

Abstract
Benzodiazepines are the most widely prescribed class of psychoactive drugs in current therapeutic use, despite the important unwanted side effects that they produce, such as sedation, myorelaxation, ataxia, amnesia, and ethanol and barbiturate potentiation and tolerance. They exert their therapeutic effects via binding to the benzodiazepine binding site of gamma-aminobutyric acid (GABA) type A receptors, and allosterically modulating the chloride flux through the ion channel complex. First isolated from plants used as tranquilizers in folkloric medicine, some natural flavonoids have been shown to possess selective affinity for the benzodiazepine binding site with a broad spectrum of central nervous system effects. Since the initial search for alternative benzodiazepine ligands amongst the flavonoids, a list of successful synthetic derivatives has been generated with enhanced activities. This review provides an update on research developments that have established the activity of natural and synthetic flavonoids on GABA type A receptors. Flavonoids are prominent drugs in the treatment of mental disorders, and can also be used as tools to study modulatory sites at GABA type A receptors and to develop GABA type A selective agents further.
Keywords: flavonoids, GABA type A receptors, benzodiazepine binding site
Flavonoids
Flavonoids may have existed in nature for almost one billion years, and over 9000 chemically unique flavonoids have been identified in plant sources. These compounds are low molecular weight substances, found in all vascular plants, and are phenylbenzopyrones (Figure 1) with an assortment of basic structures.
Figure 1.

Chemical structures of some representative flavonoids.
Flavonoids occur as aglycones, glycosides, and methylated derivatives. In plants, flavonoid aglycones occur in a variety of structural forms. For convenience, the rings are labeled A, B, and C (Figure 1). The individual carbon atoms are based on a numbering system which uses ordinary numerals for A and C, and “primed” numerals for the B ring. The different ways of closing this ring associated with the different oxidation degrees of ring A define the various classes of flavonoids. Most flavonoids occur in natural association with sugar in conjugated form and, within any one class, may be characterized as, eg, monoglycosidic or diglycosidic. The glycosidic linkage is normally located at position 3 or 7 and the carbohydrate unit can be L-rhamnose, D-glucose, glucorhamnose, galactose, or arabinose.1 The chemical diversity, size, three-dimensional shape, and physical and biochemical properties of flavonoids allow them to interact with targets in different subcellular locations to influence biological activity in plants, animals, and microbes.2
Apart from being important dietary components, many therapeutic benefits of flavonoids are known in animal systems. Flavonoids have antioxidant, antiproliferative, antitumor, anti-inflammatory, and proapoptotic activities, and some molecular targets have been identified.3–7 The health-promoting effects of flavonoids may relate to interactions with key enzymes, signaling cascades involving cytokines and transcription factors, or antioxidant systems.8
Because flavonoids can be found ubiquitously in plants, they are major constituents of a variety of fruit and vegetables, beverages, such as tea and wine, and seeds such as cocoa beans and grape seeds. Flavonoids undergo extensive biotransformation and conjugation that occur during their absorption from the gastrointestinal tract, in the liver, and finally in cells.9–11 Dietary flavonoids are substrates for phase I and phase II enzymes in the small intestine and liver. They are deglycosylated and metabolized into glucuronides, sulfates, and O-methylated derivatives.12
Flavonoid absorption from the intestine occurs by several different pathways. Flavonoid aglycones can be easily absorbed into the intestinal cells because their lipophilicity facilitates their passage across the mucosal phospholipid bilayer of cells. Lactase phlorizin hydrolase has a crucial role in the absorption of flavonoids bearing β-glycoside linkages.13 On the other hand, flavonoid monoglycosides can be transported by the sodium glucose transporter-114,15 on the brush border membrane of intestinal cells. Most flavonoid glycosides entering enterocytes are deglycosylated by β-glucosidases, namely, broad-specificity cytosolic β-glucosidase.16 The flavonoids appear to be subjected to glucuronidation, sulfation, and methylation in the intestinal epithelial cells before entering circulation.17 The flavonoid conjugates then gain access into hepatocytes where they are further methylated, glucuronidated, or sulfated.18 These flavonoid conjugates are excreted into the urine and also into bile fluid, thereby returning to the intestinal lumen.19 Subsequently, they may be reabsorbed again, mainly in the large intestine. Further metabolism occurs in the colon, where enzymes of the gut microflora induce the breakdown of flavonoids to phenolic acids which may undergo absorption and be further metabolized in the liver.11,20 The present review focuses on advances in our knowledge pertaining to the action of flavonoids on the central nervous system, more precisely their effect on gamma amino butyric acid (GABA) type A receptors.
GABA type A receptor
Neuropharmacology is based simply on the fundamental balance between chemical excitation and inhibition. These processes are indispensable in the networks of neurons, and all neurons have receptors for inhibitory and excitatory neurotransmitters. The GABA system is one of the mechanisms that takes care of chemical inhibition in the brain, and has been widely used for pharmacological modulation of brain function. Most brain neurons express GABA type A receptors, which are considered to be the most important for pharmacological modulation. These receptors are heteropentameric GABA-gated chloride channels belonging to the Cys-loop ligand-gated ion channel superfamily that also includes the nicotinic acetylcholine receptors, glycine receptors, and 5-HT3 receptors.21,22 The subunits of all these receptors share a common ancestral structure. In addition to the rapid actions of GABA via GABA type A receptors, GABA also modulates neural activity, albeit on a slower time scale, via activation of GABA type A receptors belonging to the G protein-coupled receptor superfamily. GABA type A receptor subunits are encoded by 19 different genes that have been grouped into eight subclasses based on sequence homology (α1–6, β1–3, γ1–3, δ, ε, θ, π, ρ1–3). Alternative splicing contributes to additional receptor diversity.23,24 Knowing the number of subunits and potential combinations, the quantity of possible receptor subtypes could be enormous. However, only 11 structurally and functionally distinct receptor subtypes have been conclusively identified and are reasonably abundant in at least parts of the brain. They represent combinations of 2α and 2β subunits, together with a single γ2 or δ subunit. A further 15 receptor subtypes exist with high probability and a more limited distribution.25 These numbers do not account for additional heterogeneity based on two different types of α or β subunits in one receptor complex,26 or due to alternative splicing of subunits. GABA type A receptors with different subunit compositions exhibit different pharmacology and channel gating properties, are differentially expressed during development and in the adult brain, accumulate at different neuronal cell surfaces, and are subject to differential regulation by extracellular cues.27
GABA type A receptors can be allosterically modulated by benzodiazepines, barbiturates, steroids, anesthetics, anticonvulsants, and many other drugs, the number of which is constantly increasing.28,29 These compounds do not interact directly with the GABA binding site, but exert their actions by binding to allosteric sites at GABA type A receptors that influence the binding properties of other binding sites present on these receptors and so modulate GABA-induced chloride ion influx (Figure 2).
Figure 2.

Schematic model of the GABA type A receptors.
Notes: As shown in this model, GABA type A receptors exhibit a GABA binding site that mediates the effects of agonists and competitive antagonists, a Cl− channel, and modulatory binding sites for benzodiazepiness, barbiturates, picrotoxin, and anesthetic steroids. The model is not meant to reflect the subunit structure of the receptor.
Benzodiazepines are anticonvulsive, sedative-hypnotic, and anxiolytic compounds in clinical use. They produce allosteric changes that enhance the action of GABA on GABA type A receptors, increasing the GABA-induced frequency of opening of the chloride channels and the apparent affinity of the receptor for GABA. Benzodiazepines represented a major advance in psychopharmacology in the 1960s and have been some of the most widely prescribed drugs. They are sedating and, more importantly, produce physical dependence, such that significant withdrawal symptoms are observed on treatment cessation. In addition, benzodiazepines can be drugs of abuse. This inspired the development of subtype-selective agonists, which might retain the beneficial effects of an antianxiety and anticonvulsant profile, while no longer being sedative, ataxic, or having dependence liability.30
The location of the benzodiazepine binding site at the α and γ subunit interface indicates that the pharmacology of the benzodiazepine receptor subtypes is mainly determined by the α and γ isoforms forming this site, whereas β subunits, although needed to construct a channel, do not greatly affect the sensitivity of the GABA type A receptors to benzodiazepine ligands. The traditional benzodiazepine agonists (such as diazepam) are active at GABA type A receptors containing a γ subunit, a β subunit, and one of the α subunits, ie, α 1, 2, 3, or 5.31 The benzodiazepine-sensitive GABA type A receptors can be further subdivided, in that receptors containing the α1 subunit have a higher sensitivity to a subpopulation of benzodiazepine ligands, such as quazepam or zolpidem (an imidazopyridine).28,31,32 Furthermore, receptors containing the α2 or α3 subunit have an intermediate affinity for zolpidem, whereas those containing α5 have very low affinity for this drug.
Receptors containing the α4 or α6 subunits, together with β and γ2, do not bind the traditional benzodiazepine agonists, including zolpidem, but demonstrate high affinity for some ligands, such as flumazenil and Ro15-4513, or bretazenil.28 Both the potency and efficacy for benzodiazepine ligands depend on the nature of the α subunit.
The benzodiazepine binding site ligands so far identified do not distinguish well between the α2 and α3 or between the α4 and α6 subunits. However, all four subunits can produce functional channels in vitro when coexpressed with other subunits, and their differential distribution in the brain suggests that they modulate different behavioral circuitry.
The functional relevance of the receptor subtypes was revealed using a combined molecular genetic and pharmacological approach.33,34 Mouse lines were generated in which each of the benzodiazepine-sensitive GABA type A receptors (containing α1, α2, α3, or α5 subunits) was rendered insensitive to diazepam by a point mutation in the benzodiazepine binding site (eg, α1 H101R).35 A comparison of drug-induced behavioral responses in the mutated and wild-type mice then allowed identification of diazepam effects that were missing or reduced in the mutant mice. It was demonstrated that α1βγ2 receptors mediate the sedative, antegrade amnestic, and some anticonvulsant actions of diazepam.35,36 The anxiolytic activity of diazepam is mediated mostly by GABA type A receptors composed of α2βγ2 subunits37 and also by α3 GABA type A receptors.38 This allowed sedation and anxiolysis to be separated in molecular terms, mediated by different pathways that were characterized by the presence of α1- and α2-GABA type A receptors, respectively. The α2βγ2 receptors are also implicated in some of the muscle relaxant activities of diazepam.37 Receptors containing the α3 subunit seem to mediate the antiabsence effects of clonazepam. The α5βγ2 receptors seem to influence learning and memory.39
The action of benzodiazepines can be blocked by flumazenil. However, flumazenil-insensitive positive modulation of GABA type A receptors has been described in receptors lacking a γ subunit by benzodiazepines at µM concentrations. It was found that recombinant α1β1 GABA type A receptors from the rat brain were sensitive to potentiation by benzodiazepine binding site ligands, with both diazepam and flumazenil acting as positive modulators.40 It was also found that classical benzodiazepines produce biphasic potentiation at rat recombinant α1β2γ2 GABA type A receptors via two distinct mechanisms.41 This biphasic potentiation is believed to be mediated via two sites, referred to as high-affinity and low-affinity benzodiazepine binding sites. Both sites are present on receptors composed of α1β2γ2 GABA type A subunits, and low affinity potentiation can be selectively observed at receptor combinations lacking a γ subunit, such as α1β2 GABA type A receptors. Furthermore, low affinity potentiation at both receptor combinations is insensitive to flumazenil.
