

Abstract
Purpose
Although the translocation t(4;14) is supposed to be a primary event in multiple myeloma, we have been surprised to observe that in large relapse series of patients, the t(4;14) can be observed only in subpopulations of plasma cells, in contrast to what is seen at diagnosis. This observation raised the question of possible subclones harboring the translocation that would be observable only at the time of relapse.
Experimental Design
To address this issue, we analyzed by FISH a cohort of 306 patients for whom we had at least two samples obtained at different disease phases.
Results
We observed a “gain” of the t(4;14) in 14 patients, and conversely, a “loss” of the translocation in 11 patients. Two hypotheses were raised: either an acquisition of the translocation during evolution, or the existence of small t(4;14)-positive subclones at the time of diagnosis. To address this question, we had (1) the opportunity to analyze two patients at the time of diagnosis by RT-PCR to look for the chimeric Eμ-MMSET transcript, and one patient positive at diagnosis, but negative at relapse. The samples were positive, supporting the second hypothesis. Furthermore, the IGH sequences of two patients who “lose” the t(4;14) were identical at diagnosis and relapse, confirming the existence of a common ancestral clone.
Conclusion
Thus, the conclusion of this study is that the t(4;14) is not a primary event in multiple myeloma, and that it can be present in silent subclones at diagnosis, but also at relapse.
Introduction
Multiple myeloma is characterized by a huge heterogeneity at all the levels, clinically, biologically, and for outcome. This heterogeneity is supposed to be supported by a wide variability of genetic lesions observed within the malignant plasma cells (1). Several models have been proposed to try to separate multiple myeloma in several entities. Even though none of them is perfect, the most achieved model has been proposed by Bergsagel and Kuëhl in 2005 (2). This model suggests primary events, like trisomies of odd chromosomes, and rearrangements involving the IGH gene. In contrast, other abnormalities are considered as secondary events, like loss of chromosome 13, 17p deletions or MYC rearrangements. In agreement with this theory, IGH translocations are observed in the very large majority of the plasma cells at diagnosis, whereas monosomy 13 or del(17p) are frequently found only in subclones.
Very recently, we and others did show that myeloma was not a totally clonal disease (3–5). Using different approaches, all these reports showed that relapse can be due to a subclone partially different from the one observed at diagnosis, suggesting the existence of an “ancestral” clone. In this model, all the subclones are genetically related, but treatment may select one or the others at different stages of the disease history.
This latter model raises the question whether IGH translocations could be present at diagnosis only in minor subclones, but present in the major clone at progression. To even more support this hypothesis, we have been surprised to observe in large relapse trials that the t(4;14) translocation could be observed only in subpopulations, in contrast to what is observed at diagnosis. To definitely address this issue, we decided to screen for t(4;14) a large cohort of patients for whom diagnostic and relapse samples were available.
Patients, Materials, and Methods
We first searched in the IFM database for patients with at least two samples obtained at different times of the disease history. We found 306 patients responding to this criterion. They were 38% females and 62% males, with a median age of 57 years (range 47–74). Eighty-five % of them were treated at diagnosis with intensive approaches at diagnosis. Eighty-two % received a VAD induction (Vincristin-Adriamycin-Dexamethasone), and 18% received a bortezomib-based induction. Fifteen % of the patients (median age = 72 years, range = 66–74) received a melphalan-prednisone (MP)-based treatment, combined with either thalidomide (19 patients), or with bortezomib (25 patients). Among these 306 patients, 38 presented a t(4;14) at diagnosis.
