

Abstract
Introduction
Anaplastic lymphoma kinase (ALK), an oncogenic receptor tyrosine kinase, has emerged as a therapeutic target in solid and hematologic tumors. Although several ALK inhibitors have gained recent approval for therapy, non-invasive indicators of target engagement or for use as predictive biomarkers in vivo are lacking. Therefore, we designed and synthesized a radiolabeled analogue of the ALK inhibitor ceritinib, [18F]fluoroethyl-ceritinib ([18F]-FEC), for use with positron emission tomography (PET).
Methods
We used two methods to synthesize [18F]-FEC. First, [18F]fluoroethyl-tosylate was prepared, coupled with ceritinib, and the product purified to yield [18F]-FEC. Alternatively, a precursor compound was synthesized, directly fluorinated with 18F-fluoride, and purified to yield [18F]-FEC.
Results
The first method produced [18F]-FEC with an average decay-corrected yield of 24% (n=4), specific activity of 1200 mCi/μmol, and >99% purity; synthesis time was 115 min from the end of bombardment (EOB). The second method produced [18F]-FEC with an average yield of 7% (n=4), specific activity of 1500 mCi/μmol, and >99% purity; synthesis time was 65 min from the EOB.
Conclusion
The synthesis of a novel 18F-labeled analogue of ceritinib has been achieved in acceptable yields, at high purity, and with high specific activity. The compound is a potential PET imaging agent for the detection of ALK-overexpressing solid tumors, such as lung cancer.
Keywords: Lung cancer, tyrosine kinase, ceritinib, 18F, PET
Introduction
The discovery of a variety of molecular and genetic alterations in different malignancies leading to oncogenesis has provided insight into the complexity of tumorigenesis and the opportunity to target specific markers with the objective of improving patient outcomes [1]. In this context, tyrosine kinase (TK) inhibitors have become a mainstream molecular-targeted therapy for many solid tumors, including several subtypes of lung cancer [2, 3]. Many TKs, especially receptor TKs, are found upstream or downstream of epidemiologically relevant oncogenes [4]. One TK receptor, anaplastic lymphoma kinase (ALK), was first identified in 1994 as a part of the nucleophosmin-ALK fusion gene, which occurs in 60% of anaplastic large-cell lymphomas. ALK is also part of the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion gene, which occurs in 3–7% of non-small cell lung cancers (NSCLCs) [5, 6]. ALK, which is grouped with leukocyte TK in a subfamily of the insulin receptor superfamily, is an attractive therapeutic target in cancers expressing activating mutations or fusions of the gene; as a result, a variety of ALK inhibitors, including crizotinib, ceritinib, and alectinib, have been developed and examined in clinical trials (Fig. 1).
Figure 1.
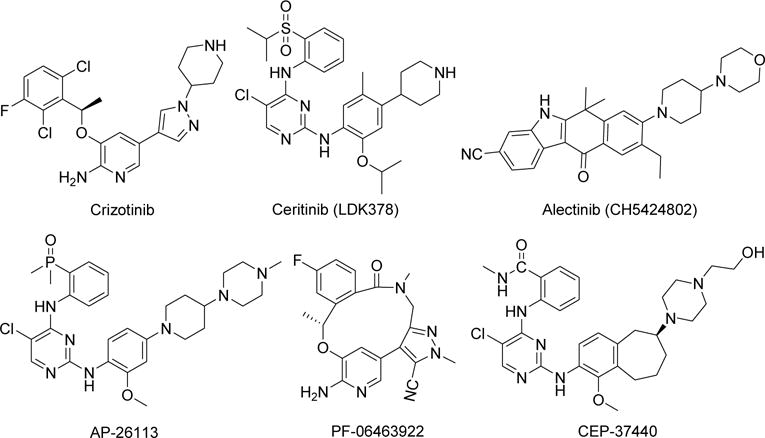
Structures of ALK inhibitors.
Among these inhibitors, crizotinib, in 2011, was the first small molecule inhibitor the U.S. Food and Drug Administration (FDA) approved for the treatment of NSCLC containing the EML4-ALK fusion gene [7, 8]. Although crizotinib is very effective against ALK-positive NSCLC overall, some patients with the disease have ALK point mutations that cause resistance to the drug [9, 10]. In an effort to circumvent the drug resistance caused by ALK mutations, investigations have advanced significantly into assessing second-generation ALK inhibitors in crizotinib-resistant ALK-positive NSCLC. For example, ceritinib and alectinib have been approved by the FDA for the treatment of patients with ALK-positive metastatic NSCLC whose disease progressed during treatment with ceritinib or who were intolerant to crizotinib [11, 12]. Other potent ALK inhibitors, including X-396, ASP3026, AP26113, PF-06463922, CEP-37440 and TSR-011, some with enhanced specificity, are also in phase 1 and 2 clinical trials [13–20]. Most recently, the FDA approved ceritinib for the treatment of patients with certain lung cancers that relapsed after first-line therapy [21, 22].