Natural flavonoids as GABA type A receptor ligands
Nature provides science and society with a virtually unlimited supply of structurally diverse and biologically active molecules. While some are directly useful in commercial applications, others are valuable for studying and understanding biological phenomena at the molecular level. Flavonoids are only a modest example.
The first report of flavonoids as central nervous system ligands was described by Roche researchers. In their study, they were looking for the presence of “diazepam-like” endogenous ligands in bovine urine, in which they isolated a few isoflavan derivatives: (S-7,4′-dihydroxyisoflavan (equol); dl-3′,7-dihydroxyisoflavan; dl-4′-hydroxy-7-methoxyisoflavan; dl-7-hydroxy-4′-methoxyisoflavan; 7-hydroxy-4′-methoxyisoflavone (formononetin); and 4′-hydroxy-7-methoxyisoflavone; 3′,7-dihydroxyisoflavone) (Figure 3) with low affinity for the benzodiazepine binding site.42 Some years later, using a radioreceptor-guided purification protocol, we were able to isolate one of these isoflavan ligands (equol) from bovine rumen contents, an important natural source of “diazepam-like” compounds.43 These isoflavans were most probably derived from plant sources in the bovine diet.
Figure 3.

Molecular structures of (A) flavans and (B) isoflavones.
The next valuable antecedent in this story of flavonoids active in the central nervous system was the discovery of the biflavonoid, amentoflavone (Figure 4) as a high affinity ligand for the benzodiazepine binding site. This natural compound (Karmelitter Geist®) was isolated from an extract of several commercially available medicinal plants and used to treat nervous disorders.44
Figure 4.
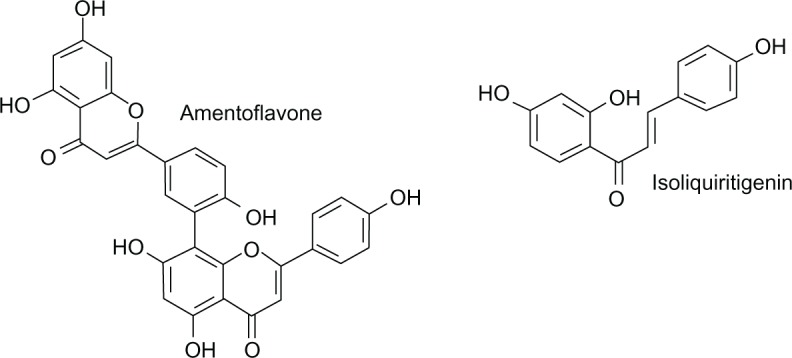
Molecular structures of amentoflavone and isoliquiritigenin.
Amentoflavone is one of the most potent flavonoids in displacing benzodiazepine binding to rat brain membranes, but is inactive in vivo. It binds, in vitro, in a mixed-type competitive and noncompetitive manner to brain receptors, with an affinity comparable with that of diazepam. Studies on subtype specificity showed that amentoflavone had little or no affinity for α4-containing or α6-containing receptors.45 This biflavonoid can be extracted from Ginkgo biloba but removed from herbal preparations such as EGb 761.46 Using a functional assay employing recombinant GABA type A receptors expressed in oocytes, amentoflavone has been shown to be a weak negative allosteric modulator of GABA action, acting independently of classical flumazenil-sensitive benzodiazepine-modulatory sites.46 It was also reported that amentoflavone influences a variety of G protein-coupled receptors for serotonin, dopamine, and opioids at nM concentrations, while having no effect on the binding of muscimol, a GABA type A agonist to GABA type A receptors.47
Since then and contemporary with the work described above, research in our laboratories was devoted to the identification of natural ligands for the benzodiazepine binding site in plants. In particular, we were looking for benzodiazepines or benzodiazepine-like compounds, and the discovery of the properties of amentoflavone44 drove our investigation to similar ligands in plants known to contain flavonoids and we also extended this study to other plants traditionally used as tranquillizers.48–51
In vivo and in vitro studies with the principal flavonoid and flavonol derivatives (Table 1) have clarified their pharmacological actions on the binding of benzodiazepines. The first monoflavonoid described as a specific ligand for the benzodiazepine binding site was chrysin (5,7-dihydroxyflavone).52 This compound, isolated from Passiflora caerulea L, is a selective and competitive inhibitor of [3H]flunitrazepam binding to the benzodiazepine binding site. Chrysin is almost equipotent to diazepam as an anxiolytic, but does not exhibit sedative or myorelaxant effects.53 It was postulated that this natural monoflavonoid is a partial agonist of the central benzodiazepine binding site. Additionally, both intraperitoneal and oral administration of chrysin in mice produced a significant hyperalgesic effect in the tail-immersion test that involved GABA type A receptors, as does flumazenil, a specific antagonist for the benzodiazepine binding site; bicuculline, a GABA type A receptor antagonist and picrotoxin, a chloride channel blocker, could antagonize the hyperalgesia of chrysin.54
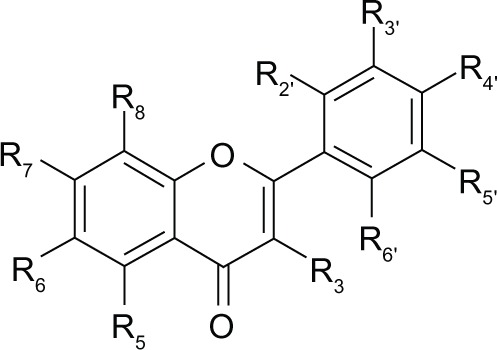
Table 1.
Flavone and flavonol derivatives
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Flavonoid | Substitution
|
|||||||||
| R5 | R6 | R7 | R8 | R3 | R2′ | R3′ | R4′ | R5′ | R6′ | |
| Flavone | H | H | H | H | H | H | H | H | H | H |
| Chrysin | OH | H | OH | H | H | H | H | H | H | H |
| Apigenin | OH | H | OH | H | H | H | H | OH | H | H |
| Kaempferol | OH | H | OH | H | OH | H | H | OH | H | H |
| Cirsiliol | OH | OCH3 | OCH3 | H | H | H | OH | OH | H | H |
| Quercetin | OH | H | OH | H | OH | H | OH | OH | H | H |
| Myricetin | OH | H | OH | H | OH | H | OH | OH | OH | H |
| Wogonin | OH | H | OH | OCH3 | H | H | H | H | H | H |
| Oroxylin A | OH | OCH3 | OH | H | H | H | H | H | H | H |
| Dinatin-hispidulin | OH | OCH3 | OH | H | H | H | H | OH | H | H |
| Skrofulein-cirsimaritin | OH | OCH3 | OCH3 | H | H | H | H | OH | H | H |
| 5,7-dimethoxyfavone | OCH3 | H | OCH3 | H | H | H | H | H | H | H |
| 5,7-dimethoxy-6-methylfavone | OCH3 | CH3 | OCH3 | H | H | H | H | H | H | H |
| 5-hydroxy-7-methoxy-6-methylfavone | OH | CH3 | OCH3 | H | H | H | H | H | H | H |
| 5-hydroxy-7-methoxy-6,8-dimethylfavone | OH | CH3 | OCH3 | CH3 | H | H | H | H | H | H |
| 6-Methylapigenin | OH | CH3 | OH | H | H | H | H | OH | H | H |
| Luteolin | OH | H | OH | H | H | H | OH | OH | H | H |
| Baicalein | OH | OH | OH | H | H | H | H | H | H | H |
| Baicalin | OH | O-glucuronide | OH | H | H | H | H | H | H | H |
| K36 | OH | OCH3 | OH | OCH3 | H | OH | H | H | H | H |