For all the patients, a bone marrow aspirate was shipped to the central laboratory using overnight courier. Upon receipt, mononuclear bone marrow cells were separated using Ficoll-Hypaque. Then, plasma cells were sorted using anti-CD138-coated magnetic beads (Miltenyi Biotec, Paris, France, or StemCell Technologies, Vancouver, Canada). Only samples with at least 90% of plasma cells were kept for further analyses. Fluorescence in situ hybridization (FISH) was performed as previously described (6). Briefly, sorted plasma cells were fixed in Carnoy’s fixative and stored at −20°C until hybridization. After slide preparation, they were denatured in 70% formamide for 5 minutes, dehydrated in 70%, 85%, 100% ethanol series. The probe specific for the t(4;14) was purchased from Abbott Molecular (Paris, France), and denatured separately for 5 minutes at 75°C. After denaturation, the probe was dropped on the plasma cells, and hybridized overnight at 37°C. Then, coverslips were removed and the slides were washed 2 minutes in 2×SSC-0.1% Triton at 75°C. All the relapse samples were analyzed for the t(4;14), independently of the diagnosis result. For RT-PCR analyses, we used specific primers for IGH and MMSET, as previously described by Malderini (7). Finally, IGH sequences were performed according to routine practice.
Results
Among the 268 patients who did not display the t(4;14) at diagnosis, we observed the translocation in 14 patients at the time of relapse. The translocation was observed in a median of 56% of the plasma cells (range = 30–88%) (positivity cut-off= 20%). For all these patients with “acquisition” of the translocation at relapse, we of course re-hybridized the diagnosis sample to verify the negativity. In all cases, the absence of t(4;14) was confirmed. On the opposite, we observed a “loss” of the t(4;14) in 11 patients. At this time, two hypotheses were possible: either the acquisition of the t(4;14) during evolution, or the presence of minor t(4;14)-positive subclones at the time of diagnosis. To address this question, we looked for non-fixed frozen plasma cells at diagnosis for these 25 patients. We found samples for two patients who “lost” the t(4;14), and for one patient who “gained” the translocation in our biobank. RT-PCR analysis for IGH-MMSET revealed the presence of the chimeric Eμ-MMSET transcript in all the samples, confirming the second hypothesis. Of note, one sample was positive only by nested RT-PCR. Furthermore, the analysis of the productive IGH rearrangements in three patients (two “losing” the t(4;14), and one gaining it) from the fixed FISH pellets showed the same clonal sequence, confirming the hypothesis of an ancestral clone.
Discussion
During the past decade, important efforts have been produced to understand the oncogenesis of multiple myeloma (8–12). Essentially based on gene expression profiling data, several classifications have been proposed (13–15). The most powerful model has been proposed by Bergsagel and Kuëhl.2 This model does identify primary and secondary genetic events. Early oncogenesis is driven by two different (almost) exclusive genetic events: gains of whole chromosomes leading to hyperdiploidy, and translocations involving the IGH gene at 14q32. These latter events are supposed to occur by errors during the VDJ rearrangements, whereas hyperdiploidy cause is not known. This model is experimentally confirmed by the observation that IGH translocations are present in the large majority of the plasma cells at diagnosis, in agreement with a primary event. Data are less demonstrated for hyperdiploidy since hyperdiploidy is not routinely analyzed at diagnosis.
During the past year, two groups (IFM and Mayo Clinic) did demonstrate another level of heterogeneity at the patient level (3–5). They showed that relapses are sometimes due to clones related, but different from the diagnosis clone. Furthermore, with time, different clones are responsible for the different relapses, maybe selected by the various treatments used at each phase. Whether these findings modify the previous oncogenetic model is not sure. It is totally conceivable that the primary events occur in the ancestral clone, as shown in Egan’s paper (4), showing that the t(4;14) was present in all subclones.
Our data significantly modify these concepts. Primarily, we have been alerted by the fact that in relapse trials including cytogenetic analyses, the t(4;14), when present, was often observed in only subclones (sometimes as few as 30%), in contrast to the situation seen at diagnosis. Since cytogenetics at diagnosis was not available, three hypotheses were raised. The first one was that the t(4;14) was present at diagnosis, but lost in some plasma cells at relapse because of subsequent chromosomal rearrangements that could affect the der(4) chromosome. The second hypothesis was the absence of the t(4;14) at diagnosis, and the acquisition of the translocation during evolution. Finally, the third hypothesis was the existence of subclones, and that the original principal clone was not the one harboring the t(4;14). Whereas the first hypothesis was compatible with the Bergsagel and Kuëhl’s model, the two others were not compatible with a primary event.
