

Abstract
RNA sequencing (RNA-Seq) uses the capabilities of high-throughput sequencing methods to provide insight into the transcriptome of a cell. Compared to previous Sanger sequencing- and microarray-based methods, RNA-Seq provides far higher coverage and greater resolution of the dynamic nature of the transcriptome. Beyond quantifying gene expression, the data generated by RNA-Seq facilitate the discovery of novel transcripts, identification of alternatively spliced genes, and detection of allele-specific expression. Recent advances in the RNA-Seq workflow, from sample preparation to library construction to data analysis, have enabled researchers to further elucidate the functional complexity of the transcription. In addition to polyadenylated messenger RNA (mRNA) transcripts, RNA-Seq can be applied to investigate different populations of RNA, including total RNA, pre-mRNA, and noncoding RNA, such as microRNA and long ncRNA. This article provides an introduction to RNA-Seq methods, including applications, experimental design, and technical challenges.
Introduction
The central dogma of molecular biology outlines the flow of information that is stored in genes as DNA, transcribed into RNA, and finally translated into proteins (Crick 1958; Crick 1970). The ultimate expression of this genetic information modified by environmental factors characterizes the phenotype of an organism. The transcription of a subset of genes into complementary RNA molecules specifies a cell's identity and regulates the biological activities within the cell. Collectively defined as the transcriptome, these RNA molecules are essential for interpreting the functional elements of the genome and understanding development and disease.
The transcriptome has a high degree of complexity and encompasses multiple types of coding and noncoding RNA species. Historically, RNA molecules were relegated as a simple intermediate between genes and proteins, as encapsulated in the central dogma of molecular biology. Therefore, messenger RNA (mRNA) molecules were the most frequently studied RNA species because they encoded proteins via the genetic code. In addition to protein-coding mRNA, there is a diverse group of noncoding RNA (ncRNA) molecules that are functional. Previously, most known ncRNAs fulfilled basic cellular functions, such as ribosomal RNAs and transfer RNAs involved in mRNA translation, small nuclear RNA (snRNAs) involved in splicing, and small nucleolar RNAs (snoRNAs) involved in the modification of rRNAs (Mattick and Makunin 2006). More recently, novel classes of RNA have been discovered, enhancing the repertoire of ncRNAs. For instance, one such class of ncRNAs is small noncoding RNAs, which include microRNA (miRNA) and piwi-interacting RNA (piRNA), both of which regulate gene expression at the posttranscriptional level (Stefani and Slack 2008). Another noteworthy class of ncRNAs is long noncoding RNAs (lncRNAs). As a functional class, lncRNAs were first described in mice during the large-scale sequencing of cDNA libraries (Okazaki et al. 2002). A myriad of molecular functions have been discovered for lncRNAs, including chromatin remodeling, transcriptional control, and posttranscriptional processing, although the vast majority are not fully characterized (Guttman et al. 2009; Mercer et al. 2009; Wilusz et al. 2009).
Initial gene expression studies relied on low-throughput methods, such as northern blots and quantitative polymerase chain reaction (qPCR), that are limited to measuring single transcripts. Over the last two decades, methods have evolved to enable genome-wide quantification of gene expression, or better known as transcriptomics. The first transcriptomics studies were performed using hybridization-based microarray technologies, which provide a high-throughput option at relatively low cost (Schena et al. 1995). However, these methods have several limitations: the requirement for a priori knowledge of the sequences being interrogated; problematic cross-hybridization artifacts in the analysis of highly similar sequences; and limited ability to accurately quantify lowly expressed and very highly expressed genes (Casneuf et al. 2007; Shendure 2008). In contrast to hybridization-based methods, sequence-based approaches have been developed to elucidate the transcriptome by directly determining the transcript sequence. Initially, the generation of expressed sequence tag (EST) libraries by Sanger sequencing of complementary DNA (cDNA) was used in gene expression studies, but this approach is relatively low-throughput and not ideal for quantifying transcripts (Adams et al. 1991, 1995; Itoh et al. 1994). To overcome these technical constraints, tag-based methods such as serial analysis of gene expression (SAGE) and cap analysis gene expression (CAGE) were developed to enable higher throughput and more precise quantification of expression levels. By quantifying the number of tagged sequences, which directly corresponded to the number of mRNA transcripts, these tag-based methods provide a distinct advantage over measuring analog-style intensities as in array-based methods (Velculescu et al. 1995; Shiraki et al. 2003). However, these assays are insensitive to measuring expression levels of splice isoforms and cannot be used for novel gene discovery. In addition, the laborious cloning of sequence tags, the high cost of automated Sanger sequencing, and the requirement for large amounts of input RNA have greatly limited its use.
The development of high-throughput next-generation sequencing (NGS) has revolutionized transcriptomics by enabling RNA analysis through the sequencing of complementary DNA (cDNA) (Wang et al. 2009). This method, termed RNA sequencing (RNA-Seq), has distinct advantages over previous approaches and has revolutionized our understanding of the complex and dynamic nature of the transcriptome. RNA-Seq provides a more detailed and quantitative view of gene expression, alternative splicing, and allele-specific expression. Recent advances in the RNA-Seq workflow, from sample preparation to sequencing platforms to bioinformatic data analysis, has enabled deep profiling of the transcriptome and the opportunity to elucidate different physiological and pathological conditions. In this article we will provide an introduction to RNA sequencing and analysis using next-generation sequencing methods and discusses how to apply these advances for more comprehensive and detailed transcriptome analyses.
Transcriptome Sequencing
The introduction of high-throughput next-generation sequencing (NGS) technologies revolutionized transcriptomics. This technological development eliminated many challenges posed by hybridization-based microarrays and Sanger sequencing-based approaches that were previously used for measuring gene expression. A typical RNA-Seq experiment consists of isolating RNA, converting it to complementary DNA (cDNA), preparing the sequencing library, and sequencing it on an NGS platform (Fig. 1). However, many experimental details, dependent on a researcher's objectives, should be considered before performing RNA-Seq. These include the use of biological and technical replicates, depth of sequencing, and desired coverage across the transcriptome. In some cases, these experimental options will have minimal impact on the quality of the data. However, in many cases the researcher must carefully design the experiment, placing a priority on the balance between high-quality results and the time and monetary investment.
Figure 1.
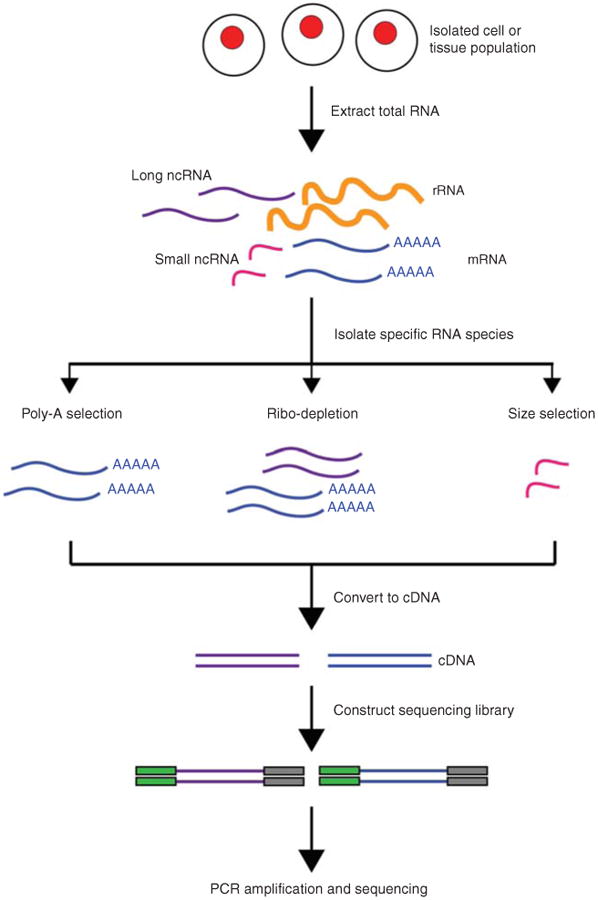
Overview of RNA-Seq. First, RNA is extracted from the biological material of choice (e.g., cells, tissues). Second, subsets of RNA molecules are isolated using a specific protocol, such as the poly-A selection protocol to enrich for polyadenylated transcripts or a ribo-depletion protocol to remove ribosomal RNAs. Next, the RNA is converted to complementary DNA (cDNA) by reverse transcription and sequencing adaptors are ligated to the ends of the cDNA fragments. Following amplification by PCR, the RNA-Seq library is ready for sequencing.
Isolation of RNA
The first step in transcriptome sequencing is the isolation of RNA from a biological sample. To ensure a successful RNA-Seq experiment, the RNA should be of sufficient quality to produce a library for sequencing. The quality of RNA is typically measured using an Agilent Bioanalyzer, which produces an RNA Integrity Number (RIN) between 1 and 10 with 10 being the highest quality samples showing the least degradation. The RIN estimates sample integrity using gel electrophoresis and analysis of the ratios of 28S to 18S ribosomal bands. Note that the RIN measures are based on mammalian organisms and certain species with abnormal ribosomal ratios (i.e., insects) may erroneously generate poor RIN numbers. Low-quality RNA (RIN < 6) can substantially affect the sequencing results (e.g., uneven gene coverage, 3′–5′ transcript bias, etc.) and lead to erroneous biological conclusions. Therefore, high-quality RNA is essential for successful RNA-Seq experiments. Unfortunately, high-quality RNA samples may not be available in some cases, such as human autopsy samples or paraffin embedded tissues, and the effect of degraded RNA on the sequencing results should be carefully considered (Tomita et al. 2004; Thompson et al. 2007; Rudloff et al. 2010).