The dried flower heads of Matricaria recutita L are used in folk medicine to prepare a spasmolytic and sedative tea. The fractionation of the aqueous extract of this plant led to the detection of several fractions with significant affinity for the benzodiazepine binding site and to the isolation and identification of apigenin (5,7,4′-trihydroxyflavone). Apigenin competitively binds to the benzodiazepine binding site of the GABA type A receptor, has clear anxiolytic activity in mice when administered intraperitoneally, without showing evidence of sedation or muscle relaxant effects at doses similar to those used for classical benzodiazepines and no anticonvulsant action.55 However, other studies, performed in rats, found that apigenin fitted the profile of an inverse benzodiazepine agonist and was sedative and mildly proconvulsant, but not anxiolytic.56,57 Meanwhile, others reported that apigenin fitted the profile of a benzodiazepine antagonist.58 The discrepancies between the in vitro and in vivo results obtained for apigenin may be due to its in vivo metabolism or the use of different species, ie, rats or mice, because mice have higher baseline levels of anxiety. Thus, apigenin appears to have diverse effects at the benzodiazepine binding site and the nature of these effects is unclear. Overall, it seems that the effects of apigenin on GABA type A receptors are complex and involve both flumazenil-sensitive and flumazenil-insensitive components, and that other receptors could be involved in the behavioral effects of this drug.59,60
The flavonoids, kaempferol (3,5,7,4′-tetrahydroxyflavone) and cirsiliol (5,3′,4′-trihydroxy-6,7-dimethoxyflavone), isolated from Tilia tomentosa Moench and Salvia guaranitica, respectively, exhibited a very low affinity for the benzodiazepine binding site and were devoid of anxiolytic actions by the intraperitoneal route. However, cirsiliol produced sedation in mice as measured by the holeboard test and had thiopental-potentiating effects.61–63 Recently, a comparative study of the anxiolytic activity of the flavonols, kaempferol, quercetin, (3,5,7,3′,4′-pentahydroxyflavone), and myricetin (3,5,7,3′,4′,5′-hexahydroxyflavone) in the elevated plus-maze after oral and intraperitoneal administration in mice showed that only kaempferol and quercetin were active after oral administration.64 The anxiolytic activity of kaempferol was also partially antagonized by concomitant administration of flumazenil.65 No anxiolytic effects were observed when kaempferol and quercetin were given via the intraperitoneal route. It was hypothesized that flavonoids could act as prodrugs which are transformed into their active hydroxyphenylacetic acid metabolites by intestinal microflora.64
Wogonin (5,7-dihydroxy-8-methoxyflavone), isolated from Scutellaria baicalensis Georgi, a Chinese medicinal herb, has been reported to have anxiolytic and anticonvulsant activity in orally treated mice, that could be blocked by coadministration of flumazenil,66,67 with no sedative or myorelaxant effects. Another flavonoid isolated from this herb, oroxylin A (5,7-dihydroxy-6-methoxyflavone), inhibits [3H] flunitrazepam binding and, orally administered in mice, acts as a neutralizing allosteric modulator blocking the anxiolytic, myorelaxant, and motor incoordination effects but not the sedative and anticonvulsant effects elicited by diazepam, a full benzodiazepine agonist.68
Dinatin, a synonym of hispidulin (4′,5,7-trihydroxy-6-methoxyflavone) and skrofulein, a synonym of cirsimaritin (4′,5-dihydroxy-6,7-dimethoxyflavone), two flavones isolated from the medicinal plant from Artemisia herba-alba Asso, were found to be antagonists or weak partial agonists of the benzodiazepine binding site, and inhibited the binding of [3H] diazepam to rat brain membranes in vitro by a mixed competitive and noncompetitive mechanism.69 Hispidulin, the 6-methoxy derivative of apigenin, was further isolated, together with apigenin from Salvia officinalis (sage) using benzodiazepine binding site assay-guided fractionation.70 This flavone has been demonstrated to have anticonvulsant activity in a model of epilepsy in seizure-prone Mongolian gerbils and to cross the blood-brain barrier.71 Unlike apigenin, hispidulin has been shown to act as a positive allosteric modulator of α1,3,5,6β2γ2S GABA type A receptor subtypes.71
Daidzein (4′,7-dihydroxyisoflavone) and its 8-C glycoside, puerarin (7,4′-dihydroxy-8-C-glucosylisoflavone) (Figure 3), two isoflavones isolated from Puerariae radix, a Chinese traditional herb used to treat drunkenness and alcoholic addiction, were found to inhibit the binding of [3H] flunitrazepam to rat brain membranes.72
Leptospermum scoparium Forst contains the lipophilic flavonoids, 5,7-dimethoxyflavone, 5,7-dimethoxy-6-methylflavone, 5-hydroxy-7-methoxy-6-methylflavone, and 5-hydroxy-7-methoxy-6,8-dimethylflavone, which interact specifically with the benzodiazepine binding site. A dry extract of the tincture prepared from this plant and containing these flavones induced sedative and anxiolytic effects in rats.73
Another apigenin derivative was further reported as a benzodiazepine binding site ligand. 6-Methylapigenin (4′,5,7-dihydroxy-6-methylflavone), the 6-methyl derivative of apigenin, was isolated from the roots and rhizomes of Valeriana wallichii, a known sedative medicinal plant, and inhibited [3H]flunitrazepam binding at 0.5 µM in a manner suggesting it may be a positive modulator of GABA type A receptors.74 6-Methylapigenin induced anxiolytic effects in mice treated by the intraperitoneal route and was able to potentiate the sleep-enhancing properties of hesperidin (hesperetin 7-rhamnoglucoside), a flavanone glycoside also isolated from V. wallichii and Valeriana officinalis.75
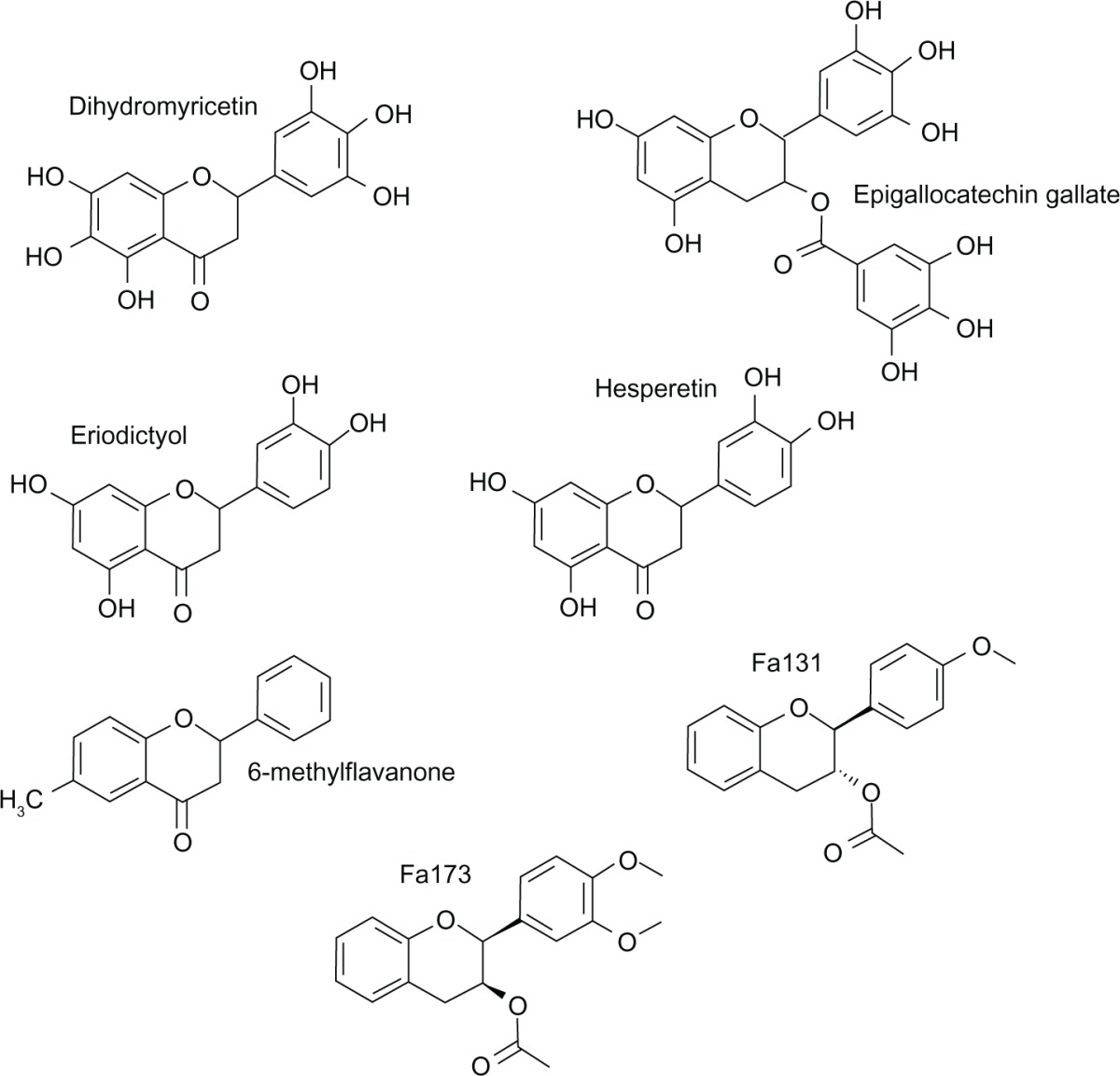
Epigallocatechin gallate (EGCG, (−)-cis-3,3′,4′,5,5′,7-hexahydroxy-flavane-3-gallate) (Figure 5), a flavanol ester and a constituent of green tea, was demonstrated to exert dose-dependent anxiolytic, sedative-hypnotic, and amnesic activities after acute intraperitoneal administration in mice, that could be mediated, at least in part, by GABA type A receptors.76,77 In vitro, this flavonoid could also inhibit activation by GABA and enhance the modulatory action of diazepam on activation by GABA of recombinant human α1β2γ2L GABA type A receptors expressed in Xenopus laevis oocytes.78
Figure 5.
Molecular structures of flavanones and flavanols.