Although extensive work on the development of therapeutic drugs for ALK inhibition has been reported, no diagnostic compound for assessing molecular-specific pharmacodynamics and target sensitivity to ALK inhibition in vivo, such as an imaging agent, has been yet reported. The current lack of diagnostic assays and markers predictive of disease sensitivity to ALK inhibition in vivo can limit the ability to properly select patients for clinical trials of drugs targeting the ALK kinase. The lack of effective markers and methods for non-invasive monitoring of the early efficacy of therapy also presents a major clinical challenge to selecting the optimal setting(s) in which to test and monitor the biological and therapeutic efficacy of these novel drugs. Positron emission tomography (PET) with ALK-specific radiolabeled agents could provide enhanced assessment of the relative levels and heterogeneity of ALK protein in tumors in individual patients and thus hone the selection criteria for the inclusion of patients in redesigned clinical trials.
Previously, [18F]-labeled imatinib and dasatinib analogues for PET imaging of Bcr-Abl, c-KIT, and Src protein kinases were reported, and furthermore, K562 leukemic cell tumor xenografts in mice have been visualized with both [18F]-labeled imatinib and dasatinib [23, 24]. Structure-activity relationship and molecular modelling analyses of ceritinib suggest that the compound’s central pyrimidine ring, which can participate in hydrogen bonds to the hinge area via the pyrimidine and amino nitrogen atoms, is critical for its ALK inhibition activity [4]. Therefore, substitutions on the nitrogen of piperidine are fairly well-tolerated; however, the properties of the substituents substantially influence the ALK inhibition activity of ceritinib [4]. To further study ALK inhibition activity, investigators have prepared ceritinib derivatives in which the nitrogen of piperidine was substituted with various phosphoramides and carbamates; with this modification, at least one analogue showed enhanced efficacy over ceritinib [4]. We hypothesized that ceritinib, minimally modified with the attachment of a radioisotope, would retain its efficacy and may be a potential imaging agent for monitoring lung cancers expressing ALK protein. We have designed and synthesized an [18F]-labeled analogue of ceritinib, [18F]fluoroethyl-ceritinib ([18F-FEC), for PET imaging of ALK protein in lung cancer. Herein, we report the chemistry and radiochemistry for the synthesis of this potential PET imaging agent.
Experimental
Reagents and instrumentation
Reagents and solvents were purchased from Sigma-Aldrich Co., LLC (St. Louis, MO) and used without further purification. Ceritinib was purchased from Pure Chemistry Scientific, Inc. (Sugar Land, TX). Silica gel solid-phase extraction cartridges (Sep-Pak, 900 mg) were purchased from Alltech Associates (Deerfield, IL, USA). K18F/kryptofix 2.2.2. was purchased from Cyclotope, Inc. (Houston, TX) as an aqueous solution.
Thin layer chromatography (TLC) was performed on pre-coated Kieselgel 60 F254 glass plates (Merck, Darmstadt, Germany). Proton, 13C, and 19F nuclear magnetic resonance (NMR) spectrometry was performed at The University of Texas MD Anderson Cancer Center using a Bruker 300-MHz or 600-MHz spectrometer with tetramethylsilane as an internal reference or hexafluorobenzene as an external reference. High-resolution mass spectrometry (HRMS) was performed on a Bruker BioTOF II mass spectrometer at the University of Minnesota using electrospray ionization.
High-performance liquid chromatography (HPLC) was performed on 1100 series pumps (Agilent Technologies, Stuttgart, Germany) with a built-in ultraviolet (UV) detector and a radioactivity detector with a single-channel analyser (Bioscan, Washington, DC). Purifications and quality control analyses were performed on an Econosil semi-preparative C18 reverse-phase column (Alltech, 10 × 250 mm) and an analytical C18 column (Alltech, 4.6 × 250 mm), respectively. Quality control analyses were performed in a 60% MeCN/40% H2O/0.5% trifluoracetic acid (TFA) solvent system.