Library Preparation Methods
Following RNA isolation, the next step in transcriptome sequencing is the creation of an RNA-Seq library, which can vary by the selection of RNA species and between NGS platforms. The construction of sequencing libraries principally involves isolating the desired RNA molecules, reverse-transcribing the RNA to cDNA, fragmenting or amplifying randomly primed cDNA molecules, and ligating sequencing adaptors. Within these basic steps, there are several choices in library construction and experimental design that must be carefully made depending on the specific needs of the researcher (Table 1). Additionally, the accuracy of detection for specific types of RNAs is largely dependent on the nature of the library construction. Although there are a few basic steps for preparing RNA-Seq libraries, each stage can be manipulated to enhance the detection of certain transcripts while limiting the ability to detect other transcripts.
Table 1. RNA-Seq library protocols.
| Library design | Usage | Description |
|---|---|---|
| Poly-A selection | Sequencing mRNA | Select for RNA species with poly-A tail and enriches for mRNA |
| Ribo-depletion | Sequencing mRNA, pre-mRNA, ncRNA | Removes ribosomal RNA and enriches for mRNA, pre-mRNA, and ncRNA |
| Size selection | Sequencing miRNA | Selects RNA species using size fractionation by gel electrophoresis |
| Duplex-specific nuclease | Reduce highly abundant transcripts | Cleaves highly abundant transcripts, including rRNA and other highly expressed genes |
| Strand-specific | De novo transcriptome assembly | Preserves strand information of the transcript |
| Multiplexed | Sequencing multiple samples together | Genetic barcoding method that enables sequencing multiple samples together |
| Short-read | Higher coverage | Produces 50–100 bp reads; generally higher read coverage and reduced error rate compared to long-read sequencing |
| Long-read | De novo transcriptome assembly | Produces >1000 bp reads; advantageous for resolving splice junctions and repetitive regions |
Selection of RNA Species
Before constructing RNA-Seq libraries, one must choose an appropriate library preparation protocol that will enrich or deplete a “total” RNA sample for particular RNA species. The total RNA pool includes ribosomal RNA (rRNA), precursor messenger RNA (pre-mRNA), mRNA, and various classes of noncoding RNA (ncRNA). In most cell types, the majority of RNA molecules are rRNA, typically accounting for over 95% of the total cellular RNA. If the rRNA transcripts are not removed before library construction, they will consume the bulk of the sequencing reads, reducing the overall depth of sequence coverage and thus limiting the detection of other less-abundant RNAs. Because the efficient removal of rRNA is critical for successful transcriptome profiling, many protocols focus on enriching for mRNA molecules before library construction by selecting for polyadenylated (poly-A) RNAs. In this approach, the 3′ poly-A tail of mRNA molecules is targeted using poly-T oligos that are covalently attached to a given substrate (e.g., magnetic beads). Alternatively, researchers can selectively deplete rRNA using commercially available kits, such as RiboMinus (Life Technologies) or RiboZero (Epicentre). This latter method facilitates the accurate quantification of noncoding RNA species, which may be polyadenylated and thus excluded from poly-A libraries. Lastly, highly abundant RNA can be removed by denaturing and re-annealing double-stranded cDNA in the presence of duplex-specific nucleases that preferentially digest the most abundant species, which re-anneal as double-stranded molecules more rapidly than less-abundant molecules (Christodoulou et al. 2011). This method can also be used to remove other highly abundant mRNA transcripts in samples, such as hemoglobin in whole blood, immunoglobulins in mature B cells, and insulin in pancreatic beta cells.
A comprehensive understanding of the technical biases and limitations surrounding each methodological approach is essential for selecting the best method for library preparation. For example, poly-A libraries are the superior choice if one is solely interested in coding RNA molecules. Conversely, ribo-depletion libraries are a more appropriate choice for accurately quantifying noncoding RNA as well as pre-mRNA that has not been posttranscriptionally modified. Furthermore, moderate differences exist between ribo-depletion protocols, such as the efficiency of rRNA removal and differential coverage of small genes, which should be investigated before selecting a method (Huang et al. 2011).
In addition to the selective depletion of specific RNA species, new approaches have been developed to selectively enrich for regions of interest. These approaches include methods employing PCR-based approaches, hybrid capture, in-solution capture, and molecular inversion probes (Querfurth et al. 2012). The hybridization-based in solution capture involves a set of biotinylated RNA baits transcribed from DNA template oligo libraries that contain sequences corresponding to particular genes of interest. The RNA baits are combined with the RNA-Seq library where they hybridize to RNA sequences that are complementary to the baits, and the bounded complexes are recovered using streptavidin-coated beads. The resulting RNA-Seq library is now enriched for sequences corresponding to the baits and yet retains its gene expression information despite the removal of other RNA species (Levin et al. 2009). The approach enables researchers to reduce sequencing costs by sequencing selected regions in a greater number of samples.
Selection of Small RNA Species
Complementing the library preparation protocols discussed above, more specific protocols have been developed to selectively target small RNA species, which are key regulators of gene expression. Small RNA species include microRNA (miRNA), small interfering RNA (siRNA), and piwi-interacting RNA (piRNA). Because small RNAs are lowly abundant, short in length (15–30 nt), and lack polyadenylation, a separate strategy is often preferred to profile these RNA species (Morin et al. 2010). Similar to total RNA isolation, commercially available extraction kits have been developed to isolate small RNA species. Most kits involve isolation of small RNAs by size fractionation using gel electrophoresis. Size fractionation of small RNAs requires involves running the total RNA on a gel, cutting a gel slice in the 14–30 nucleotide region, and purifying the gel slice. For higher concentrations of small RNAs, the excised gel slice can be concentrated by ethanol precipitation. An alternative to gel electrophoresis is the use of silica spin columns, which bind and elute small RNAs from a silica column. After isolation of small RNAs species from total RNA, the RNA is ready for cDNA synthesis and primer ligation.
cDNA Synthesis
Universal to all RNA-Seq preparation methods is the conversion of RNA into cDNA because most sequencing technologies require DNA libraries. Most protocols for cDNA synthesis create libraries that were uniformly derived from each cDNA strand, thus representing the parent mRNA strand and its complement. In this conventional approach, the strand orientation of the original RNA is lost as the sequencing reads derived from each cDNA strand are indistinguishable in an effort to maximize efficiency of reverse transcription. However, strand information can be particularly valuable for distinguishing overlapping transcripts on opposite strands, which is critical for de novo transcript discovery (Parkhomchuk et al. 2009; Vivancos et al. 2010; Mills et al. 2013). Therefore, alternative library preparation protocols have since been developed that yield strand-specific reads. One strategy to preserve strand information is to ligate adapters in predetermined directions to single-stranded RNA or the first-strand of cDNA (Lister et al. 2008). Unfortunately, this approach is laborious and results in coverage bias at both the 5′ and 3′ ends of cDNA molecules. The preferred strategy to preserve strandedness is to incorporate a chemical label such as deoxy-UTP (dUTP) during synthesis of the second-strand cDNA that can be specifically removed by enzymatic digestion (Parkhomchuk et al. 2009). During library construction, this facilitates distinguishing the second-strand cDNA from the first strand. Although this approach is favored, the validity of antisense transcripts near highly expressed genes should be measured with caution because a small amount of reads (∼1%) have been observed from the opposite strand (Zeng and Mortazavi 2012).
Multiplexing
Another consideration for constructing cost-effective RNA-Seq libraries is assaying multiple indexed samples in a single sequencing lane. The large number of reads that can be generated per sequencing run (e.g., a single lane of an Illumina HiSeq 2500 generates up to 750 million paired-end reads) permits the analysis of increasingly complex samples. However, increasingly high sequencing depths provide diminishing returns for lower complexity samples, resulting in oversampling with minimal improvement in data quality (Smith et al. 2010). Therefore, an affordable and efficient solution is to introduce unique 6-bp indices, also known as “barcodes,” to each RNA-Seq library. This enables the pooling and sequencing of multiple samples in the same sequencing reaction because the barcodes identify which sample the read originated from. Depending on the application, adequate transcriptome coverage can be attained for 2–20 samples (Birney et al. 2007; Blencowe et al. 2009). To detect transcripts of moderate to high abundance, ∼30–40 million reads are required to accurately quantify gene expression. To obtain coverage over the full-sequence diversity of complex transcript libraries, including rare and lowly-expressed transcripts, up to 500 million reads is required (Fu et al. 2014). As such, for any given study it is important to consider the level of sequencing depth required to answer experimental questions with confidence while efficiently using NGS resources.