Some natural and synthetic flavanones have been investigated in benzodiazepine binding studies. Examples are eriodictyol (5,7,3′,4′-tetrahydroxyflavanone), hesperetin (5,7,3′-trihydroxy-4′-methoxyflavanone), and flavanone itself (Figure 5), among others. All of them were found to be inactive or only weakly active in vitro.73,79,80 However, it was recently reported that dihydromyricetin (3,5,7,3′,4′,5′-hexahydroxyflavanone) (Figure 5) is a positive modulator of GABA type A receptors at benzodiazepine binding sites that competitively inhibited [3H]flunitrazepam binding with moderate affinity. Dihydromyricetin is a flavonoid component of Hovenia dulcis, an herbal medicine listed among the premier antihangover plants in China that ameliorates alcohol-induced liver injuries and relieves hangover. This flavanone, administered intraperitoneally in rats, is highly effective in counteracting acute alcohol intoxication, alcohol exposure/withdrawal-induced GABA type A receptor plasticity and alcohol withdrawal syndrome symptoms, as well as reducing excessive alcohol consumption.81
Luteolin (5,7,3′,4′-tetrahydroxyflavone) is a flavonoid aglycon found in a wide variety of plants. It has been reported to displace [3H]flunitrazepam from the benzodiazepine binding site in vitro, with low affinity, and it has demonstrated anxiolytic-like effects administered orally in mice. Despite the need to analyze the interaction of luteolin with the benzodiazepine binding site further, these results suggested that by itself this interaction does not seem to explain the results observed in vivo fully, thus prompting renewed interest in the analysis of possible interactions with other receptors.82,83
Baicalein (5,6,7-trihydroxyflavone) and baicalin (baicalein 7-O-D-glucuronide), together with wogonin and oroxylin A, are the major bioactive components in S. baicalensis. Baicalin was reported to induce an anxiolytic-like effect devoid of sedation and myorelaxation in mice when administered orally, acting via the benzodiazepine binding site. It showed significant preference for α2-containing and α3-containing subtypes compared with α1-containing and α5-containing subtypes in whole-cell patch clamp studies.84 Baicalein and baicalin also showed neuroprotective and anticonvulsant effects in rats injected intraperitoneally and the anticonvulsant effect of baicalein was inhibited by flumazenil.85 Other authors reported that baicalein showed anxiolytic and sedative effects when administered via the intracerebroventricular route in mice, and that this central effect were blocked by pentylenetetrazole but not by flumazenil. Therefore, they concluded that the in vivo actions of baicalein could be mediated by GABAergic nonbenzodiazepine binding sites.86 Another naturally occurring flavonoid isolated from S. baicalensis, ie, K36 (5,7,2′-trihydroxy-6,8-dimethoxyflavone), exhibited the highest affinity for the benzodiazepine binding site, comparable with that of the synthetic anxiolytic, diazepam (Ki 6.05 nM). In electrophysiological experiments, K36 potentiated currents mediated by the recombinant rat α1β2γ2 GABA type A receptor expressed in X. laevis oocytes and this enhancement was demonstrated to act via the benzodiazepine binding site. Oral administration of K36 produced significant benzodiazepine binding site-mediated anxiolysis in the mouse elevated plus-maze, which was abolished upon coadministration of flumazenil. Sedation, myorelaxation, and motor incoordination were not observed. Structure-activity relationships utilizing synthetic flavonoids on the flavone backbone supported that 2′-hydroxyl-substitution is a critical moiety on flavonoids with regard to benzodiazepine binding site affinity.87
Chalcones are unique in the flavonoid family in lacking a heterocyclic C ring and exhibit the basic structure with two benzene rings linked through an α, β-unsaturated carbonyl group. Isoliquiritigenin (2′,4′,4′-trihydroxychalcone) (Figure 4) is a chalcone compound found in Glycyrrhiza uralensis (licorice), Allium ascalonicum, Sinofranchetia chinensis, Dalbergia odorifera, and Glycine max L. This chalcone showed anxiolytic effects in the elevated plus-maze test administered intraperitoneally in mice and rats. It also significantly potentiated pentobarbital-induced sleep in a dose-dependent manner and this effect was fully inhibited by flumazenil. The binding affinity of isoliquiritigenin was 0.453 µM and it potentiated GABA-evoked currents on isolated dorsal raphe neurons. Thus, these results suggest that this natural chalcone produces hypnotic effects by positive allosteric modulation of the benzodiazepine binding site of the GABA type A receptor.88,89
Synthetic derivatives: importance of 6-substitutions on the flavonoid nucleus
The results obtained with natural flavonoids encouraged us to attempt to increase their potency as benzodiazepine binding site ligands by introducing electronegative substituents into their molecules, because this feature was shown to be essential for the activity of the classical benzodiazepines.90
Based on structure-activity relationship studies, incorporation of electronegative groups into the C6 and C3′ on the flavone backbone was found to yield significant increases in the binding affinities for the benzodiazepine binding site.79,80 It was also shown that 2′-hydroxyl was also a critical moiety on flavonoids with regard to benzodiazepine binding.87 These have guided the generation of several synthetic flavonoids with high benzodiazepine binding affinity and in vivo activity, and further quantitative structure-activity relationship studies resulted in the development of several pharmacophore models.
The first report of an active derivative of flavone was a brominated compound, 6-bromoflavone. This derivative showed a high affinity for the benzodiazepine binding site and resulted in a competitive ligand for these receptors. Its pharmacological profile, when administered intraperitoneally in mice, was quite similar to that observed for diazepam.91 The data to date strongly suggest that 6-bromoflavone is a full agonist of the central benzodiazepine binding site. 6-Bromo-3′-nitroflavone exhibited a different affinity for benzodiazepine binding site subtypes containing the α1 and α2/3 receptors. The plus-maze test revealed that it is anxiolytic at intraperitoneal doses of 10–300 µg/kg in mice.92,93 In contrast, 6-chloro-3′-nitroflavone and 6-methyl-3′-bromoflavone had no anxiolytic effects, and their pharmacological profile manifested antagonistic actions at the benzodiazepine binding site.94,95 In turn, 6,3′-dibromoflavone and 6-nitro-3′-bromoflavone had anxiolytic effects when administered intraperitoneally in mice, with partial agonistic behavior (Table 2).96
Table 2.
Synthetic flavone derivatives
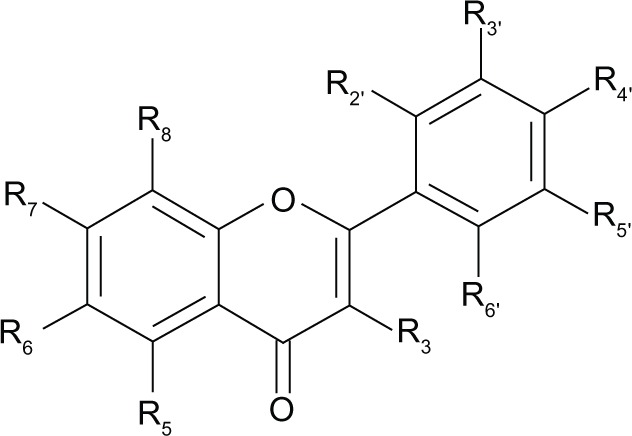
| |||||
|---|---|---|---|---|---|
| Flavonoid derivative | Kia (μM) | GABA ratiob | Pharmacological profile
|
||
| Predictedb | In vivo | In vitro | |||
| 6-bromo | 0.07091 | 1.6–2.0 | Full agonist | Full agonist | Positive modulator101 |
| 6-methyl | 0.125112 | ND | ND | ND | Positive modulator (at sites independent of flumazenil-sensitive benzodiazepine binding site)103 |
| 6-chloro | 0.164113 | ND | ND | Antagonist101 | Neutralizing modulator101 |
| 6-nitro | 0.275113 | ND | ND | ND | ND |
| 6-hydroxy | 0.580112 | ND | ND | Partial agonist102 | Positive modulator102 |
| 6-methoxy | 0.860112 | ND | ND | ND | ND |
| 6-fluoro | 4.5113 | ND | ND | Antagonist102 | Neutralizing modulator102 |
| Flavone | 191 | ND | ND | Partial agonist | ND |
| 6-bromo-3′-nitro | 0.00192 | 1.38 | Partial agonist | Partial agonist93 | ND |
| 6-methyl-3′-nitro | 0.0056114 | 0.72 | Inverse agonist | ND | ND |
| 6-chloro-3′-nitro | 0.008113 | 1.16 | Antagonist | Antagonist95 | ND |
| 6,3′-dinitro | 0.02697 | 1.30 | Partial agonist | Partial agonist98 | Low efficacy modulator99 |
| 6-fluoro-3′-nitro | 0.180113 | ND | ND | ND | ND |
| 3′-nitro | 0.285113 | ND | ND | ND | ND |
| 6,3′-dibromo | 0.019113 | 1.29 | Partial agonist | Partial agonist96 | ND |
| 6-methyl-3′-bromo | 0.01394 | 1.03 | Antagonist | Antagonist94 | ND |
| 6-chloro-3′-bromo | 0.023115 | 1.10 | Antagonist | ND | ND |
| 6-nitro-3′-bromo | 0.025113 | 1.19 | Partial agonist | Partial agonist96 | ND |
| 6-fluoro-3′-bromo | 0.236115 | ND | ND | ND | ND |
| 3′-bromo | 0.413113 | ND | ND | ND | ND |
| 6-hydroxy-3′-bromo | 1116 | ND | ND | ND | ND |
| 6-methoxy-3′-bromo | 1116 | ND | ND | ND | ND |
| 6-bromo-3′-chloro | 0.017115 | 1.23 | Partial agonist | ND | ND |
| 6,3′-dichloro | 0.023115 | 1.10 | Antagonist | ND | ND |
| 6-fluoro-3′-chloro | 0.199115 | ND | ND | ND | ND |
| 3′-chloro | 0.614113 | ND | ND | ND | ND |
| 6,3′-dimethyl | 29114 | 1.63 | Full agonist | ND | ND |
| 6-bromo-3′-methyl | 0.154116 | ND | ND | ND | ND |
| 6,3′-dimethyl | 0.208116 | ND | ND | ND | ND |
| 3′-methyl | 10116 | ND | ND | ND | ND |
| 3′-methoxy | 2.4115 | ND | ND | ND | ND |
| 3′-fluoro | 3.55115 | ND | ND | ND | ND |
| 6-bromo-3′-fluoro | 0.042115 | 0.97 | Antagonist-inverse agonist | ND | ND |
| 6-bromo-3′-methoxy | 0.609115 | ND | ND | ND | ND |
| 6-chloro-3′-fluoro | 0.117115 | ND | ND | ND | ND |
| 6-chloro-3′-methoxy | 0.848115 | ND | ND | ND | ND |
| 6,3′-difluoro | 0.920115 | ND | ND | ND | ND |
| 6-fluoro-3′-methoxy | 2.5115 | ND | ND | ND | ND |
| 3-bromo | >7591 | ND | ND | ND | ND |
| 6,3-dibromo | >7591 | ND | ND | ND | ND |
| 3-bromo-3′-nitro | >20113 | ND | ND | ND | ND |
| 2′-hydroxy | 0.31101 | ND | ND | Antagonist101 | Neutralizing modulator101 |
| 6,2′-dihydroxy | 0.04117 | ND | ND | Partial inverse agonist117 | Negative modulator117 |
| 2′-methoxy-6-methyl | >100105 | ND | ND | Partial agonist105 | Positive modulator (α1 and α2β1γ2L subtypes) Allosteric activator (α2β2/3 and α2β2/3γ2L containing subtypes)105 |
| 3-hydroxy-2′-methoxy-6-methyl | >300106 | ND | ND | Partial agonist106 | Positive allosteric modulator (α2β2/3γ2L subtypes) Direct activation (α4β2/3γ subtypes)106 |
Notes:
Ki ± standard error of the mean values are means of 3–5 independent determinations and estimate the inhibition of 3H-flunitrazepam binding to rat cerebral cortical synaptosomal membranes. The standard error of the mean varies between 6% and 13% of the absolute values listed.
The pharmacological profile of ligands interacting in vitro with the benzodiazepine binding site can be predicted through the GABA ratios obtained in binding assays. These values are the ratio of Ki values of a competitive benzodiazepine binding site ligand measured in the presence or absence of GABA: ratios > 1 indicate compounds with agonistic profiles, ratios < 1 point to compounds with inverse agonistic profiles and ratios of about 1 indicate antagonistic profiles.110,111
Abbreviations: GABA, gamma aminobutyric acid; ND, not determined.
