG, Seelall V, Rapoport DM. Validation of a self-applied unattended monitor for sleep disordered breathing. J Clin Sleep Med. 2008;4:26–37. [PMC free article] [PubMed] [Google Scholar]
- 41.Tiihonen P, Hukkanen T, Tuomilehto H, Mervaala E, Toyras J. Evaluation of a novel ambulatory device for screening of sleep apnea. Telemed J E Health. 2009;15:283–9. doi: 10.1089/tmj.2008.0118. [DOI] [PubMed] [Google Scholar]
- 42.Ayappa I, Norman RG, Suryadevara M, Rapoport DM. Comparison of limited monitoring using a nasal-cannula flow signal to full polysomnography in sleep-disordered breathing. Sleep. 2004;27:1171–9. doi: 10.1093/sleep/27.6.1171. [DOI] [PubMed] [Google Scholar]
- 43.Lauderdale DS, Knutson KL, Yan LL, Liu K, Rathouz PJ. Self-reported and measured sleep duration: how similar are they? Epidemiology. 2008;19:838–45. doi: 10.1097/EDE.0b013e318187a7b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deuschle M, Schilling C, Leweke FM, et al. Hypocretin in cerebrospinal fluid is positively correlated with Tau and pTau. Neurosci Lett. 2014;561:41–5. doi: 10.1016/j.neulet.2013.12.036. [DOI] [PubMed] [Google Scholar]
- 45.Fronczek R, van GS, Frolich M, et al. Hypocretin (orexin) loss in Alzheimer's disease. Neurobiol Aging. 2012;33:1642–50. doi: 10.1016/j.neurobiolaging.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 46.Mavanji V, Perez-Leighton CE, Kotz CM, et al. Promotion of Wakefulness and Energy Expenditure by Orexin A in the Ventrolateral Preoptic Area. Sleep. 2015;38:1361–70. doi: 10.5665/sleep.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/ hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–20. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mehta R, Khanday MA, Mallick BN. REM sleep loss associated changes in orexin-A levels in discrete brain areas in rats. Neurosci Lett. 2015;590:62–7. doi: 10.1016/j.neulet.2015.01.067. [DOI] [PubMed] [Google Scholar]
- 49.Espiritu JR. Aging-related sleep changes. Clin Geriatr Med. 2008;24:1–14. v. doi: 10.1016/j.cger.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 50.Benedetti F, Serretti A, Colombo C, Lorenzi C, Tubazio V, Smeraldi E. A glycogen synthase kinase 3-beta promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neurosci Lett. 2004;368:123–6. doi: 10.1016/j.neulet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 51.Lim AS, Yu L, Kowgier M, Schneider JA, Buchman AS, Bennett DA. Modification of the relationship of the apolipoprotein E epsilon4 allele to the risk of Alzheimer disease and neurofibrillary tangle density by sleep. JAMA Neurol. 2013;70:1544–51. doi: 10.1001/jamaneurol.2013.4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davies J, Chen J, Pink R, et al. Orexin receptors exert a neuroprotective effect in Alzheimer's disease (AD) via heterodimerization with GPR103. Sci Rep. 2015;5:12584. doi: 10.1038/srep12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171–81. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 55.Braak H, Thal DR, Ghebremedhin E, Del TK. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–9. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 56.Aston-Jones G, Cohen JD. An integrative theory of locus coeruleusnorepinephrine function: adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–50. doi: 10.1146/annurev.neuro.28.061604.135709. [DOI] [PubMed] [Google Scholar]
- 57.Bourgin P, Huitron-Resendiz S, Spier AD, et al. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–5. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trivedi P, Yu H, MacNeil DJ, Van der Ploeg LH, Guan XM. Distribution of orexin receptor mRNA in the rat brain. FEBS Lett. 1998;438:71–5. doi: 10.1016/s0014-5793(98)01266-6. [DOI] [PubMed] [Google Scholar]
- 59.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–86. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: application to asthma genetics. Genet Epidemiol. 2004;26:61–9. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- 61.Aston-Jones G, Bloom FE. Norepinephrine-containing locus coeruleus neurons in behaving rats exhibit pronounced responses to non-noxious environmental stimuli. J Neurosci. 1981;1:887–900. doi: 10.1523/JNEUROSCI.01-08-00887.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropath Exp Neurol. 1999;58:188–97. doi: 10.1097/00005072-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 63.Mann DM, Yates PO, Hawkes J. The noradrenergic system in Alzheimer and multi-infarct dementias. J Neurol Neurosurg Psychiatry. 1982;45:113–9. doi: 10.1136/jnnp.45.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bondareff W, Mountjoy CQ, Roth M. Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology. 1982;32:164–8. doi: 10.1212/wnl.32.2.164. [DOI] [PubMed] [Google Scholar]