Quantitative Standards
Although RNA-Seq is a widely used technique for transcriptome profiling, the rapid development of sequencing technologies and methods raises questions about the performance of different platforms and protocols. Variation in RNA-Seq data can be attributed to an assortment of factors, ranging from the NGS platform used to the quality of input RNA to the individual performing the experiment. To control for these sources of technical variability, many laboratories use positive controls or “spike-ins” for sequencing libraries. The External RNA Controls Consortium (ERCC) developed a set of universal RNA synthetic spike-in standards for microarray and RNA-Seq experiments (Jiang et al. 2011; Zook et al. 2012). The spike-ins consist of a set of 96 DNA plasmids with 273–2022 bp standard sequences inserted into a vector of ∼2800 bp. The spike-in standard sequences are added to sequencing libraries at different concentrations to assess coverage, quantification, and sensitivity. These RNA standards serve as an effective quality control tool for separating technical variability from biological variability detected in differential transcriptome profiling studies.
Selection of Tissue or Cell Populations
When beginning an RNA-Seq experiment, one of the initial considerations is the choice of biological material to be used for library construction and sequencing. This choice is not trivial considering there are hundreds of cell types in over 200 different tissues that make up greater than 50 unique organs in humans alone. In addition to spatial (e.g., cell- and tissue-type) specificity, gene expression shows temporal specificity, such that different developmental stages will show unique expression signatures. Ultimately, the biological material chosen will be dependent on both the experimental goals and feasibility. For example, the tissue of choice for an investigation of unique gene expression signatures in colon cancer, the tissue choice is clear. However, for research studies investigating variation in gene expression across individuals in a population, the choice of biological material is less apparent and will likely depend on the feasibility of obtaining the biological samples (e.g., blood draws are less invasive and easier to perform than tissue biopsies).
Handling Tissue Heterogeneity
Another consideration when selecting the biological source of RNA is the heterogeneity of tissues. The accuracy of gene expression quantification is dependent on the purity of samples. In fact, the heterogeneity can substantially impact estimations of transcript abundances in samples composed of multiple cell types. Most tissue samples isolated from the human body are heterogeneous by nature. Furthermore, pathological tissue samples are often composed of disease-state cells surrounded by normal cells. To isolate distinct cell types, experimental methods have been developed, including laser-capture microdissection and cell purification. Laser-capture microdissection enables the isolation of cell types that are morphologically distinguishable under direct microscopic visualization (Emmert-Buck et al. 1996). Although this technique yields high-quality RNA, the total yield is low and requires PCR amplification, thereby introducing amplification biases and creating less distinguishable expression profiles across different cell types (Kube et al. 2007). Cell purification and enrichment protocols are also available, such as differential centrifugation and fluorescence-activated cell sorting (Cantor et al. 1975). In conjunction with RNA-Seq, these experimental methods have overcome previous technical limitations and enable researchers to uncover unique expression signatures across specific cell-types and developmental stages (Moran et al. 2012; Nica et al. 2013). In addition to these experimental methods, in silico probabilistic models can be applied in downstream analysis to differentiate the transcript abundances of distinct cells from RNA-Seq data of heterogeneous tissue samples (Erkkila et al. 2010; Li and Xie 2013). Interestingly, in some cases, the sample heterogeneity can have advantages in transcriptome profiling by identifying novel pathways, implicating cellular origins of disease, or identifying previously unknown pathological sites (Alizadeh et al. 2000; Khan et al. 2001; Sorlie et al. 2001).
Single-Cell Transcriptomics
Beyond tissue heterogeneity, considerable evidence indicates that cell-to-cell variability in gene expression is ubiquitous, even within phenotypically homogeneous cell populations (Huang 2009). Unfortunately, conventional RNA-Seq studies do not capture the transcriptomic composition of individual cells. The transcriptome of a single cell is highly dynamic, reflecting its functionality and responses to ever-changing stimuli. In addition to cellular heterogeneity resulting from regulation, individual cells show transcriptional “noise” that arises from the kinetics of mRNA synthesis and decay (Yang et al. 2003; Sun et al. 2012). Furthermore, genes that show mutually exclusive expression in individual cells may be observed as genes showing co-expression in expression analyses of bulk cell populations.
To uncover cell-to-cell variation within populations, significant efforts have been invested in developing single-cell RNA-Seq methods. The biggest challenge has been extending the limits of library preparation to accommodate extremely low input RNA. A human cell contains <1 pg of mRNA (Kawasaki 2004), whereas most sequencing protocols such as Illumina's TruSeq RNA-Seq kit recommends 400 ng to 1 μg of input RNA material. Various single-cell RNA amplification methods have been developed to accommodate less input RNA (Tang et al. 2009, 2010; Hashimshony et al. 2012; Islam et al. 2012; Picelli et al. 2013; Sasagawa et al. 2013; Shalek et al. 2013). The key limiting factors in the detection of transcripts in single cells are cDNA synthesis and PCR amplification. The efficiency of RNA-to-cDNA conversion is imperfect, estimated to be as low as 5%–25% of all transcripts (Islam et al. 2012). In addition, PCR amplification methods do not linearly amplify transcript and are prone to introduce biases based on the nucleic acid composition of different transcripts, ultimately altering the relative abundance of these transcripts in the sequencing library. Methods that avoid PCR amplification steps, such as CEL-Seq, through linear in vitro amplification of the transcriptome can avoid these biases (Hashimshony et al. 2012). In addition, the use of nanoliter-scale reaction volumes with microfluidic devices as opposed to microliter-scale reactions can reduce biases that arise during sample preparation (Wu et al. 2014). Although single-cell methods are still under active development, quantitative assessments of these techniques indicate that obtaining accurate transcriptome measurements by single-cell RNA-Seq is possible after accounting for technical noise (Brennecke et al. 2013; Wu et al. 2014). These methods will undoubtedly be important for uncovering oscillatory and heterogeneous gene expression within single-cell types, as well as identifying cell-specific biomarkers that further our understanding of biology across many physiological and pathological conditions.
Sequencing Platforms for Transcriptomics
When designing an RNA-Seq experiment, the selection of a sequencing platform is important and dependent on the experimental goals. Currently, several NGS platforms are commercially available and other platforms are under active technological development (Metzker 2010). The majority of high-throughput sequencing platforms use a sequencing-by-synthesis method to sequence tens of millions of sequence clusters in parallel. The NGS platforms can often be categorized as either ensemble-based (i.e. sequencing many identical copies of a DNA molecule) or single-molecule-based (i.e. sequencing a single DNA molecule). The differences between these sequencing techniques and platforms can affect downstream analysis and interpretation of the sequencing data.
In recent years, the sequencing industry has been dominated by Illumina, which applies an ensemble-based sequencing-by-synthesis approach (Bentley et al. 2008). Using fluorescently labeled reversible-terminator nucleotides, DNA molecules are clonally amplified while immobilized on the surface of a glass flowcell. Because molecules are clonally amplified, this approach provides the relative RNA expression levels of genes. To remove potential PCR-amplification biases, PCR controls and specific steps in the downstream computational analysis are required. One major benefit of ensemble-based platforms is low sequencing error rates (<1%) dominated by single mismatches. Low error rates are particularly important for sequencing miRNAs, whose relatively small sizes result in misalignment or loss of reads if error rates are too high. Currently, the Illumina HiSeq platform is the most commonly applied next-generation sequencing technology for RNA-Seq and has set the standard for NGS sequencing. The platform has two flow cells, each providing eight separate lanes for sequencing reactions to occur. The sequencing reactions can take between 1.5 and 12 d to complete, depending on the total read length of the library. Even more recently, Illumina released the MiSeq, a desktop sequencer with lower throughput but faster turnaround (generates ∼30 million paired-end reads in 24 h). The simplified workflow of the MiSeq instrument offers rapid turnaround time for transcriptome sequencing on a smaller scale.
Single-molecule-based platforms such as PacBio enable single-molecule real-time (SMRT) sequencing (Eid et al. 2009). This approach uses DNA polymerase to perform uninterrupted template-directed synthesis using fluorescently labeled nucleosides. As each base is enzymatically incorporated into a growing DNA strand, a distinctive pulse of fluorescence is detected in real-time by zero-mode waveguide nanostructure arrays. An advantage of SMRT is that it does not include a PCR amplification step, thereby avoiding amplification bias and improving uniform coverage across the transcriptome. Another advantage of this sequencing approach is the ability to produce extraordinarily long reads with average lengths of 4200 to 8500 bp, which greatly improves the detection of novel transcript structures (Au et al. 2013; Sharon et al. 2013). A critical disadvantage of SMRT is a high rate of errors (∼5%) that are predominately characterized by insertions and deletions (Carneiro et al. 2012); the high error rate results in misalignment and loss of sequencing reads due to the difficulty of matching erroneous reads to the reference genome.
Another important consideration for choosing a sequencing platform is transcriptome assembly. Transcriptome assembly, which is discussed in greater detail later, is necessary to transform a collection of short sequencing reads into a set of full-length transcripts. In general, longer sequencing reads make it simpler to accurately and unambiguously assemble transcripts, as well as identify splicing isoforms. The extremely long reads generated by the PacBio platform are ideal for de novo transcriptome assembly in which the reads are not aligned to a reference transcriptome. The longer reads will facilitate an accurate detection of alternative splice isoforms, which may not be discovered with shorter reads. Moleculo, a company acquired by Illumina, has developed long-read sequencing technology capable of producing 8500 bp reads. Although it has yet to be widely adopted for transcriptome sequencing, the long reads aid transcriptome assembly. Lastly, Illumina has developed protocols for its desktops MiSeq to sequence slightly longer reads (up to 350 bp). Although much shorter than PacBio and Moleculo reads, the longer MiSeq reads can also be used to improve both de novo and reference transcriptome assembly.