The most active anxiolytic flavone derivative prepared in our laboratory was 6,3′-dinitroflavone. A 1–30 µg/kg intraperitoneal injection of this compound in mice produced anxiolytic-like effects. It is at least 30 times more potent by weight than diazepam and 3000 times more potent than flavone. It also has a very favorable separation index, being the separation index the ratio between the minimal sedative dose and the maximal anxiolytic one. Its magnitude qualifies the pharmacological selectivity of the compound.97,98 Inhibition of [3H]flunitrazepam binding to recombinant GABA type A receptors in transiently transfected HEK293 indicated that 6,3′dinitroflavone exhibited the highest affinity for GABA type A receptors composed of α1β2γ2 subunits and a 2–20-fold lower affinity for homologous receptors containing α2, α3, or α5 subunits. 6,3′-dinitroflavone did not elicit currents in the absence of GABA in X. laevis oocytes expressing any of the recombinant GABA type A receptors tested. However, 6,3′-dinitroflavone was slightly able to modulate GABA-elicited currents, similar to other benzodiazepine site ligands.99
Relatively extensive pharmacological studies on our synthetic flavonoids have been conducted by other authors, and the results obtained confirmed and extended our findings.100,101 Recently, flavone analogs, each varying only in the 6-position substituent, were compared. Whole-cell patch-clamp and animal behavior experiments demonstrated 6-bromoflavone to be a positive modulator at GABA type A receptors, acting via a flumazenil-sensitive high-affinity benzodiazepine binding site. In contrast, 6-fluoroflavone and 6-chloroflavone were found to be neutralizing modulators. In patch-clamp studies, 6-hydroxyflavone displayed a significant preference for α2-containing and α3-containing subtypes, which were thought to mediate the anxiolytic effect, compared with α1-and α5-containing subtypes expressed in HEK 293T cells. In mice, 6-hydroxyflavone exhibited anxiolytic-like effects unaccompanied by the sedative, cognitive impairment, myorelaxant, motor incoordination, and anticonvulsant effects commonly associated with classical benzodiazepines.102 In addition, in vitro electrophysiological and in vivo animal experiments showed that 2′-hydroxyflavone was an antagonist, different in efficacy from its structural analog, 6,2′-dihydroxyflavone, a negative modulator of GABA type A receptors. The fact that flavone derivatives differing only at position 6 showed drastically different pharmacological properties clearly points to 6-substitution being an important determinant of efficacy. All the results suggest that a large width of the first atom on the 6-substituent favors a high binding affinity of the 6-substituted flavone, whereas a large overall volume of the 6-substituent favors positive modulator activity, which could be modified by, eg, 2′-hydroxyl substitution.101
6-Methylflavone was found to be a positive allosteric modulator at α1β2γ2L and α1β2 GABA type A receptors, with no significant difference between the enhancement seen at either receptor subtype, at ionotropic GABA type A receptors expressed in X. laevis oocytes and at sites independent of the flumazenil-sensitive benzodiazepine binding site.103 Subsequently, 6-methylflavanone (Figure 5) was also found to be a flumazenil-insensitive positive modulator of recombinant GABA type A receptors that, unlike 6-methylflavone, was subtype-selective, being a more efficacious positive modulator at α2β2γ2L receptors that at α1β2γ2L and α1β2 receptors.104 6-Methylflavanone differs from 6-methylflavone in having a single rather than a double bond at C2–C3; hence, this bond is crucial to the observed subtype-selective efficacy of 6-methylflavanone. Two new 6-methylflavone derivatives were recently reported, ie, 2′-methoxy-6-methylflavone105 and 3-hydroxy-2′-methoxy-6-methylflavone.106 2′-Methoxy-6-methylflavone potentiated GABA at α2β1γ2L and all α1-containing GABA type A receptor subtypes. However, it directly activated α2β2/3γ2L GABA type A receptors without potentiating GABA. This activation was attenuated by bicuculline and gabazine, but not flumazenil, indicating a novel site. In mice, when administered intraperitoneally, it displayed anxiolytic-like effects, and at higher doses induced sedation. Like the in vitro data, the anxiolytic effects of 2′-methoxy-6-methylflavone were not blocked by flumazenil, a benzodiazepine antagonist, but were attenuated by pentylenetetrazole, a GABA type A channel blocker. These data imply that the anxiolytic effects are mediated via the GABAergic system, but not through the classical benzodiazepine binding site, despite 2′-methoxy-6-methylflavone weakly displacing flunitrazepam binding. These data would indicate that 2′-methoxy-6-methylflavone could bind to a novel as yet unidentified site on α2β2/3γ2L GABA type A receptors. 2′-Methoxy-6-methylflavone could serve as a tool to study the complex nature of the activation and modulation of GABA type A receptor subtypes. In addition, the synthetic flavonoid, 3-hydroxy-2′-methoxy-6-methylflavone was reported to be an anxiolytic without sedative and myorelaxant effects when administered intraperitoneally in mice, acting through positive allosteric modulation of the α2β2/3γ2L and direct activation of α4β2/3δ GABA type A receptor subtypes (Table 2).
Using 6-methylflavanone and EGCG as lead compounds, structure-activity studies led to the discovery of Fa131 (2S,3R-3-acetoxy-4′-methoxyflavan) (Figure 5) as the most efficacious compound in a series of flavonol esters with positive modulatory activity of GABA type A receptors.107,108 Interestingly, similar to barbiturates, Fa131 also acts as a weak partial agonist α1β2γ2L receptor and at higher doses exerts a negative modulatory effect.107 Fa131 is the first positive modulator to distinguish between the α2-subunit and α3-subunit containing GABA type A receptors, highlighting the potential of targeting flumazenil-insensitive allosteric sites in the search for new anxioselective drugs. In mice, when administered intraperitoneally, it induced an anxiolytic-like action with no sedative or myorelaxant effects, and only weak barbiturate-potentiating effects on the loss of the righting reflex test. Recently there has been a report of a new flavan, Fa173 (2S;3S-3 acetoxy-3′,4′-dimethoxyflavan) (Figure 5), which neutralizes the potentiating actions of Fa131, etomidate, and loreclezole at α1β2 and α1β2γ2L GABA type A receptors expressed in X. laevis oocytes. Furthermore potentiation of high, but not low, concentrations of diazepam can be blocked by Fa173.109
A series of isoflavone derivatives was recently synthesized, and their modulatory effect was evaluated on the α1β2γ2L GABA type A receptors expressed in X. laevis oocytes. This set of isoflavones acted as positive modulators of GABA type A receptors and it was demonstrated that substitution of the A, B, and C rings plays an important role in determining GABA type A modulation activity. Flumazenil-insensitive modulation by the isoflavones suggested that these compounds might not bind to the benzodiazepine binding site.118
Since the first discovery of a flavonoid as a benzodiazepine binding site ligand almost 30 years ago, an extensive collection of natural and synthetic flavonoids has been reported providing potential leads for new GABA type A receptor agents and they have already become drugs with beneficial effects in the central nervous system. The in vivo and in vitro actions of some flavonoids are more complex than a single action at the benzodiazepine binding site, and some evidence of a flavonoid site in GABA type A receptors is emerging. More studies are required in order to determine the precise site of action of these bioactive molecules on GABA type A receptors and to understand fully their mechanisms of action as modulators of brain function. Also, further investigations of type specificity of flavonoids might lead to identification of flavonoids with selective pharmacological activity, thus providing a clinically interesting lead structure. Further, the concentrations of dietary flavonoids encountered in vivo are high enough to have pharmacological activity at receptors, and the evidence also supports localization of flavonoids within the brain.
In summary, flavonoids are prominent drugs in the treatment of mental disorders, and can also be used as tools to study modulatory sites at GABA type A receptors and to develop GABA subtype-selective agents further.