- 65.Bondareff W. Neuropathology of nucleus basalis and locus ceruleus in Alzheimer's disease. In: Brazier MA, editor. The Biological Substrates of Alzheimer's Disease. Cambridge, UK: Academic Press; 1986. pp. 101–13. [Google Scholar]
- 66.Bondareff W, Mountjoy CQ, Roth M, et al. Neuronal degeneration in locus ceruleus and cortical correlates of Alzheimer disease. Alzheimer Dis Assoc Disord. 1987;1:256–62. doi: 10.1097/00002093-198701040-00005. [DOI] [PubMed] [Google Scholar]
- 67.Bondareff W, Mountjoy CQ, Roth M. Selective loss of neurones of origin of adrenergic projection to cerebral cortex (nucleus locus coeruleus) in senile dementia. Lancet. 1981;1:783–4. doi: 10.1016/s0140-6736(81)92657-x. [DOI] [PubMed] [Google Scholar]
- 68.Burke WJ, Chung HD, Huang JS, et al. Evidence for retrograde degeneration of epinephrine neurons in Alzheimer's disease. Ann Neurol. 1988;24:532–6. doi: 10.1002/ana.410240409. [DOI] [PubMed] [Google Scholar]
- 69.Busch C, Bohl J, Ohm TG. Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol Aging. 1997;18:401–6. doi: 10.1016/s0197-4580(97)00035-3. [DOI] [PubMed] [Google Scholar]
- 70.Chan-Palay V, Asan E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. 1989;287:373–92. doi: 10.1002/cne.902870308. [DOI] [PubMed] [Google Scholar]
- 71.Forno LS, Norville RL. Synaptic morphology in the human locus ceruleus. Acta Neuropathol. 1981;53:7–14. doi: 10.1007/BF00697178. [DOI] [PubMed] [Google Scholar]
- 72.German DC, Manaye KF, White CL, III, et al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992;32:667–76. doi: 10.1002/ana.410320510. [DOI] [PubMed] [Google Scholar]
- 73.Hardy J, Adolfsson R, Alafuzoff I, et al. Transmitter deficits in Alzheimer's disease. Neurochem Int. 1985;7:545–63. doi: 10.1016/0197-0186(85)90050-6. [DOI] [PubMed] [Google Scholar]
- 74.Hoogendijk WJ, Pool CW, Troost D, van ZE, Swaab DF. Image analyser-assisted morphometry of the locus coeruleus in Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis. Brain. 1995;118:131–43. doi: 10.1093/brain/118.1.131. [DOI] [PubMed] [Google Scholar]
- 75.Ichimiya Y, Arai H, Kosaka K, Iizuka R. Morphological and biochemical changes in the cholinergic and monoaminergic systems in Alzheimer-type dementia. Acta Neuropathol. 1986;70:112–6. doi: 10.1007/BF00691428. [DOI] [PubMed] [Google Scholar]
- 76.Iversen LL, Rossor MN, Reynolds GP, et al. Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci Lett. 1983;39:95–100. doi: 10.1016/0304-3940(83)90171-4. [DOI] [PubMed] [Google Scholar]
- 77.Marcyniuk B, Mann DM, Yates PO. The topography of cell loss from locus caeruleus in Alzheimer's disease. J Neurol Sci. 1986;76:335–45. doi: 10.1016/0022-510x(86)90179-6. [DOI] [PubMed] [Google Scholar]
- 78.Strong R, Huang JS, Huang SS, Chung HD, Hale C, Burke WJ. Degeneration of the cholinergic innervation of the locus ceruleus in Alzheimer's disease. Brain Res. 1991;542:23–8. doi: 10.1016/0006-8993(91)90992-5. [DOI] [PubMed] [Google Scholar]
- 79.Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28:327–35. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 80.Gannon M, Che P, Chen Y, Jiao K, Roberson ED, Wang Q. Noradrenergic dysfunction in Alzheimer's disease. Front Neurosci. 2015;9:220. doi: 10.3389/fnins.2015.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bright J, Hussain S, Dang V, et al. Human secreted tau increases amyloid-beta production. Neurobiol Aging. 2015;36:693–709. doi: 10.1016/j.neurobiolaging.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 82.Reiman EM, Quiroz YT, Fleisher AS, et al. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–56. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alcolea D, Martinez-Lage P, Sanchez-Juan P, et al. Amyloid precursor protein metabolism and inflammation markers in preclinical Alzheimer disease. Neurol. 2015;85:626–33. doi: 10.1212/WNL.0000000000001859. [DOI] [PubMed] [Google Scholar]
- 84.Osorio RS, Ayappa I, Mantua J, et al. The interaction between sleep-disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol Aging. 2013;35:1318–24. doi: 10.1016/j.neurobiolaging.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]