Preparation of fluoroethyl-tosylate: 1
Ethylene glycol di-tosylate (50 mg, 1 equiv.) was dissolved in dry MeCN (3 mL); then a solution of Bu4NF in tetrahydrofuran (150 μL, 1M, 1.1 equiv.) was added, and the reaction mixture was heated at 95°C for 1 h. The crude reaction mixture was concentrated, purified by column chromatography and eluted with 20% ethyl acetate in hexane. After evaporation of the solvent, 150 mg of the compound was obtained as a liquid in 60% yield. 1H NMR (CDCl3, 600 MHz) δ: 7.81 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H), 4.57 (dt, J = 47.1 Hz and 4.1 Hz, 2H), 4.26 (dt, J = 27.1 Hz and 4.1 Hz, 2H), 2.46 (s, 3H). 19F NMR (CDCl3, 300 MHz) δ: −224.65 (s, decoupled).
Preparation of fluoroethyl-ceritinib (FEC): 2
Ceritinib (23 mg, 0.041 mmol) was dissolved in dry acetonitrile (2.0 mL), and then triethylamine (300 μL) was added. To this mixture, fluoroethyl-tosylate 1 (10 mg, 0.045 mmol, 1.1 equiv.) was added and the reaction mixture was refluxed for 4 h. The mixture was concentrated under vacuum, then the crude product purified by flash chromatography on a silica gel column, and eluted with 5% MeOH/CH2Cl2 to obtain fluoroethyl-ceritinib 2 as a pale yellow solid (16 mg, 64% yield). 1H NMR (CDCl3, 600 MHz) δ: 9.49 (s, 1H, NH), 8.58 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 8.00 (s, 1H), 7.93 (dd, J = 7.9 Hz and 1.5 Hz, 1H), 7.62 (td, J = 7.8 Hz and 1.6 Hz, 1H), 7.54 (s, 1H, NH), 7.26 (td, J = 7.6 Hz, and 1.0 Hz, 1H), 6.81 (s, 1H), 4.62 (dt, J = 47.8 Hz and 4.8 Hz, 2H), 4.52 (septet, J = 6.1 Hz, 1H), 3.26 (septet, J = 6.7 Hz, 1H), 3.12 (d, J = 11.8 Hz, 2H), 2.77 (dt, J = 28.4 Hz and 4.8 Hz, 2H), 2.68 (m, 1H), 2.20 (td, J = 11.2 Hz and 3.6 Hz, 2H), 2.16 (s, 3H), 1.79 (m, 4H), 1.36 (d, J = 6.1 Hz, 6H), 1.32 (d, J = 6.8 Hz, 6H). 13C NMR decoupled (CDCl3, 125 MHz) δ: 157.49, 155.36, 155.30, 144.69, 138.48, 137.68 9, 134.66, 131.23, 127.37, 126.80, 124.86, 123.69, 123.08, 120.60, 110.73, 105.68, 81.86 (d, 1JF, C=167.3 Hz), 71.34, 58.68 (d, 2JF,C=19.6 Hz), 55.42, 54.90, 37.84, 32.82, 22.26, 18.93, 15.36. 19F NMR, decoupled (CDCl3, 300 MHz) δ: −217.9 (s). HRMS: m/z [M+H]+ calculated for C30H40ClFN5O3S, 604.1788; found, 604.2540.
Preparation of hydoxyethyl-ceritinib: 3
Ceritinib (200 mg, 0.36 mmol) was dissolved in acetonitrile (4.0 mL), and then triethylamine (2.0 mL) was added. To this mixture, hydroxyethyl-tosylate (196 mg, 0.91 mmol, 2.5 equiv.) was added and the reaction mixture refluxed at 85°C for 4 h. The mixture was concentrated under vacuum, then the crude product purified by flash chromatography on a silica gel column, and eluted with 4% MeOH/CH2Cl2. After solvent evaporation, hydoxyethyl-ceritinib was obtained as an off-white solid (190 mg, 88% yield). 1HNMR (CDCl3, 600 MHz), δ: 9.49 (s, 1H, NH), 8.58 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 8.01 (s, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.62 (t, J = 7.7 Hz, 1H), 7.54 (s, 1H, NH), 7.26 (t, J = 7.9 Hz, 1H), 6.79 (s, 1H), 4.57 (septet, J = 6.0 Hz, 1H), 3.67 (t, J = 5.1 Hz, 2H), 3.26 (septet, J = 6.9 Hz, 1H), 3.08 (d, J = 10.2 Hz, 2H), 2.69 (m, 1H), 2.61 (t, J = 5.1, 2H), 2.23 (m, 2H), 2.16 (s, 3H), 1.77 (m, 4H), 1.38 (d, J = 6.1 Hz, 6H), 1.32 (d, J = 6.8 Hz, 6H).