Acknowledgments
We are grateful to the International Foundation for Science, Stockholm, Sweden, the National Research Council of Argentina, the University of Buenos Aires, and the Ministry of Health and the National Agency for Promotion of Science and Technology, Argentina, for financial support.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Tapas AR, Sakarkar DM, Kakde RB. Flavonoids as nutraceuticals: a review. Trop J Pharm Res. 2008;7:1089–1099. [Google Scholar]
- 2.Buer CS, Imin N, Djordjevic MA. Flavonoids: new roles for old molecules. J Integr Plant Biol. 2010;52:98–111. doi: 10.1111/j.1744-7909.2010.00905.x. [DOI] [PubMed] [Google Scholar]
- 3.Spencer JPE. Beyond antioxidants: the cellular and molecular interactions of flavonoids and how these underpin their actions on the brain. Proc Nutr Soc. 2010;69:244–260. doi: 10.1017/S0029665110000054. [DOI] [PubMed] [Google Scholar]
- 4.Taylor LP, Grotewold E. Flavonoids as developmental regulators. Curr Opin Plant Biol. 2005;8:317–323. doi: 10.1016/j.pbi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 5.González R, Ballester I, López-Posadas R, et al. Effects of flavonoids and other polyphenols on inflammation. Crit Rev Food Sci Nutr. 2011;51:331–362. doi: 10.1080/10408390903584094. [DOI] [PubMed] [Google Scholar]
- 6.Shanmugam MK, Kannaiyan R, Sethi G. Targeting cell signaling and apoptotic pathways by dietary agents: role in the prevention and treatment of cancer. Nutr Cancer. 2011;63:161–173. doi: 10.1080/01635581.2011.523502. [DOI] [PubMed] [Google Scholar]
- 7.Xiao ZP, Peng ZY, Peng MJ, Yan WB, Ouyang YZ, Zhu HL. Flavonoids health benefits and their molecular mechanism. Mini Rev Med Chem. 2011;11:169–177. doi: 10.2174/138955711794519546. [DOI] [PubMed] [Google Scholar]
- 8.Tuñón MJ, García-Mediavilla MV, Sánchez-Campos S, González-Gallego J. Potential of flavonoids as anti-inflammatory agents: modulation of pro-inflammatory gene expression and signal transduction pathways. Curr Drug Metab. 2009;10:256–271. doi: 10.2174/138920009787846369. [DOI] [PubMed] [Google Scholar]
- 9.Williamson G, Manach C. Bioavailability and bioefficacy of polyphenols in humans. II. Review of 93 intervention studies. Am J Clin Nutr. 2005;81:243S–255S. doi: 10.1093/ajcn/81.1.243S. [DOI] [PubMed] [Google Scholar]
- 10.Manach C, Williamson G, Morand C, Scalbert A, Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81:230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 11.Spencer JPE, Abd El Mohsen MM, Minihane AM, Mathers JC. Biomarkers of the intake of dietary polyphenols: strengths, limitations and application in nutrition research. Br J Nutr. 2008;99:12–22. doi: 10.1017/S0007114507798938. [DOI] [PubMed] [Google Scholar]
- 12.Spencer JPE. Metabolism of tea flavonoids in the gastrointestinal tract. J Nutr. 2003;133(Suppl):3255S–3261S. doi: 10.1093/jn/133.10.3255S. [DOI] [PubMed] [Google Scholar]
- 13.Day AJ, Canada FJ, Diaz JC, et al. Dietary flavonoid and isoflavone glycosides are hydrolysed by the lactase site of lactase phlorizin hydrolase. FEBS Lett. 2000;468:166–170. doi: 10.1016/s0014-5793(00)01211-4. [DOI] [PubMed] [Google Scholar]
- 14.Walgren RA, Lin JT, Kinne RK, Walle T. Cellular uptake of dietary flavonoid quercetin 4′-beta-glucoside by sodium-dependent glucose transporter SGLT1. J Pharmacol Exp Ther. 2000;294:837–843. [PubMed] [Google Scholar]
- 15.Wolffram S, Block M, Ader P. Quercetin-3-glucoside is transported by the glucose carrier SGLT1 across the brush border membrane of rat small intestine. J Nutr. 2002;132:630–635. doi: 10.1093/jn/132.4.630. [DOI] [PubMed] [Google Scholar]
- 16.Nemeth K, Plumb GW, Berrin JG, et al. Deglycosylation by small intestinal epithelial cell beta-glucosidases is a critical step in the absorption and metabolism of dietary flavonoid glycosides in humans. Eur J Nutr. 2003;42:29–42. doi: 10.1007/s00394-003-0397-3. [DOI] [PubMed] [Google Scholar]
- 17.Radominska-Pandya A, Little JM, Pandya JT, et al. UDP-glucuronosyltransferases in human intestinal mucosa. Biochim Biophys Acta. 1998;1394:199–208. doi: 10.1016/s0005-2760(98)00115-5. [DOI] [PubMed] [Google Scholar]
- 18.O’Leary KA, Day AJ, Needs PW, Mellon FA, O’Brien NM, Williamson G. Metabolism of quercetin-7- and quercetin-3-glucuronides by an in vitro hepatic model: the role of human beta-glucuronidase, sulfotransferase, catechol-O-methyltransferase and multi-resistant protein 2 (MRP2) in flavonoid metabolism. Biochem Pharmacol. 2003;65:479–491. doi: 10.1016/s0006-2952(02)01510-1. [DOI] [PubMed] [Google Scholar]
- 19.Matsukawa N, Matsumoto M, Hara H. High biliary excretion levels of quercetin metabolites after administration of a quercetin glycoside in conscious bile duct cannulated rats. Biosci Biotechnol Biochem. 2009;73:1863–1865. doi: 10.1271/bbb.90031. [DOI] [PubMed] [Google Scholar]
- 20.Scheline RR. Handbook of Mammalian Metabolism of Plant Compounds. Boca Raton, FL: CRC Press Inc; 1999. Metabolism of oxygen heterocyclic compounds. [Google Scholar]
- 21.Unwin N. The structure of ion channels in membranes of excitable cells. Neuron. 1989;3:665–676. doi: 10.1016/0896-6273(89)90235-3. [DOI] [PubMed] [Google Scholar]
- 22.Barnard EA, Skolnick P, Olsen RW, et al. International Union of Pharmacology. XV. Subtypes of γ-aminobutyric acid A receptors: Classification on the basis of subunit structure and receptor function. Pharmacol Rev. 1998;50:291–313. [PubMed] [Google Scholar]
- 23.Whiting P, McKernan RM, Iversen LL. Another mechanism for creating diversity in gamma-aminobutyrate type A receptors: RNA splicing directs expression of two forms of gamma 2 phosphorylation site. Proc Natl Acad Sci U S A. 1990;87:9966–9970. doi: 10.1073/pnas.87.24.9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinley DD, Lennon DJ, Carter DB. Cloning, sequence analysis and expression of two forms of mRNA coding for the human beta 2 subunit of the GABAA receptor. Mol Brain Res. 1995;28:175–179. doi: 10.1016/0169-328x(94)00228-7. [DOI] [PubMed] [Google Scholar]
- 25.Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: Classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Benke D, Fakitsas P, Roggenmoser C, Michel C, Rudolph U, Mohler H. Analysis of the presence and abundance of GABAA receptors containing two different types of alpha subunits in murine brain using point-mutated alpha subunits. J Biol Chem. 2004;279:43654–43660. doi: 10.1074/jbc.M407154200. [DOI] [PubMed] [Google Scholar]
- 27.Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korpi ER, Gründer G, Lüddens H. Drug interactions at GABA(A) receptors. Prog Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 29.Sieghart W. Structure and pharmacology of gamma-aminobutyric acid A receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- 30.Iversen L. GABA pharmacology – what prospects for the future? Biochem Pharmacol. 2004;68:1537–1540. doi: 10.1016/j.bcp.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 31.Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263. doi: 10.1016/s1054-3589(06)54010-4. [DOI] [PubMed] [Google Scholar]
- 32.Olsen RW, Sieghart W. GABAA receptors: Subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rudolph U, Mohler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol. 2004;44:475–498. doi: 10.1146/annurev.pharmtox.44.101802.121429. [DOI] [PubMed] [Google Scholar]
- 34.Whiting PJ. GABAA-receptors: a viable target for novel anxiolytics? Curr Opin Pharmacol. 2006;6:24–29. doi: 10.1016/j.coph.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Rudolph U, Crestani F, Benke D, et al. H. Benzodiazepine actions mediated by specific gammaaminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- 36.McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- 37.Low K, Crestani F, Keist R, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- 38.Dias R, Sheppard WF, Fradley RL, et al. Evidence for a significant role of alpha3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci. 2005;25:10682–10688. doi: 10.1523/JNEUROSCI.1166-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collinson N, Kuenzi FM, Jarolimek W, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malherbe P, Draguhn A, Multhaup G, Beyreuther K, Möhler H. GABAA-receptor expressed from rat brain α- and β-subunit cDNAs displays potentiation by benzodiazepine receptor ligands. Mol Brain Res. 1990;8:199–208. doi: 10.1016/0169-328x(90)90017-8. [DOI] [PubMed] [Google Scholar]
- 41.Walters RJ, Hadley SH, Morris DW, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci. 2000;3:1274–1281. doi: 10.1038/81800. [DOI] [PubMed] [Google Scholar]
- 42.Luk KC, Stern L, Weigele M, O’Brien RA, Spirst N. Isolation and identification of “diazepam-like” compounds from bovine urine. J Nat Prod. 1983;46:852–861. doi: 10.1021/np50030a005. [DOI] [PubMed] [Google Scholar]
- 43.Medina JH, Danelon JL, Wasowski C, Levi de Stein M, Paladini AC. Production of benzodiazepine-like compounds in bovine rumen. Biochem Biophys Res Commun. 1991;181:1048–1055. doi: 10.1016/0006-291x(91)92043-j. [DOI] [PubMed] [Google Scholar]
- 44.Nielsen M, Frokjaer S, Braestrup C. High affinity of naturally-occurring biflavonoid, amentoflavone, to brain benzodiazepine receptors in vitro. Biochem Pharmacol. 1988;37:3285–3287. doi: 10.1016/0006-2952(88)90640-5. [DOI] [PubMed] [Google Scholar]
- 45.Hansen RS, Paulsen I, Davies M. Determinants of amentoflavone interaction at the GABA(A) receptor. Eur J Pharmacol. 2005;519:199–207. doi: 10.1016/j.ejphar.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 46.Hanrahan JR, Chebib M, Davucheron NM, Hall BJ, Johnston GAR. Semisynthetic preparation of amentoflavone: A negative modulator at GABAA receptors. Bioorg Med Chem Lett. 2003;13:2281–2284. doi: 10.1016/s0960-894x(03)00434-7. [DOI] [PubMed] [Google Scholar]