Transcriptome Analysis
Gene expression profiling by RNA-Seq provides an unprecedented high-resolution view of the global transcriptional landscape. As the sequencing technologies and protocol methodologies continually evolve, new informatics challenges and applications develop. Beyond surveying gene expression levels, RNA-Seq can also be applied to discover novel gene structures, alternatively spliced isoforms, and allele-specific expression (ASE). In addition, genetic studies of gene expression using RNA-Seq have observed genetically correlated variability in expression, splicing, and ASE (Montgomery et al. 2010; Pickrell et al. 2010; Battle et al. 2013; Lappalainen et al. 2013). This section will introduce how expression data are analyzed to provide greater insight into the extensive complexity of transcriptomes.
RNA-Sequencing Data Analysis Workflow
The conventional pipeline for RNA-Seq data includes generating FASTQ-format files contains reads sequenced from an NGS platform, aligning these reads to an annotated reference genome, and quantifying expression of genes (Fig. 2). Although basic sequencing analysis tools are more accessible than ever, RNA-Seq analysis presents unique computational challenges not encountered in other sequencing-based analyses and requires specific consideration to the biases inherent in expression data.
Figure 2.
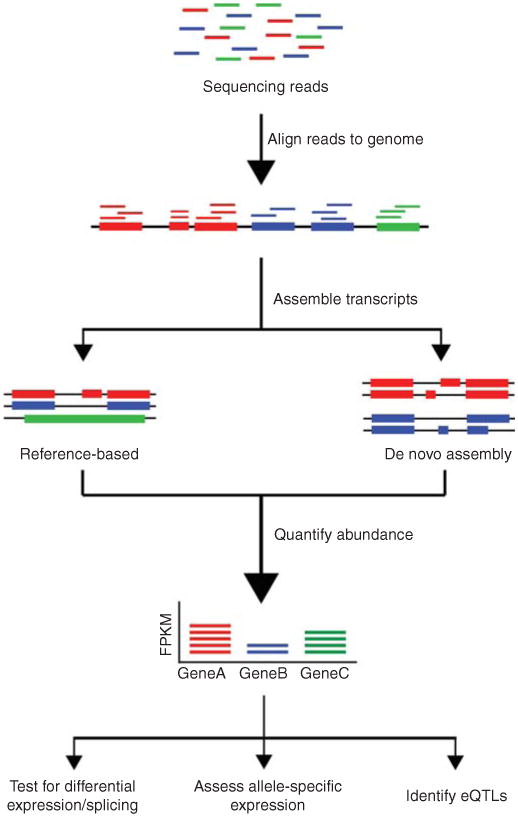
Overview of RNA-Seq data analysis. Following typical RNA-Seq experiments, reads are first aligned to a reference genome. Second, the reads may be assembled into transcripts using reference transcript annotations or de novo assembly approaches. Next, the expression level of each gene is estimated by counting the number of reads that align to each exon or full-length transcript. Downstream analyses with RNA-Seq data include testing for differential expression between samples, detecting allele-specific expression, and identifying expression quantitative trait loci (eQTLs).
Read Alignment
Mapping RNA-Seq reads to the genome is considerably more challenging than mapping DNA sequencing reads because many reads map across splice junctions. In fact, conventional read mapping algorithms, such as Bowtie (Langmead et al. 2009) and BWA (Li and Durbin 2009), are not recommended for mapping RNA-Seq reads to the reference genome because of their inability to handle spliced transcripts. One approach to resolving this problem is to supplement the reference genome with sequences derived from exon–exon splice junctions acquired from known gene annotations (Mortazavi et al. 2008). A preferred strategy is to map reads with a “splicing-aware” aligner that can recognize the difference between a read aligning across an exon–intron boundary and a read with a short insertion. As RNA-Seq data have become more widely used, a number of splicing-aware mapping tools have been developed specifically for mapping transcriptome data. The more commonly used RNA-Seq alignment tools include GSNAP (Wu and Nacu 2010), MapSplice (Wang et al. 2010a), RUM (Grant et al. 2011), STAR (Dobin et al. 2013), and TopHat (Trapnell et al. 2009) (Table 2). Each aligner has different advantages in terms of performance, speed, and memory utilization. Selecting the best aligner to use depends on these metrics and the overall objectives of the RNA-Seq study. Efforts to systematically evaluate the performance of RNA-Seq aligners have been initiated by GENCODE's RNA-Seq Genome Annotation Assessment Project3 (RGASP3), which has found major performance difference between alignments tools on numerous benchmarks, including alignment yield, basewise accuracy, mismatch and gap placement, and exon junction discovery (Engstrom et al. 2013).
Table 2. Widely used RNA-Seq software packages.
| Primary category | Tool name | Notes |
|---|---|---|
| Splice-aware read alignment | GEM | Filtration-based approach to approximate string matching for alignment |
| GSNAP | Based on seed and extend alignment algorithm aware of complex variants | |
| MapSplice | Based on Burrows-Wheeler Transform (BWT) algorithm | |
| RUM | Integrates alignment tools Blat and Bowtie to increase accuracy | |
| STAR | Based on seed searching in an uncompressed suffix arrays followed by seed clustering and stitching procedure; fast but memory-intensive | |
| TopHat | Uses Bowtie, based on BWT, to align reads; resolves spliced reads using exons by split read mapping | |
| Transcript assembly and quantification | Cufflinks | Assembles transcripts to reference annotations or de novo and quantifies abundance |
| FluxCapacitor | Quantifies transcripts using reference annotations | |
| iReckon | Models novel isoforms and estimates their abundance | |
| Differential expression (DE) | BaySeq | Count-based approach using empirical Bayesian method to estimate posterior likelihoods |
| Cuffdiff2 | Isoform-based approach based on beta negative binomial distribution | |
| DESeq | Exon-based approach using the negative binomial model | |
| DEGSeq | Isoform-based approach using the Poisson model | |
| EdgeR | Count-based approach using empirical Bayes method based on the negative binomial model | |
| MISO | Isoform-based model using Bayes factors to estimate posterior probabilities | |
| Other tools | HCP | Normalizes expression data by inferring known and hidden factors with prior knowledge |
| PEER | Normalizes expression data by inferring known and hidden factors using a probabilistic estimation based on the Bayesian framework | |
| Matrix eQTL | Fast eQTL detection tool that uses linear models (linear regression or ANOVA) |
Transcript Assembly and Quantification
After RNA-Seq reads are aligned, the mapped reads can be assembled into transcripts. The majority of computational programs infer transcript models from the accumulation of read alignments to the reference genome (Trapnell et al. 2010; Li et al. 2011; Roberts et al. 2011a; Mezlini et al. 2013) (Table 2). An alternative approach for transcript assembly is de novo reconstruction, in which contiguous transcript sequences are assembled with the use of a reference genome or annotations (Robertson et al. 2010; Grabherr et al. 2011; Schulz et al. 2012). The reconstruction of transcripts from short-read data is a major challenge and a gold standard method for transcript assembly does not exist. The nature of the transcriptome (e.g., gene complexity, degree of polymorphisms, alternative splicing, dynamic range of expression), common technological challenges (e.g., sequencing errors), and features of the bioinformatics workflow (e.g., gene annotation, inference of isoforms) can substantially affect transcriptome assembly quality. RGASP3 has initiated efforts to evaluate computational methods for transcriptome reconstruction and has found that most algorithms can identify discrete transcript components, but the assembly of complete transcript structures remains a major challenge (Steijger et al. 2013).
A common downstream feature of transcript reconstruction software is the estimation of gene expression levels. Computational tools such as Cufflinks (Trapnell et al. 2010), FluxCapacitor (Montgomery et al. 2010; Griebel et al. 2012), and MISO (Katz et al. 2010), quantify expression by counting the number of reads that map to full-length transcripts (Table 2). Alternative approaches, such as HTSeq, can quantify expression without assembling transcripts by counting the number of reads that map to an exon (Anders et al. 2013). To accurately estimate gene expression, read counts must be normalized to correct for systematic variability, such as library fragment size, sequence composition bias, and read depth (Oshlack and Wakefield 2009; Roberts et al. 2011b). To account for these sources of variability, the reads per kilobase of transcripts per million mapped reads (RPKM) metric normalizes a transcript's read count by both the gene length and the total number of mapped reads in the sample. For paired end-reads, a metric that normalizes for sources of variances in transcript quantification is the paired fragments per kilobase of transcript per million mapped reads (FPKM) metric, which accounts for the dependency between paired-end reads in the RPKM estimate (Trapnell et al. 2010). Another technical challenge for transcript quantification is the mapping of reads to multiple transcripts that are a result of genes with multiple isoforms or close paralogs. One solution to correct for this “read assignment uncertainty” is to exclude all reads that do not map uniquely, as in Alexa-Seq (Griffith et al. 2010). However, this strategy is far from ideal for genes lacking unique exons. An alternative strategy used by Cufflinks (Trapnell et al. 2012), and MISO (Katz et al. 2010) is to construct a likelihood function that models the sequencing experiment and estimates the maximum likelihood that a read maps to a particular isoform.