To address this important question, we did select patients for whom at least two samples were available in the IFM biobank. We identified 306 patients responding to this condition. All of them have been tested at diagnosis for the t(4;14), which was found in 12.4% of the patients. We performed FISH for the t(4;14) on all the relapse samples. The data confirmed our first hypothesis that the t(4;14) can be observed only at relapse, but also that it can disappear at relapse, at least at the FISH level. Thus, only the two last hypotheses were still plausible. The only way to answer this question was to use a sensitive method to test the presence of the t(4;14) in minor subclones at diagnosis. We found two patients for whom non-fixed frozen plasma cells from the diagnosis were available. Using sensitive RT-PCR, we detected the chimeric Eμ-MMSET transcript specific of the t(4;14), confirming the third hypothesis, at least in these patients. We also had the opportunity to test one patient in whom the t(4;14) was not detected at the time of relapse, although present at diagnosis. The RT-PCR analysis for IGH-MMSET confirmed the presence of the rearrangement in some cells in the relapse sample. Finally, to definitely demonstrate this subclonal hypothesis, and to eliminate the possibility of the occurrence of a second myeloma (and also a possible mix in the FISH slides), we did show in three patients that the IGH sequences were exactly the same at both time points.
Our data have important implications. First, they clearly show that the t(4;14) is not a “primary” event, in the genetic sense. In these cases with occurrence at relapse, or disappearance at relapse, the translocation is not present in the ancestral clone. So the model should be reconsidered (Figure 1). Second, although the t(4;14) is supposed to confer aggressiveness, it can become apparent only at the time of relapse. What are the mechanisms that control this supposedly aggressive clone is not known. In contrast, the t(4;14) can be present at diagnosis, but “lost” at the time of relapse. Whether the t(4;14) subclone has been eliminated by the chemotherapy or just silenced until next relapse is an unanswered question. In one patient, we showed that the t(4;14) subclone was still present at relapse, even if not detectable by FISH. These issues may have important consequences for the management of the patients. For instance, are these “secondary” clones as aggressive as the ones seen at diagnosis? To conclude, our data bring another level of complexity in an already complex disease.
Figure 1.
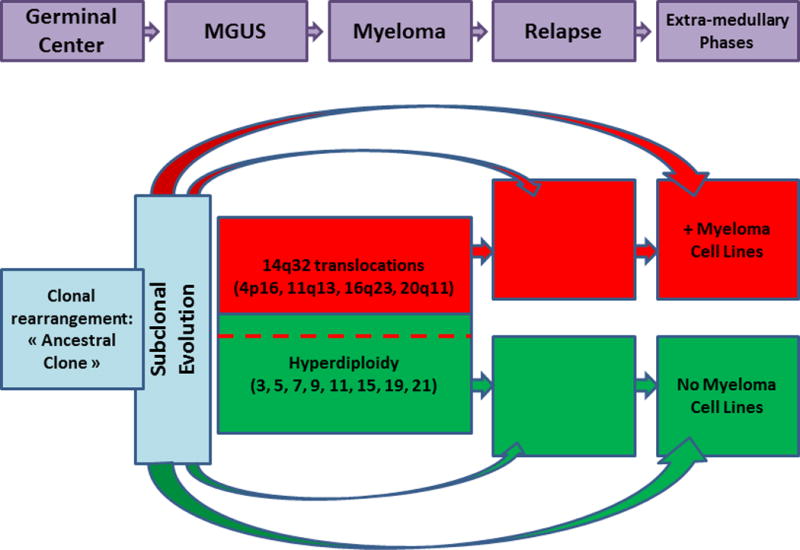
Model for myeloma oncogenesis
In this model, the first oncogenic rearrangements occur in the lymph node germinal centers, leading to immortalization of the clone, known as the “ancestral clone”. This clone will evolve to related subclones displaying different molecular changes. Then, two oncogenic pathways are observed, one based on translocations involving the IGH gene, and the second based on the acquisition of multiple chromosomal gains leading to hyperdiplidy. These two pathways are not totally exclusive, few patients with IGH translocations presenting also hyperdiploidy. At the time of relapse or subsequent extramedullary evolution, different scenarios can be observed: either relapse from the same clone observed at diagnosis, or clonal evolution from this clone, or relapse from an ancestral subclone. Of note, only patients engaged in the IGH translocation pathway can generate human myeloma cell lines.