- 47.Butterweck V, Nahrstedt A, Evans J, et al. In vitro receptor screening of pure constituents of St John’s wort reveals novel interactions with a number of GPCRs. Psychopharmacology. 2002;162:193–202. doi: 10.1007/s00213-002-1073-7. [DOI] [PubMed] [Google Scholar]
- 48.Medina JH, Peña C, Levi de Stein M, Wolfman C, Paladini AC. Benzodiazepine-like molecules as well as other ligands for the brain benzodiazepine receptor are relatively common constituents of plants. Biochem Biophys Res Commun. 1989;165:547–553. doi: 10.1016/s0006-291x(89)80001-4. [DOI] [PubMed] [Google Scholar]
- 49.Medina JH, Viola H, Wolfman C, et al. Overview – flavonoids: a new family of benzodiazepine receptor ligands. Neurochem Res. 1997;22:419–425. doi: 10.1023/a:1027303609517. [DOI] [PubMed] [Google Scholar]
- 50.Paladini AC, Marder M, Viola H, Wolfman C, Wasowski C, Medina JH. Flavonoids and the central nervous system: from forgotten factors to potent anxiolytic compounds. J Pharm Pharmacol. 1999;51:519–526. doi: 10.1211/0022357991772790. [DOI] [PubMed] [Google Scholar]
- 51.Marder M, Paladini AC. GABA(A)-receptor ligands of flavonoid structure. Curr Top Med Chem. 2002;2:853–867. doi: 10.2174/1568026023393462. [DOI] [PubMed] [Google Scholar]
- 52.Medina JH, Paladini AC, Wolfman C, et al. Chrysin (5,7 di-OH flavone) a naturally-occurring ligand for benzodiazepine receptors, with anticonvulsant properties. Biochem Pharmacol. 1990;40:2227–2232. doi: 10.1016/0006-2952(90)90716-x. [DOI] [PubMed] [Google Scholar]
- 53.Wolfman C, Viola H, Paladini AC, Dajas F, Medina JH. Possible anxiolytic effects of chrysin, a central benzodiazepine receptor ligand isolated from Passiflora coerulea. Pharmacol Biochem Behav. 1994;47:1–4. doi: 10.1016/0091-3057(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 54.Zhai K, Hu L, Chen J, Fu CY, Chen Q. Chrysin induces hyperalgesia via the GABAA receptor in mice. Planta Med. 2008;74:1229–1234. doi: 10.1055/s-2008-1081288. [DOI] [PubMed] [Google Scholar]
- 55.Viola H, Wasowski C, Levi de Stein M, et al. Apigenin, a component of Matricaria recutita flowers, is a central benzodiazepine receptors-ligand with anxiolytic effects. Planta Med. 1995;61:213–216. doi: 10.1055/s-2006-958058. [DOI] [PubMed] [Google Scholar]
- 56.Avallone R, Zanoli P, Puia G, Kleinschnitz M, Schreier P, Baraldi M. Pharmacological profile of apigenin, a flavonoid isolated from Matricaria chamomilla. Biochem Pharmacol. 2000;59:1387–1394. doi: 10.1016/s0006-2952(00)00264-1. [DOI] [PubMed] [Google Scholar]
- 57.Zanoli P, Avallone R, Baraldi M. Behavioral characterization of the flavonoids apigenin and chrysin. Fitoterapia. 2000;71:S117–S123. doi: 10.1016/s0367-326x(00)00186-6. [DOI] [PubMed] [Google Scholar]
- 58.Dekermendjian K, Kahnberg P, Witt MR, Sterner O, Nielsen M, Liljefors T. Structure–activity relationships and molecular modeling analysis of flavonoids binding to the benzodiazepine site of the rat brain GABAA receptor complex. J Med Chem. 1999;42:4343–4350. doi: 10.1021/jm991010h. [DOI] [PubMed] [Google Scholar]
- 59.Goutman JD, Waxemberg MD, Donate-Oliver F, Pomata PE, Calvo DJ. Flavonoid modulation of ionic currents mediated by GABAA and GABAC receptors. Eur J Pharmacol. 2003;461:79–87. doi: 10.1016/s0014-2999(03)01309-8. [DOI] [PubMed] [Google Scholar]
- 60.Campbell EL, Chebib M, Johnston GAR. The dietary flavonoids apigenin and (−)-epigallocatechin gallate enhance the positive modulation by diazepam of the activation by GABA of recombinant GABAA receptors. Biochem Pharmacol. 2004;68:1631–1638. doi: 10.1016/j.bcp.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 61.Viola H, Wolfman C, Levi de Stein M, et al. Isolation of pharmacologically active benzodiazepine receptor ligands from Tília tomentosa (Tiliaceae) J Ethnopharmacol. 1994;44:47–53. doi: 10.1016/0378-8741(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 62.Viola H, Marder M, Wolfman C, Wasowski C, Medina JH, Paladini AC. Sedative and hypnotic properties of Salvia guaranítica St Hil and of its active principle, cirsiliol. Phytomedicine. 1997;4:45–50. doi: 10.1016/S0944-7113(97)80027-X. [DOI] [PubMed] [Google Scholar]
- 63.Marder M, Viola H, Wasowski C, et al. Cirsiliol and caffeic acid ethyl ester isolated from Salvia guaranitica, are competitive ligands for the central benzodiazepine receptors. Phytomedicine. 1996;3:29–31. doi: 10.1016/S0944-7113(96)80006-7. [DOI] [PubMed] [Google Scholar]
- 64.Vissiennona C, Nieber K, Kelber O, Butterweck V. Route of administration determines the anxiolytic activity of the flavonols kaempferol, quercetin and myricetin – are they prodrugs? J Nutr Biochem. 2011 Aug 11; doi: 10.1016/j.jnutbio.2011.03.017. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 65.Grundmann O, Nakajima J, Kamata K, Seo S, Butterweck V. Kaempferol from the leaves of Apocynum venetum possesses anxiolytic activities in the elevated plus maze test in mice. Phytomedicine. 2009;16:295–302. doi: 10.1016/j.phymed.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 66.Hui KM, Huen MS, Wang HY, et al. Anxiolytic effect of wogonin, a benzodiazepine receptor ligand isolated from Scutellaria baicalensis Georgi. Biochem Pharmacol. 2002;64:1415–1424. doi: 10.1016/s0006-2952(02)01347-3. [DOI] [PubMed] [Google Scholar]
- 67.Park HG, Yoon SY, Choi JY, et al. Anticonvulsant effect of wogonin isolated from Scutellaria baicalensis. Eur J Pharmacol. 2007;574:112–119. doi: 10.1016/j.ejphar.2007.07.011. [DOI] [PubMed] [Google Scholar]
- 68.Huen MS, Leung JW, Ng W, et al. 5,7-Dihydroxy-6-methoxyflavone, a benzodiazepine site ligand isolated from Scutellaria baicalensis Georgi, with selective antagonistic properties. Biochem Pharmacol. 2003;66:125–1132. doi: 10.1016/s0006-2952(03)00233-8. [DOI] [PubMed] [Google Scholar]
- 69.Shen XL, Nielsen M, Witt MR, Sterner O, Bergendorff O, Khayyal M. Inhibition of [methyl-3H]diazepam binding to rat brain membranes in vitro by dinatin and skrofulein. Zhongguo Yao Li Xue Bao. 1994;15:385–388. [PubMed] [Google Scholar]
- 70.Kavvadias D, Monschein V, Sand P, Riederer P, Schreier P. Constituents of sage (Salvia officinalis) with in vitro affinity to human brain benzodiazepine receptor. Planta Med. 2003;69:113–117. doi: 10.1055/s-2003-37712. [DOI] [PubMed] [Google Scholar]
- 71.Kavvadias D, Sand P, Youdim KA, et al. The flavone hispidulin, a benzodiazepine receptor ligand with positive allosteric properties, traverses the blood-brain barrier and exhibits anticonvulsive effects. Br J Pharmacol. 2004;142:811–820. doi: 10.1038/sj.bjp.0705828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shen XL, Witt MR, Nielsen M, Sterner O. Inhibition of [3H] flunitraze-pam binding to rat brain membranes in vitro by puerarin and daidzein. Yao Xue Xue Bao. 1996;31:59–62. [PubMed] [Google Scholar]
- 73.Häberlein H, Tschiersch KP, Schäfer HL. Flavonoids from Leptospermum scoparium with affinity to the benzodiazepine receptor characterized by structure activity relationships and in vivo studies of a plant extract. Pharmazie. 1994;49:912–922. [PubMed] [Google Scholar]
- 74.Wasowski C, Marder M, Viola H, Medina JH, Paladini AC. Isolation and identification of 6-methylapigenin, a competitive ligand for the brain GABA(A) receptors, from Valeriana wallichii. Planta Med. 2002;68:934–936. doi: 10.1055/s-2002-34936. [DOI] [PubMed] [Google Scholar]
- 75.Marder M, Viola H, Wasowski C, Fernandez S, Medina JH, Paladini AC. 6-Methylapigenin and hesperidin: new valeriana flavonoids with activity on the CNS. Pharmacol Biochem Behav. 2003;75:537–545. doi: 10.1016/s0091-3057(03)00121-7. [DOI] [PubMed] [Google Scholar]
- 76.Adachi N, Tomonaga S, Tachibana T, Denbow DM, Furuse M. (−)-Epigallocatechin gallate attenuates acute stress responses through GABAergic system in the brain. Eur J Pharmacol. 2006;531:171–175. doi: 10.1016/j.ejphar.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 77.Vignes M, Maurice T, Lanté F, et al. Anxiolytic properties of green tea polyphenol (−)-epigallocatechin gallate (EGCG) Brain Res. 2006;1110:102–115. doi: 10.1016/j.brainres.2006.06.062. [DOI] [PubMed] [Google Scholar]
- 78.Campbell EL, Chebib M, Johnston GA. The dietary flavonoids apigenin and (−)-epigallocatechin gallate enhance the positive modulation by diazepam of the activation by GABA of recombinant GABA(A) receptors. Biochem Pharmacol. 2004;68:1631–1638. doi: 10.1016/j.bcp.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 79.Paladini AC, Marder M, Viola H, Wolfman C, Wasowski C, Medina JH. Flavonoids and the central nervous system: from forgotten factors to potent anxiolytic compounds. J Pharm Pharmacol. 1999;51:519–526. doi: 10.1211/0022357991772790. [DOI] [PubMed] [Google Scholar]
- 80.Marder M, Paladini AC. GABA(A)-receptor ligands of flavonoid structure. Curr Top Med Chem. 2002;2:853–867. doi: 10.2174/1568026023393462. [DOI] [PubMed] [Google Scholar]
- 81.Shen Y, Lindemeyer AK, Gonzalez C, et al. Dihydromyricetin as a novel anti-alcohol intoxication medication. J Neurosci. 2012;32:390–401. doi: 10.1523/JNEUROSCI.4639-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coleta M, Campos MG, Cotrim MD, Lima TC, Cunha AP. Assessment of luteolin (3′,4′,5,7-tetrahydroxyflavone) neuropharmacological activity. Behav Brain Res. 2008;189:75–82. doi: 10.1016/j.bbr.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 83.Raines T, Jones P, Moe N, Duncan R, McCall S, Ceremuga TE. Investigation of the anxiolytic effects of luteolin, a lemon balm flavonoid in the male Sprague-Dawley rat. AANA J. 2009;77:33–36. [PubMed] [Google Scholar]