Considerations for miRNA Sequencing Analysis
The general approach for analysis of miRNA sequencing data is similar to approaches discussed for mRNA. To identify known miRNAs, the sequencing reads can be mapped to a specific database, such as miRBase, a repository containing over 24,500 miRNA loci from 206 species in its latest release (v21) in June 2014 (Kozomara and Griffiths-Jones 2014). In addition, several tools have been developed to facilitate analysis of miRNAs including the commonly used tools miRanalyzer (Hackenberg et al. 2011) and miRDeep (An et al. 2013). MiRanalyzer can detect known miRNAs annotated on miRBase as well as predict novel miRNAs using a machine-learning approach based on the random forest method with a broad range of features. Similarly, miRDeep is able to identify known miRNAs and predict novel miRNAs using properties of miRNA biogenesis to score the compatibility of the position and frequency of sequenced RNA from the secondary structure of precursor miRNAs. Although miRDeep and miRanalyzer contain modules for target prediction, expression quantification, and differential expression, the methods developed for mRNA quantification and differential expression can also be applied to miRNA data (Eminaga et al. 2013).
Quality Assessment and Technical Considerations
At each stage in the RNA-Seq analysis pipeline, careful consideration should be applied to identifying and correcting for various sources of bias. Bias can arise throughout the RNA-Seq experimental pipeline, including during RNA extraction, sample preparation, library construction, sequencing, and read mapping (Kleinman and Majewski 2012; Lin et al. 2012; Pickrell et al. 2012; 't Hoen et al. 2013). First, the quality of the raw sequence data in FASTQ-format files should be evaluated to ensure high-quality reads. User-friendly software tools designed to generate quality overviews include the FASTX-toolkit (http://hannonlab.cshl.edu/fastx_toolkit), the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), and the RobiNA package (Lohse et al. 2012). Several important parameters that should be evaluated include the sequence diversity of reads, adaptor contamination, base qualities, nucleotide composition, and percentage of called bases. These technical artifacts can arise at the sequencing stage or during the construction of the RNA-Seq. For example, the 5′ read end, derived from either end of a double-stranded cDNA fragment, shows higher error rate due to mispriming events introduced by the random oligos during the RNA-Seq library construction protocol (Lin et al. 2012). If possible, actions to correct for these biases should be performed, such as trimming the ends of reads, to expedite the speed and improve the quality of the read alignments.
After aligning the reads, additional parameters should be assessed to account for biases that arise at the read mapping stage. These parameters include the percentage of reads mapped to the transcriptome, the percentage of reads with a mapped mate pair, the coverage bias at the 5′- and 3′-ends, and the chromosomal distribution of reads. One of the most common sources of mapping errors for RNA-Seq data occurs when a read spans the splicing junction of an alternatively spliced gene. A misalignment can be easily introduced due to ambiguous mapping of the read end to one of the two (or more) possible exons and is especially common when reads are mapped to a reference transcriptome that contains an incomplete annotation of isoforms (Kleinman and Majewski 2012; Pickrell et al. 2012). If genotype information is available, the integrity of the samples should also be evaluated by investigating the correlation of single-nucleotide variants (SNVs) between the DNA and RNA reads ('t Hoen et al. 2013). The concordance between the DNA and RNA sequencing data may provide insight into sample swaps or sample mixtures caused accidentally as a result of personnel or equipment error. In the case of a swapped sample, more discordant variants would be observed between the DNA and RNA sequencing data. In the case of a mixture of samples, more significant patterns of allele-specific expression would be observed than expected for a single individual as a result of more combinations of heterozygous and homozygous sites that would skew the alleles beyond the expected 1:1 allelic ratio.
Differential Gene Expression
A primary objective of many gene expression experiments is to detect transcripts showing differential expression across various conditions. Extensive statistical approaches have been developed to test for differential expression with microarray data, where the continuous probe intensities across replicates can be approximated by a normal distribution (Cui and Churchill 2003; Smyth 2004; Grant et al. 2005). Although in principle these approaches are also applicable to RNA-Seq data, different statistical models must be considered for discrete read counts that do not fit a normal distribution. Early RNA-Seq studies suggested that the distribution of read counts across replicates fit a Poisson distribution, which formed the basis for modeling RNA-Seq count data (Marioni et al. 2008). However, further studies indicated that biological variability is not captured by the Poisson assumption, resulting in high false-positive rates due to underestimation of sampling error (Anders and Huber 2010; Langmead et al. 2010; Robinson and Oshlack 2010). Hence, negative binomial distribution models that take into account overdispersion or extra-Poisson variation have been shown to best fit the distribution of read counts across biological replicates.
To model the count-based nature of RNA-Seq data, complex statistical models have been developed to handle sources of variability that model overdispersion across technical and biological replicates. One source of variability is differences in sequencing read depth, which can artificially create differences between samples. For instance, differences in read depth will result in the samples appearing more divergent if raw read counts between genes are compared. To correct for this, it is advantageous to transform raw read count data to FPKM or RPKM values in differential expression analyses. Although this correction metric is commonly used in place of read counts, the presence of several highly expressed genes in a particular sample can significantly alter the RPKM and FPKM values. For example, a highly expressed gene can “absorb” many reads, consequently repressing the read counts for other genes and artificially inflating gene expression variation. To account for this bias, several statistical models have been proposed that use the highly expressed genes as model covariates (Robinson and Oshlack 2010). Another source of variability that has been observed is that the distribution of sequencing reads is unequal across genes. Therefore, a two-parameter generalized Poisson model that simultaneously considers read depth and sequencing bias as independent parameters was developed and shown to improve RNA-Seq analysis (Srivastava and Chen 2010). More complex normalization methods have also been developed to account for hidden covariates without removing significant biological variability. For example, the probabilistic estimation of expression residuals (PEER) framework (Stegle et al. 2012) and the hidden covariates with prior (HCP) framework (Mostafavi et al. 2013) are methods that use a Bayesian approach to infer hidden covariates and remove their effects from expression data.
To detect differential expression, a variety of statistical methods have been designed specifically for RNA-Seq data. A popular tool to detect differential expression is Cuffdiff, which is part of the Tuxedo suite of tools (Bowtie, Tophat, and Cufflinks) developed to analyze RNA-Seq data (Trapnell et al. 2013). In addition to Cuffdiff, several other packages support testing differential expression, including baySeq (Hardcastle and Kelly 2010), DESeq (Anders and Huber 2010), DEGseq (Wang et al. 2010b), and edgeR (Robinson et al. 2010) (Table 2). Although these packages can assign significance to differentially expressed transcripts, the biological observations should be carefully interpreted. Each model makes specific assumptions that may be violated in the context of the observed data; therefore, an understanding of the model parameters and their constraints is critical for drawing meaningful and accurate biological conclusions (Bullard et al. 2010). Furthermore, replicates in RNA-Seq experiments are crucial for measuring variability and improving estimations for the model parameters (Tarazona et al. 2011; Glaus et al. 2012). Biological replicates (e.g., cells grown on two different plates under the same conditions) are preferred to technical replicates (e.g., one RNA-Seq library sequenced on two different lanes), which show little variation. Although the number of replicates required per condition is an open research question, a minimum of three replicates per sample has been suggested (Auer and Doerge 2010). In many cases, multiplexed RNA-Seq libraries can be used to add biological replicates without increasing sequencing costs (if sequenced at a lower depth) and will greatly improve the robustness of the experimental design (Liu et al. 2014). Additionally, the accuracy of measurements of differential gene expression can be further improved by using ERCC spike-in controls to distinguish technical variation from biological variation.
Allele-Specific Expression
A major advantage of RNA-Seq is the ability to profile transcriptome dynamics at a single-nucleotide resolution. Therefore, the sequenced transcript reads can provide coverage across heterozygous sites, representing transcription from both the maternal and paternal alleles. If a sufficient number of reads cover a heterozygous site within a gene, the null hypothesis is that the ratio of maternal to paternal alleles is balanced. Significant deviation from this expectation suggests allele-specific expression (ASE). Potential mechanisms for ASE include genetic variation (e.g., single-nucleotide polymorphism in a cis-regulatory region upstream of a gene) and epigenetic effects (e.g., genomic imprinting, methylation, histone modifications, etc.). Early studies showed that allele-specific differences can affect up to 30% of loci within an individual (Ge et al. 2009) and are caused by both common and rare genetic variants (Pastinen 2010). Studies have also applied ASE to identify expression modifiers of protein-coding variation (Lappalainen et al. 2011; Montgomery et al. 2011), effects of loss-of-function variation (MacArthur et al. 2012), and differences between pathogenic and healthy tissues (Tuch et al. 2010). Furthermore, ASE studies using single-cell transcriptomics have uncovered a stochastic pattern of allelic expression that may contribute to variable expressivity, a novel perspective which may have fundamental implications for variable disease penetrance and severity (Deng et al. 2014).