Preparation of chloroethyl-ceritinib: 4
Hydroxyethyl-ceritinib 3 (41 mg, 0.068 mmol) was dissolved in dry pyridine (3.0 mL), cooled to 0°C under argon, followed by the drop-wise addition of a solution of tosyl chloride (55 mg, 0.29 mmol, 4.2 equiv.) in dry dichloromethane (1.0 mL). This mixture was allowed to slowly warm to room temperature over 3 h followed by continued stirring for another 15 h. The crude reaction mixture was concentrated under vacuum, re-dissolved in dicholoromethane (5 mL) and subsequently washed with 0.1M NaHCO3 solution (aq., 3×3 mL). The organic layer was isolated, dried under MgSO4, concentrated, chromatographed over a silica gel column, and eluted with 3% MeOH/CH2Cl2. After solvent evaporation, chloroethyl-ceritinib was obtained as a white solid (20 mg, 48% yield). 1H NMR (CDCl3, 600 MHz), δ: 9.49 (s, 1H, NH), 8.58 (d, J = 8.3 Hz, 1H), 8.15 (s, 1H), 8.00 (s, 1H), 7.92 (dd, J = 7.9 Hz and 1.3 Hz, 1H), 7.62 (td, J = 7.9 Hz and 1.5 Hz, 1H), 7.53 (s, 1H, NH), 7.26 (t, J = 7.9 Hz, 1H), 6.80 (s, 1H), 4.52 (septet, J = 6.1 Hz, 1H), 3.64 (t, J = 7.0 Hz, 2H), 3.26 (septet, J = 6.9 Hz, 1H), 3.07 (d, J = 11.3 Hz, 2H), 2.79 (t, J = 7.0, 2H), 2.66 (m, 1H), 2.22 (m, 2H), 2.15 (s, 3H), 1.77 (m, 4H), 1.36 (d, J = 6.0 Hz, 6H), 1.32 (d, J = 6.8 Hz 6H). 13C NMR decoupled (CDCl3, 125 MHz) δ: 157.61, 155.50, 155.45, 144.83, 138.63, 134.78, 131.39, 127.63, 126.96, 125.02, 123.82, 123.22, 120.73, 110.90, 105.87, 71.56, 68.29, 60.40, 55.57, 54.79, 38.00, 32.83, 22.40, 19.08, 15.51. HRMS: m/z [M+H]+ calculated for C30H39Cl2N5O3S, 620.2229; found, 620.2207.
Preparation of [18F]fluoroethyl-ceritinib ([18F]-FEC): [18F]-2
Method 1
The aqueous solution of K18F/kryptofix [2.2.2] (110 mCi in 0.5 ml) was transferred into a V-vial. Water was removed by azeotropic evaporation with acetonitrile (1.0 mL) at 100°C under a stream of argon. A solution of ethylene glycol ditosylate (5–7 mg) in anhydrous acetonitrile (0.5 mL) was added to the dry K18F/kryptofix [2.2.2]. The reaction mixture was heated at 110°C for 15 min, an aliquot was assessed by analytical HPLC, which showed a high conversion to the desired product [18F]-1. The crude reaction mixture was passed through a silica Sep-Pak cartridge followed by elution with two portions of ethyl acetate (2.5 mL, total), and the solvent was evaporated at 80°C under a stream of argon. The residue was dissolved in 60% acetonitrile/water (1.0 mL) and injected onto the HPLC connected to a semipreperative C18 column. The product [18F]-1 was eluted with 55% acetonitrile/water at a flow of 4 mL/min. The appropriate fraction (radioactive) was collected, and an aliquot of the product [18F]-1 was analysed on an analytical HPLC column to verify its identity and purity by co-injection with the nonradioactive authentic sample 1. The solvent from the rest of the product was partially evaporated under reduced pressure, and then water (10 mL) was added. The solution was passed through a C18 reverse-phase cartridge; the cartridge was dried by vacuum for 5 min followed by flashing with argon for 5 min. The product was eluted with 1.5 mL of dry acetonitrile and collected into a V-vial containing ceritinib (10 mg).