Statement of Translational Relevance.
This study clearly shows that the t(4;14), a well-known poor prognosis factor, can be observed only at the time of relapse, or, conversely, be the major clone at diagnosis, but not at relapse. These findings have major implications for the oncogenesis of multiple myeloma, but also for clinical practice. Actually, since bortezomib has been shown to improve the outcome of these patients, the translocation has to be sought at diagnosis, but also at relapse, whatever the result at diagnosis.
Acknowledgments
This study is partially supported by a grant from the French Institut National du Cancer (INCa), and by a grant from the American NIH (PO1-155258).
Footnotes
Disclosures: We have no conflict of interest to disclose.
References
- 1.Avet-Loiseau H, Magrangeas F, Moreau P, Attal M, Facon T, Anderson K, et al. Molecular heterogeneity of multiple myeloma: Pathogenesis, prognosis, and therapeutic implications. J Clin Oncol. 2011;29:1893–7. doi: 10.1200/JCO.2010.32.8435. [DOI] [PubMed] [Google Scholar]
- 2.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J., Jr Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106:296–303. doi: 10.1182/blood-2005-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magrangeas F, Avet-Loiseau H, Gouraud W, Lodé L, Decaux O, Godmer P, et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia. 2012 doi: 10.1038/leu.2012.226. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Egan JB, Shi CX, Tembe W, Christoforides A, Kurdoglu A, Sinari S, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060–6. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120:1067–76. doi: 10.1182/blood-2012-01-405985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109:3489–95. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 7.Malgeri U, Baldini L, Perfetti V, Fabris S, Vignarelli MC, Colombo G, et al. Detection of t(4;14)(p16.3;q32) chromosomal translocation in multiple myeloma by reverse transcription-polymerase chain reaction analysis of IGH-MMSET fusion transcripts. Cancer Res. 2000;60:4058–61. [PubMed] [Google Scholar]
- 8.Kuehl M, Bergsagel PL. Multiple myeloma: evolving genetic events and host interactions. Nat Rev Cancer. 2002;2:175–87. doi: 10.1038/nrc746. [DOI] [PubMed] [Google Scholar]
- 9.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–31. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210–21. doi: 10.1038/leu.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Barlogie B, Shaughnessy JD., Jr The molecular characterization and clinical management of multiple myeloma in the post-genome era. Leukemia. 2009;23:1941–56. doi: 10.1038/leu.2009.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335–48. doi: 10.1038/nrc3257. [DOI] [PubMed] [Google Scholar]
- 13.Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–84. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 14.Zhan F, Barlogie B, Mulligan G, Shaughnessy JD, Jr, Bryant B. High-risk myeloma: a gene expression based risk-stratification model for newly diagnosed multiple myeloma treated with high-dose therapy is predictive of outcome in relapsed disease treated with single-agent bortezomib or high-dose dexamethasone. Blood. 2008;111:968–9. doi: 10.1182/blood-2007-10-119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broyl A, Hose D, Lokhorst H, de Knegt Y, Peeters J, Jauch A, et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood. 2010;116:2543–53. doi: 10.1182/blood-2009-12-261032. [DOI] [PubMed] [Google Scholar]