Conventional workflows to detect ASE involve counting reads containing each allele at heterozygous sites and applying a statistical test, such as the binomial test or the Fisher's exact test (Degner et al. 2009; Rozowsky et al. 2011; Wei and Wang 2013). However, more rigorous statistical approaches are necessary to overcome technical challenges involved in ASE detection. These challenges include read-mapping bias, sampling variance, overdispersion at extreme read depths, alternatively spliced alleles, insertions and deletions (indels), and genotyping errors. To account for overdispersion, one approach is to model allelic read counts using a beta-binomial distribution at individual loci (Sun 2012); however, accurate estimation of the overdispersion parameter requires replicates and, in our experience, major source of bias come from site-specific mapping differences. Another strategy is to use a hierarchical Bayesian model that combines information across loci, as well as across replicates and technologies, to make global and site-specific inferences for ASE (Skelly et al. 2011). To assess reference-allele mapping bias, the number of mismatches in reads containing the nonreference allele should be assessed as increased bias is observed with greater sequence divergence between alleles (Stevenson et al. 2013). To correct for read-mapping bias, an enhanced reference genome can be constructed that masks all SNP positions or includes the alternative alleles at polymorphic loci (Degner et al. 2009; Satya et al. 2012). Statistical methods to better address these technical biases are under active development and are expected to foster further improvements in ASE detection.
Expression Quantitative Trait Loci
Another prominent direction of RNA-Seq studies has been the integration of expression data with other types of biological information, such as genotyping data. The combination of RNA-Seq with genetic variation data has enabled the identification of genetic loci correlated with gene expression variation, also known as expression quantitative trait loci (eQTLs). This expression variation caused by common and rare variants is postulated to contribute to phenotypic variation and susceptibility to complex disease across individuals (Majewski and Pastinen 2011). The goal of eQTL analysis is to identify associations that will uncover underlying biological processes, discover genetic variants causing disease, and determine causal pathways. Initial eQTL studies using RNA-Seq data identified a greater number of statistically significant eQTLs than had been identified by microarray studies (Montgomery et al. 2010; Pickrell et al. 2010). Most of the eQTLs identified directly influenced gene expression in an allele-specific manner and were located near transcriptional start sites, indicating that eQTLs could modulate expression directly, or in cis. Later studies identified trans-eQTLs, which are variants that affect the expression of a distant gene (>1 Mb) by modifying the activity or expression of upstream factors that regulate the gene (Fehrmann et al. 2011; Battle et al. 2013; Westra et al. 2013). Although trans-eQTLs show weaker effects and present validation difficulties, they can potentially reveal previously unknown pathways in gene regulation networks.
RNA-Seq has revolutionized QTL analyses because it enables association analyses of more than just gene expression levels alone. For example, RNA-Seq provides unprecedented opportunity to investigate variations in splicing by profiling alternately spliced isoforms of a gene. This has enabled the identification of variants influencing the quantitative expression of alternatively spliced isoforms commonly referred to as splicing-QTLs (sQTLs) (Lalonde et al. 2011). In addition, specific RNA-Seq library constructions (e.g., ribo-depleted) have enabled the detection of eQTLs affecting other RNA species; recent studies have identified variants affecting the expression of various ncRNAs, including long intergenic noncoding RNAs (Montgomery et al. 2010; Gamazon et al. 2012; Kumar et al. 2013; Popadin et al. 2013). The expanding potential of RNA-Seq to associate phenotypic variations with genetic variation offers an enhanced understanding of gene regulation.
Traditional eQTL mapping methods that were developed for microarray data use linear models such as linear regression and ANOVA to associate genetic variants with gene expression (Kendziorski and Wang 2006). These methods have been directly applied to RNA-Seq data following appropriate normalization of total read counts. Most eQTL studies perform separate testing for each transcript-SNP pair using linear regression and ANOVA models to detect significant association. Nonlinear approaches have also been developed to test associations, such as generalized linear and mixed models, Bayesian regression (Servin and Stephens 2007). Alternative models, such as Merlin, have also been developed to detect eQTLs from expression data that include related individuals using pedigree data (Abecasis et al. 2002). In addition, several methods have been developed to simultaneously test the effect of multiple SNPs on the expression of a single gene using Bayesian methods (Lee et al. 2008). To further improve on the detection of causal regulatory variants, several studies have integrated ASE information with eQTL analysis. These studies showed that genetic variants showing allele-specific effects and identified as eQTLs show higher enrichment in functional annotations and provide stronger evidence of cis-regulatory impact (Battle et al. 2013; Lappalainen et al. 2013; Sun and Hu 2013). Because high-throughput sequencing has created genotype data sets featuring millions of SNPs and expression data sets featuring tens of thousands of transcripts, the task of testing billions of transcript-SNP pairs in eQTL analysis can be computationally intensive. To mitigate this computational burden, software has been developed such as Matrix eQTL to efficiently test the associations by modeling the effect of genotype as either additive linear (least squares model) or categorical (ANOVA model) (Shabalin 2012). Because of the large number of tests performed, it is important to correct for multiple-testing by calculating the false discovery rate (Benjamini and Hochberg 1995; Yekutieli and Benjamini 1999) or resampling using bootstrap or permutation procedures (Karlsson 2006; Zhang et al. 2012).
However, the design and interpretation of eQTL studies is not straightforward. Many complications result from the complexity of gene regulation, which shows both spatial (cell and tissue location) specificity as well as temporal (developmental stage) specificity. For instance, several studies have performed eQTL analysis across multiple tissues, indicating that genetic regulatory elements can have tissue-specific effects (Petretto et al. 2006; Schadt et al. 2008; Dimas et al. 2009; Kwan et al. 2009; Grundberg et al. 2012; Flutre et al. 2013). Therefore, future eQTL analyses should test for SNP-transcript associations in well-defined cell types that are relevant to the trait of interest (Lonsdale et al. 2013). For example, a study detecting eQTLs in cardiovascular disease should use heart tissue while a study interested in autoimmune disease should use whole blood. Another major consideration for eQTL studies is accounting for population structure and elucidating the causal variants (Stranger et al. 2012). The structure of genomic variation can vary significantly between populations and will influence the resolution of any genetic association study (Frazer et al. 2007; Altshuler et al. 2010). Furthermore, if substantial linkage disequilibrium (LD) exists within the genome, the associated genetic variant is often “tagging” the causal variant rather than acting as the causal regulatory variant itself. As eQTL studies integrate data across different populations and use population-scale genome sequencing, the ability to elucidate causal variants will greatly improve (Montgomery et al. 2010; Lappalainen et al. 2013).
Future Prospects
As sequencing technologies advance, computational tools will need to evolve in parallel to solve new technical challenges and support novel applications. For example, as the ability of sequencing platforms to produce longer reads becomes a reality, new mapping methods are required to accurately and efficiently align long reads. Because longer reads can span multiple exon–exon junctions, the identification and quantification of alternative isoforms will improve significantly with the extra information encoded in longer reads. Furthermore, as laboratory methods mature to enable sequencing of minute quantities of RNA, complex statistical approaches will be needed to discriminate between technical noise and meaningful biological variation. These progresses will facilitate the analysis of transcriptomes in rare cell types and cell states, enabling researchers to reconstruct biological networks active at the cellular level. In addition, these advancements will allow transcriptome analysis to move into the field of clinical diagnostics; for example, earlier monitoring of cancer screening and pregnancy could be accomplished by sequencing cancerous RNA or fetal RNA in the maternal blood. Furthermore, the integration of whole-genome sequencing with RNA-Seq in larger samples will provide greater insight into genetic regulatory variation. These experimental and bioinformatic advances will provide a powerful toolbox for fully characterizing the transcriptome as it relates to basic biological questions, as well as its rising impact on personalized medicine.
Acknowledgments
The authors gratefully acknowledge their colleagues, Tuuli Lappalainen and Jin Billy Li, as well as fellow laboratory members, including Zach Zappala, Kevin Smith, Marianne DeGorter and Mauro Pala, for their valuable comments. K.R.K. is supported by the National Defense Science and Engineering Graduate (NDSEG) Fellowship from the U.S. Department of Defense, and S.B.M. is funded by the Edward Mallinckrodt, Jr. Foundation.