- 84.Wang F, Xu Z, Ren L, Tsang SY, Xue H. GABA A receptor subtype selectivity underlying selective anxiolytic effect of baicalin. Neuropharmacology. 2008;55:1231–1237. doi: 10.1016/j.neuropharm.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 85.Yoon SY, de la Peña IC, Shin CY, et al. Convulsion-related activities of Scutellaria flavones are related to the 5,7-dihydroxyl structures. Eur J Pharmacol. 2011;659:155–160. doi: 10.1016/j.ejphar.2011.03.012. [DOI] [PubMed] [Google Scholar]
- 86.de Carvalho RS, Duarte FS, de Lima TC. Involvement of GABAergic non-benzodiazepine sites in the anxiolytic-like and sedative effects of the flavonoid baicalein in mice. Behav Brain Res. 2011;221:75–82. doi: 10.1016/j.bbr.2011.02.038. [DOI] [PubMed] [Google Scholar]
- 87.Huen MS, Hui KM, Leung JW, et al. Naturally occurring 2′-hydroxyl-substituted flavonoids as high-affinity benzodiazepine site ligands. Biochem Pharmacol. 2003;66:2397–2407. doi: 10.1016/j.bcp.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 88.Cho S, Kim S, Jin Z, et al. Isoliquiritigenin, a chalcone compound, is a positive allosteric modulator of GABAA receptors and shows hypnotic effects. Biochem Biophys Res Commun. 2011;413:637–642. doi: 10.1016/j.bbrc.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 89.Jamal H, Ansari WH, Rizvi SJ. Evaluation of chalcones – a flavonoid subclass, for, their anxiolytic effects in rats using elevated plus maze and open field behaviour tests. Fundam Clin Pharmacol. 2008;22:673–681. doi: 10.1111/j.1472-8206.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- 90.Sternbach LH. The benzodiazepine story. Prog Drug Res. 1978;22:229–266. doi: 10.1007/978-3-0348-7102-0_5. [DOI] [PubMed] [Google Scholar]
- 91.Marder M, Viola H, Wasowski C, et al. 6 Bromoflavone, a high affinity ligand for the benzodiazepine receptors is a member of a family of active flavonoids. Biochem Biophys Res Commun. 1996;223:384–389. doi: 10.1006/bbrc.1996.0903. [DOI] [PubMed] [Google Scholar]
- 92.Viola H, Marder M, Wolfman C, Wasowski C, Medina JH, Paladini AC. 6-Bromo-3′-nitroflavone, a new high affinity benzodiazepine receptor agonist recognizes two populations of cerebral cortical binding sites. Bioorg Med Chem Lett. 1997;7:373–378. [Google Scholar]
- 93.Wolfman C, Viola H, Marder M, et al. Pharmacological characterization of 6-bromo-3′-nitroflavone, a synthetic flavonoid with high affinity for the benzodiazepine receptor. Pharmacol Biochem Behav. 1998;61:239–246. doi: 10.1016/s0091-3057(98)00088-4. [DOI] [PubMed] [Google Scholar]
- 94.Viola H, Marder M, Nuñez J, et al. 6-Methyl-3′-bromoflavone, a high-affinity ligand for the benzodiazepine binding site of the GABAA receptor with some antagonistic properties. Biochem Biophys Res Commun. 1999;262:643–646. doi: 10.1006/bbrc.1999.1273. [DOI] [PubMed] [Google Scholar]
- 95.Viola H, Wolfman C, Marder M, et al. 6-Chloro-3′-nitroflavone is a potent ligand for the benzodiazepine binding site of the GABA(A) receptor devoid of intrinsic activity. Pharmacol Biochem Behav. 2000;65:313–320. doi: 10.1016/s0091-3057(99)00199-9. [DOI] [PubMed] [Google Scholar]
- 96.Viola H, Marder M, Wasowski C, Giorgi O, Paladini AC, Medina JH. 6,3′-Dibromoflavone and 6-nitro-3′-bromoflavone: new additions to the 6,3′-disubstituted flavone family of high-affinity ligands of the brain benzodiazepine binding site with agonistic properties. Biochem Biophys Res Commun. 2000;273:694–698. doi: 10.1006/bbrc.2000.2979. [DOI] [PubMed] [Google Scholar]
- 97.Marder M, Viola H, Wasowski C, et al. 6,3′-Dinitroflavone, a novel high affinity ligand for the benzodiazepine receptor with potent anxiolytic properties. Bioorg Med Chem Lett. 1995;5:2717–2720. [Google Scholar]
- 98.Wolfman C, Viola H, Marder M, et al. Anxioselective properties of 6,3′-dinitroflavone, a high-affinity benzodiazepine receptor ligand. Eur J Pharmacol. 1996;318:23–29. doi: 10.1016/s0014-2999(96)00784-4. [DOI] [PubMed] [Google Scholar]
- 99.Furtmueller R, Furtmueller B, Ramerstorfer J, et al. 6,3′-Dinitroflavone is a low efficacy modulator of GABAA receptors. Eur J Pharmacol. 2008;591:142–146. doi: 10.1016/j.ejphar.2008.06.093. [DOI] [PubMed] [Google Scholar]
- 100.Griebel G, Perrault G, Tan S, Schoemaker H, Sanger DJ. Pharmacological studies on synthetic flavonoids: comparison with diazepam. Neuropharmacology. 1999;38:965–977. doi: 10.1016/s0028-3908(99)00026-x. [DOI] [PubMed] [Google Scholar]
- 101.Ren L, Chan WM, Wang F, et al. Effects of flavone 6-substitutions on GABAA receptors efficacy. Eur J Pharmacol. 2011;670:121–129. doi: 10.1016/j.ejphar.2011.08.021. [DOI] [PubMed] [Google Scholar]
- 102.Ren L, Wang F, Xu Z, Chan WM, Zhao C, Xue H. GABA(A) receptor subtype selectivity underlying anxiolytic effect of 6-hydroxyflavone. Biochem Pharmacol. 2010;79:1337–1344. doi: 10.1016/j.bcp.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 103.Hall BJ, Chebib M, Hanrahan JR, Johnston GA. Flumazenil-independent positive modulation of gamma-aminobutyric acid action by 6-methylflavone at human recombinant alpha1beta2gamma2L and alpha1beta2 GABAA receptors. Eur J Pharmacol. 2004;491:1–8. doi: 10.1016/j.ejphar.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 104.Hall BJ, Chebib M, Hanrahan JR, Johnston GA. 6-Methylflavanone, a more efficacious positive allosteric modulator of gamma-aminobutyric acid (GABA) action at human recombinant alpha2beta2gamma2L than at alpha1beta2gamma2L and alpha1beta2 GABA(A) receptors expressed in Xenopus oocytes. Eur J Pharmacol. 2005;512:97–104. doi: 10.1016/j.ejphar.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 105.Karim N, Curmi J, Gavande N, et al. 2′-Methoxy-6-methylflavone: a novel anxiolytic and sedative with subtype selective activating and modulating actions at GABA(A) receptors. Br J Pharmacol. 2012;165(4):880–896. doi: 10.1111/j.1476-5381.2011.01604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karim N, Gavande N, Wellendorph P, Johnston GA, Hanrahan JR, Chebib M. 3-Hydroxy-2′-methoxy-6-methylflavone: a potent anxiolytic with a unique selectivity profile at GABA(A) receptor subtypes. Biochem Pharmacol. 2011;82:1971–1983. doi: 10.1016/j.bcp.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 107.Fernandez SP, Mewett KN, Hanrahan JR, Chebib M, Johnston GA. Flavan-3-ol derivatives are positive modulators of GABA(A) receptors with higher efficacy for the alpha(2) subtype and anxiolytic action in mice. Neuropharmacology. 2008;55:900–907. doi: 10.1016/j.neuropharm.2008.06.069. [DOI] [PubMed] [Google Scholar]
- 108.Mewett KN, Fernandez SP, Pasricha AK, et al. Synthesis and biological evaluation of flavan-3-ol derivatives as positive modulators of GABAA receptors. Bioorg Med Chem. 2009;17:7156–7173. doi: 10.1016/j.bmc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 109.Fernandez SP, Karim N, Mewett KN, Chebib M, Johnston GA, Hanrahan JR. Flavan-3-ol esters: new agents for exploring modulatory sites on GABA(A) receptors. Br J Pharmacol. 2012;165:965–977. doi: 10.1111/j.1476-5381.2011.01615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Facklam M, Schoch P, Haefely WE. Relationship between benzodiazepine receptor occupancy and potentiation of gamma-aminobutyric acid-stimulated chloride flux in vitro of four ligands of differing intrinsic efficacies. J Pharmacol Exp Ther. 1992;261:1106–1112. [PubMed] [Google Scholar]
- 111.Braestrup C, Nielsen M, Honore T, Jensen L, Petersen E. Benzodiazepine receptor ligands with positive and negative efficacy. Neuropharmacology. 1983;22:1451–1457. doi: 10.1016/0028-3908(83)90113-2. [DOI] [PubMed] [Google Scholar]
- 112.Ai J, Dekermendjian K, Wang X, Nielsen M, Witt MR. 6-Methylflavone, a benzodiazepine receptor ligand with antagonistic properties on rat brain and human recombinant GABAA receptors in vitro. Drug Dev Res. 1997;41:99–108. [Google Scholar]
- 113.Marder M, Zinczuk J, Colombo MI, et al. Synthesis of halogenated/nitrated flavone derivatives and evaluation of their affinity for the central benzodiazepine receptor. Bioorg Med Chem Lett. 1997;7:2003–2008. [Google Scholar]
- 114.Dekermendjian K, Kahnberg P, Witt MR, Sterner O, Nielsen M, Liljerfors T. Structure-activity relationships and molecular modeling analysis of flavonoids binding to the benzodiazepine site of the rat brain GABA(A) receptor complex. J Med Chem. 1999;42:4343–4350. doi: 10.1021/jm991010h. [DOI] [PubMed] [Google Scholar]
- 115.Marder M, Viola H, Bacigaluppo JA, et al. Detection of benzodiazepine receptor ligands in small libraries of flavone derivatives synthesized by solution phase combinatorial chemistry. Biochem Biophys Res Commun. 1998;249:481–485. doi: 10.1006/bbrc.1998.9146. [DOI] [PubMed] [Google Scholar]
- 116.Marder M, Estiu G, Blanch LB, et al. Molecular modeling and QSAR analysis of the interaction of flavone derivatives with the benzodiazepine binding site of the GABA(A) receptor complex. Bioorg Med Chem. 2001;9:323–335. doi: 10.1016/s0968-0896(00)00250-9. [DOI] [PubMed] [Google Scholar]
- 117.Wang F, Xu Z, Yuen CT, et al. 6,2′-Dihydroxyflavone, a subtype-selective partial inverse agonist of GABAA receptor benzodiazepine site. Neuropharmacology. 2007;53:574–582. doi: 10.1016/j.neuropharm.2007.06.018. [DOI] [PubMed] [Google Scholar]
- 118.Gavande N, Karim N, Johnston GA, Hanrahan JR, Chebib M. Identification of benzopyran-4-one derivatives (isoflavones) as positive modulators of GABA(A) receptors. Chem Med Chem. 2011;6:1340–1346. doi: 10.1002/cmdc.201100120. [DOI] [PubMed] [Google Scholar]