References
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde B, Moreno RF, et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science. 1991;252:1651–1656. doi: 10.1126/science.2047873. [DOI] [PubMed] [Google Scholar]
- Adams MD, Kerlavage AR, Fleischmann RD, Fuldner RA, Bult CJ, Lee NH, Kirkness EF, Weinstock KG, Gocayne JD, White O, et al. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature. 1995;377:3–174. [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu FL, Bonnen PE, de Bakker PIW, Deloukas P, Gabriel SB, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Lai J, Lehman ML, Nelson CC. miRDeep*: An integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 2013;41:727–737. doi: 10.1093/nar/gks1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, McCarthy DJ, Chen Y, Okoniewski M, Smyth GK, Huber W, Robinson MD. Count-based differential expression analysis of RNA sequencing data using R and bioconductor. Nature protocols. 2013;8:1765–1786. doi: 10.1038/nprot.2013.099. [DOI] [PubMed] [Google Scholar]
- Au KF, Sebastiano V, Afshar PT, Durruthy JD, Lee L, Williams BA, van Bakel H, Schadt EE, Reijo-Pera RA, Underwood JG, et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proc Natl Acad Sci. 2013;110:E4821–E4830. doi: 10.1073/pnas.1320101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–416. doi: 10.1534/genetics.110.114983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R, et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2013;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos Ja, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe BJ, Ahmad S, Lee LJ. Current-generation high-throughput sequencing: Deepening insights into mammalian transcriptomes. Genes Dev. 2009;23:1379–1386. doi: 10.1101/gad.1788009. [DOI] [PubMed] [Google Scholar]
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor H, Simpson E, Sato VL, Fathman CG, Herzenberg LA. Characterization of subpopulations of T lymphocytes. I. Separation and functional studies of peripheral T-cells binding different amounts of fluorescent anti-Thy 1.2 (theta) antibody using a fluorescence-activated cell sorter (FACS) Cell Immunol. 1975;15:180–196. doi: 10.1016/0008-8749(75)90174-4. [DOI] [PubMed] [Google Scholar]
- Carneiro MO, Russ C, Ross MG, Gabriel SB, Nusbaum C, DePristo MA. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics. 2012;13:375. doi: 10.1186/1471-2164-13-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casneuf T, Van de Peer Y, Huber W. In situ analysis of cross-hybridisation on microarrays and the inference of expression correlation. BMC Bioinformatics. 2007;8:461. doi: 10.1186/1471-2105-8-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulou DC, Gorham JM, Herman DS, Seidman JG. Construction of normalized RNA-seq libraries for next-generation sequencing using the crab duplex-specific nuclease. Current Protocols in Molecular Biology/edited by Frederick M Ausubel, [et al] 2011;Chapter 4 doi: 10.1002/0471142727.mb0412s94. Unit 4 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Crick FH. On protein synthesis. Symp Soc Exp Biol. 1958;12:138–163. [PubMed] [Google Scholar]
- Cui X, Churchill GA. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003;4:210. doi: 10.1186/gb-2003-4-4-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Gutierrez Arcelus M, Sekowska M, et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Eminaga S, Christodoulou DC, Vigneault F, Church GM, Seidman JG. Quantification of microRNA expression with next-generation sequencing. Current Protocols in Molecular Biology/edited by Frederick M Ausubel [et al] 2013;Chapter 4 doi: 10.1002/0471142727.mb0417s103. Unit 4 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Laser capture microdissection. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- Engstrom PG, Steijger T, Sipos B, Grant GR, Kahles A, Consortium R, Alioto T, Behr J, Bertone P, Bohnert R, et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkkila T, Lehmusvaara S, Ruusuvuori P, Visakorpi T, Shmulevich I, Lah-desmaki H. Probabilistic analysis of gene expression measurements from heterogeneous tissues. Bioinformatics. 2010;26:2571–2577. doi: 10.1093/bioinformatics/btq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann RSN, Jansen RC, Veldink JH, Westra HJ, Arends D, Bonder MJ, Fu JY, Deelen P, Groen HJM, Smolonska A, et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011;7:e1002197. doi: 10.1371/journal.pgen.1002197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flutre T, Wen X, Pritchard J, Stephens M. A statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet. 2013;9:e1003486. doi: 10.1371/journal.pgen.1003486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:U851–U853. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu GK, Xu W, Wilhelmy J, Mindrinos M, Davis RW, Xiao W, Fodor SPA. Molecular indexing enables quantitative targeted RNA sequencing and reveals poor efficiencies in standard library preparations. Proc Natl Assoc Sci. 2014;111:1891–1896. doi: 10.1073/pnas.1323732111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Ziliak D, Im HK, LaCroix B, Park DS, Cox NJ, Huang RS. Genetic architecture of microRNA expression: Implications for the transcriptome and complex traits. Am J Hum Genet. 2012;90:1046–1063. doi: 10.1016/j.ajhg.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge B, Pokholok DK, Kwan T, Grundberg E, Morcos L, Verlaan DJ, Le J, Koka V, Lam KC, Gagne V, et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat Genet. 2009;41:1216–1222. doi: 10.1038/ng.473. [DOI] [PubMed] [Google Scholar]
- Glaus P, Honkela A, Rattray M. Identifying differentially expressed transcripts from RNA-seq data with biological variation. Bioinformatics. 2012;28:1721–1728. doi: 10.1093/bioinformatics/bts260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adi-conis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, Stoeckert CJ, Hogenesch JB, Pierce EA. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant GR, Liu J, Stoeckert CJ., Jr A practical false discovery rate approach to identifying patterns of differential expression in microarray data. Bioinformatics. 2005;21:2684–2690. doi: 10.1093/bioinformatics/bti407. [DOI] [PubMed] [Google Scholar]
- Griebel T, Zacher B, Ribeca P, Raineri E, Lacroix V, Guigo R, Sammeth M. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012;40:10073–10083. doi: 10.1093/nar/gks666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Griffith OL, Mwenifumbo J, Goya R, Morrissy AS, Morin RD, Corbett R, Tang MJ, Hou YC, Pugh TJ, et al. Alternative expression analysis by RNA sequencing. Nat Methods. 2010;7:843–847. doi: 10.1038/nmeth.1503. [DOI] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang TP, Meduri E, Barrett A, et al. Mapping cis- and transregulatory effects across multiple tissues in twins. Nat Genet. 2012;44:1084–1089. doi: 10.1038/ng.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenberg M, Rodriguez-Ezpeleta N, Aransay AM. miRanalyzer: An update on the detection and analysis of microRNAs in high-throughput sequencing experiments. Nucleic Acids Res. 2011;39:W132–W138. doi: 10.1093/nar/gkr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardcastle TJ, Kelly KA. baySeq: Empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell Reports. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Huang R, Jaritz M, Guenzl P, Vlatkovic I, Sommer A, Tamir IM, Marks H, Klampfl T, Kralovics R, Stunnenberg HG, et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS ONE. 2011;6:e27288. doi: 10.1371/journal.pone.0027288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. Non-genetic heterogeneity of cells in development: More than just noise. Development. 2009;136:3853–3862. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Highly multiplexed and strand-specific single-cell RNA 5′ end sequencing. Nat Protocols. 2012;7:813–828. doi: 10.1038/nprot.2012.022. [DOI] [PubMed] [Google Scholar]
- Itoh K, Matsubara K, Okubo K. Identification of an active gene by using large-scale cDNA sequencing. Gene. 1994;140:295–296. doi: 10.1016/0378-1119(94)90563-0. [DOI] [PubMed] [Google Scholar]
- Jiang L, Schlesinger F, Davis CA, Zhang Y, Li R, Salit M, Gingeras TR, Oliver B. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 2011;21:1543–1551. doi: 10.1101/gr.121095.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A. Review of “Permutation, parametric, and bootstrap tests of hypotheses. J R Stat SocA Stat. 2006;169:171–171. [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015. doi: 10.1038/nmeth.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki ES. Microarrays and the gene expression profile of a single cell. Ann N Y Acad Sci. 2004;1020:92–100. doi: 10.1196/annals.1310.010. [DOI] [PubMed] [Google Scholar]
- Kendziorski C, Wang P. A review of statistical methods for expression quantitative trait loci mapping. Mamm Genome. 2006;17:509–517. doi: 10.1007/s00335-005-0189-6. [DOI] [PubMed] [Google Scholar]
- Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, et al. Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med. 2001;7:673–679. doi: 10.1038/89044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinman CL, Majewski J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2012;335:1302. doi: 10.1126/science.1209658. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kube DM, Savci-Heijink CD, Lamblin AF, Kosari F, Vasmatzis G, Cheville JC, Connelly DP, Klee GG. Optimization of laser capture microdissection and RNA amplification for gene expression profiling of prostate cancer. BMC Mol Biol. 2007;8:25. doi: 10.1186/1471-2199-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Westra HJ, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, Almeida R, Zhernakova A, Reinmaa E, Vosa U, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013;9:e1003201. doi: 10.1371/journal.pgen.1003201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T, Grundberg E, Koka V, Ge B, Lam KC, Dias C, Kindmark A, Mallmin H, Ljunggren O, Rivadeneira F, et al. Tissue effect on genetic control of transcript isoform variation. PLoS Genet. 2009;5:e1000608. doi: 10.1371/journal.pgen.1000608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde E, Ha KC, Wang Z, Bemmo A, Kleinman CL, Kwan T, Pastinen T, Majewski J. RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression. Genome Res. 2011;21:545–554. doi: 10.1101/gr.111211.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Hansen KD, Leek JT. Cloud-scale RNA-sequencing differential expression analysis with Myrna. Genome Biol. 2010;11:R83. doi: 10.1186/gb-2010-11-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Montgomery SB, Nica AC, Dermitzakis ET. Epistatic selection between coding and regulatory variation in human evolution and disease. Am J Hum Genetics. 2011;89:459–463. doi: 10.1016/j.ajhg.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Sammeth M, Friedlander MR, 't Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, van der Werf JHJ, Hayes BJ, Goddard ME, Visscher PM. Predicting unobserved phenotypes for complex traits from whole-genome SNP data. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000231. doi:101371/journal.pgen.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Berger MF, Adiconis X, Rogov P, Melnikov A, Fennell T, Nusbaum C, Garraway LA, Gnirke A. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009;10:R115. doi: 10.1186/gb-2009-10-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Jiang CR, Brown JB, Huang H, Bickel PJ. Sparse linear modeling of next-generation mRNA sequencing (RNA-Seq) data for isoform discovery and abundance estimation. Proc Natl Acad Sci. 2011;108:19867–19872. doi: 10.1073/pnas.1113972108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xie X. A mixture model for expression deconvolution from RNA-seq in heterogeneous tissues. BMC Bioinformatics. 2013;14:S11. doi: 10.1186/1471-2105-14-S5-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Piskol R, Tan MH, Li JB. Response to comments on “Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2012;335:1302. doi: 10.1126/science.1210419. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou J, White KP. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics. 2014;30:301–304. doi: 10.1093/bioinformatics/btt688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. RobiNA: A user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40:W622–W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Philips R, Lo E, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski J, Pastinen T. The study of eQTL variations by RNA-seq: From SNPs to phenotypes. Trends Genet. 2011;27:72–79. doi: 10.1016/j.tig.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15 Spec No 1:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: Insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies—The next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Mezlini AM, Smith EJM, Fiume M, Buske O, Savich GL, Shah S, Aparicio S, Chiang DY, Goldenberg A, Brudno M. iReckon: Simultaneous isoform discovery and abundance estimation from RNA-seq data. Genome Res. 2013;23:519–529. doi: 10.1101/gr.142232.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills JD, Kawahara Y, Janitz M. Strand-specific RNA-Seq provides greater resolution of transcriptome profiling. Curr Genomics. 2013;14:173–181. doi: 10.2174/1389202911314030003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Lappalainen T, Gutierrez-Arcelus M, Dermitzakis ET. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, Arnes L, Nakic N, Garcia-Hurtado J, Rodriguez-Segui S, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Zhao YJ, Prabhu AL, Dhalla N, McDonald H, Pandoh P, Tam A, Zeng T, Hirst M, Marra M. Preparation and analysis of Micro-RNA libraries using the Illumina massively parallel sequencing technology. Methods Mol Biol. 2010;650:173–199. doi: 10.1007/978-1-60761-769-3_14. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Battle A, Zhu X, Urban AE, Levinson D, Montgomery SB, Koller D. Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge. PLoS ONE. 2013;8:e68141. doi: 10.1371/journal.pone.0068141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nica AC, Ongen H, Irminger JC, Bosco D, Berney T, Antonarakis SE, Halban PA, Dermitzakis ET. Cell-type, allelic, and genetic signatures in the human pancreatic beta cell transcriptome. Genome Res. 2013;23:1554–1562. doi: 10.1101/gr.150706.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Oshlack A, Wakefield MJ. Transcript length bias in RNA-seq data confounds systems biology. Biol Direct. 2009;4:14. doi: 10.1186/1745-6150-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Kro-bitsch S, Lehrach H, Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen T. Genome-wide allele-specific analysis: Insights into regulatory variation. Nat Rev Genetics. 2010;11:533–538. doi: 10.1038/nrg2815. [DOI] [PubMed] [Google Scholar]
- Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, et al. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2006;2:e172. doi: 10.1371/journal.pgen.0020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Gilad Y, Pritchard JK. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome. Science. 2012;335:1302. doi: 10.1126/science.1210484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popadin K, Gutierrez-Arcelus M, Dermitzakis ET, Antonarakis SE. Genetic and epigenetic regulation of human lincRNA gene expression. Am J Hum Genet. 2013;93:1015–1026. doi: 10.1016/j.ajhg.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth R, Fischer A, Schweiger MR, Lehrach H, Mertes F. Creation and application of immortalized bait libraries for targeted enrichment and next-generation sequencing. Biotechniques. 2012;52:375–380. doi: 10.2144/0000113877. [DOI] [PubMed] [Google Scholar]
- Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011a;27:2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011b;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. De novo assembly and analysis of RNA-seq data. Nat Methods. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. AlleleSeq: Analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudloff U, Bhanot U, Gerald W, Klimstra DS, Jarnagin WR, Brennan MF, Allen PJ. Biobanking of human pancreas cancer tissue: Impact of ex-vivo procurement times on RNA quality. Ann Surg Oncol. 2010;17:2229–2236. doi: 10.1245/s10434-010-0959-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, Ueda HR. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14:R31. doi: 10.1186/gb-2013-14-4-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satya RV, Zavaljevski N, Reifman J. A new strategy to reduce allelic bias in RNA-Seq readmapping. Nucleic Acids Res. 2012;40:e127. doi: 10.1093/nar/gks425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servin B, Stephens M. Imputation-based analysis of association studies: Candidate regions and quantitative traits. PLoS Genet. 2007;3:e114. doi: 10.1371/journal.pgen.0030114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalin AA. Matrix eQTL: Ultra-fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31:1009–1014. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J. The beginning of the end for microarrays? Nat Methods. 2008;5:585–587. doi: 10.1038/nmeth0708-585. [DOI] [PubMed] [Google Scholar]
- Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci. 2003;100:15776–15781. doi: 10.1073/pnas.2136655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Johansson M, Madeoy J, Wakefield J, Akey JM. A powerful and flexible statistical framework for testing hypotheses of allele-specific gene expression from RNA-seq data. Genome Res. 2011;21:1728–1737. doi: 10.1101/gr.119784.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Heisler LE, St Onge RP, Farias-Hesson E, Wallace IM, Bodeau J, Harris AN, Perry KM, Giaever G, Pourmand N, et al. Highly-multiplexed barcode sequencing: An efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 2010;38:e142. doi: 10.1093/nar/gkq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statist Appl Genetics Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Chen L. A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res. 2010;38:e170. doi: 10.1093/nar/gkq670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- Stegle O, Parts L, Piipari M, Winn J, Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat Protocols. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steijger T, Abril JF, Engstrom PG, Kokocinski F, Consortium R, Abril JF, Akerman M, Alioto T, Ambrosini G, Antonarakis SE, et al. Assessment of transcript reconstruction methods for RNA-seq. Nat Methods. 2013;10:1177–1184. doi: 10.1038/nmeth.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson KR, Coolon JD, Wittkopp PJ. Sources of bias in measures of allele-specific expression derived from RNA-seq data aligned to a single reference genome. BMC Genomics. 2013;14:536. doi: 10.1186/1471-2164-14-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekow-ska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Lariviere L, Maier KC, Seizl M, Tresch A, Cramer P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012;22:1350–1359. doi: 10.1101/gr.130161.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W. A statistical framework for eQTL mapping using RNA-seq data. Biometrics. 2012;68:1–11. doi: 10.1111/j.1541-0420.2011.01654.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Hu Y. eQTL mapping using RNA-seq data. Statist Biosci. 2013;5:198–219. doi: 10.1007/s12561-012-9068-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protocols. 2010;5:516–535. doi: 10.1038/nprot.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6:377–382. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011;21:2213–2223. doi: 10.1101/gr.124321.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 't Hoen PA, Friedlander MR, Almlof J, Sammeth M, Pulyakhina I, Anvar SY, Laros JF, Buermans HP, Karlberg O, Brannvall M, et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31:1015–1022. doi: 10.1038/nbt.2702. [DOI] [PubMed] [Google Scholar]
- Thompson KL, Pine PS, Rosenzweig BA, Turpaz Y, Retief J. Characterization of the effect of sample quality on high density oligonucleotide microarray data using progressively degraded rat liver RNA. BMC Biotechnol. 2007;7:57. doi: 10.1186/1472-6750-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H, Vawter MP, Walsh DM, Evans SJ, Choudary PV, Li J, Overman KM, Atz ME, Myers RM, Jones EG, et al. Effect of agonal and postmortem factors on gene expression profile: Quality control in microarray analyses of postmortem human brain. Biol Psychiatry. 2004;55:346–352. doi: 10.1016/j.biopsych.2003.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams Ba, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuch BB, Laborde RR, Xu X, Gu J, Chung CB, Monighetti CK, Stanley SJ, Olsen KD, Kasperbauer JL, Moore EJ, et al. Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations. PLoS ONE. 2010;5:e9317. doi: 10.1371/journal.pone.0009317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Vivancos AP, Guell M, Dohm JC, Serrano L, Himmelbauer H. Strand-specific deep sequencing of the transcriptome. Genome Res. 2010;20:989–999. doi: 10.1101/gr.094318.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Miecz-kowski P, Grimm SA, Perou CM, et al. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010a;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010b;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: A revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Wang X. A computational workflow to identify allele-specific expression and epigenetic modification in maize. Genomics Proteomics Bioinformatics. 2013;11:247–252. doi: 10.1016/j.gpb.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genetics. 2013;45:1238–1243. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: Functiona surprises from the RNA world. Genes Dev. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE., Jr Decay rates of human mRNAs: Correlation with functional characteristics and sequence attributes. Genome Res. 2003;13:1863–1872. doi: 10.1101/gr.1272403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekutieli D, Benjamini Y. Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J Stat Plan Infer. 1999;82:171–196. [Google Scholar]
- Zeng W, Mortazavi A. Technical considerations for functional sequencing assays. Nat Immunol. 2012;13:802–807. doi: 10.1038/ni.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang SP, Sun W, Wang W. Rapid and robust resampling-based multiple-testing correction with application in a genome-wide expression quantitative trait loci study. Genetics. 2012;190:1511–1520. doi: 10.1534/genetics.111.137737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Samarov D, McDaniel J, Sen SK, Salit M. Synthetic spike-in standards improve run-specific systematic error analysis for DNA and RNA sequencing. PLoS ONE. 2012;7:e41356. doi: 10.1371/journal.pone.0041356. [DOI] [PMC free article] [PubMed] [Google Scholar]