The reaction mixture in the V-vial was heated at 100°C for 20 min, and an aliquot was injected onto the analytical HPLC system, which showed that no radioactive starting material ([18F]-1) remained. The solvent was evaporated by a stream of argon and the product was purified by normal-phase flash chromatography and eluted with 5% MeOH in CH2Cl2. One mL fractions were collected, the appropriate fractions (radioactive) were combined, and radioactivity was counted in a dose calibrator (Capintech). The solvent was evaporated under a stream of argon, the product was dissolved in MeCN/H2O and an aliquot analysed in analytical HPLC by co-injection with standard compound 2 to verify its identity and purity.
Method 2
Chloroethyl-ceritinib 4 (5–7 mg) was placed in a V-vial, and a solution of dry K18F/kryptofix [2.2.2] in anhydrous acetonitrile (0.5 mL) was added. The reaction mixture was heated at 110°C for 20 min, an aliquot was analysed by HPLC, which showed presence of the product. The solvent from the reaction mixture was evaporated to dryness; the product was dissolved in CH2Cl2 and loaded onto a silica Sep-Pak cartridge. The product was eluted with 30% acetone in hexane and 1 mL fractions were collected. The radioactive fractions were combined and the solvent was evaporated at 40°C under a stream of argon. The product [18F]-2 was dissolved in MeCN and an aliquot was analysed on an analytical HPLC column to verify its identity and purity by co-injection with the nonradioactive authentic sample 2.
Results and Discussion
Method 1
Fluoroethyl-tosylate 1 was obtained in-house by the reaction of ethylene glycol ditosylate with Bu4NF in 60% yield. The compound was characterized by 1H and 19F NMR spectroscopy, which showed peaks consistent with those associated with the compound. Non-radioactive FEC 2 was prepared according to Scheme 1. The chemical yield of 2 was 64% after chromatographic purification in this single-step experiment. The product was fully characterized using spectroscopic methods, including 1H, 13C, and 19F NMR spectrometry and HRMS.
Scheme 1.
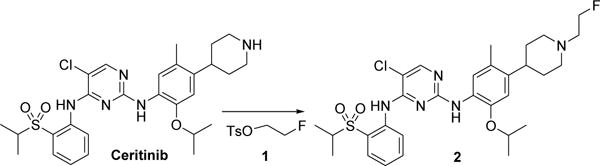
Reagents and conditions: (a) FCH2CH2OTs (1.1 equiv.), Et3N (50 equiv.), CH3CN, 85°C, 4 h, 64% yield.
Scheme 2 shows the radiosynthesis of FEC 2 ([18F]-2). All radiosyntheses were performed using 100–110 mCi of K18F/kryptofix [2.2.2]. This synthesis was achieved by a two-step process: a) radiosynthesis of [18F]-fluoroethyl-tosylate ([18F]-1) followed by b) coupling of [18F]-1 with ceritinib. The mean decay corrected radiochemical yield of [18F]fluoroethyl-tosylate was 48% (range 45–53%) after HPLC purification (n=5). The mean radiochemical yield in the coupling step was 74% (range, 65–85%) (n=4). The mean decay-corrected yield of [18F]-2 in two-steps was 24% (range, 20–28%) in four runs after purification in both steps. The product was >99% pure and had an average specific activity of 1200 mCi/μmol. The synthesis time was 115 min from the end of bombardment (EOB).
Scheme 2.
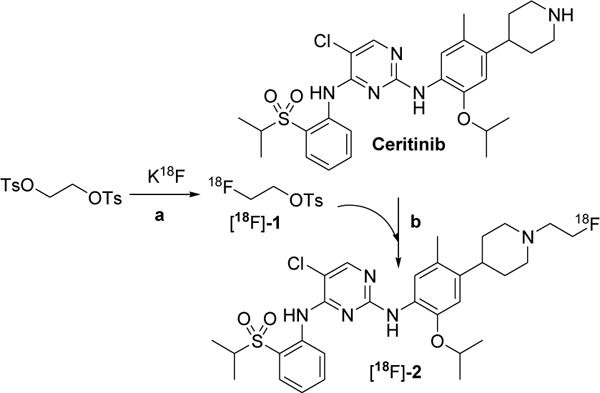
Radiosynthesis of [18F]fluoroethyl ceritinib. Reagents and conditions: (a) [18F]-KF/Kryptofix CH3CN, 110°C, 15 min, 48% yield. (b) CH3CN, 110°C, 20 min, 74% yield.
This method requires two purifications: 1) HPLC purification of [18F]fluoroethyl-tosylate and 2) a second purification after coupling ceritinib with [18F]-1 by either HPLC or flash chromatography. After the first purification with HPLC, [18F]fluoroethyl-tosylate required the removal of water from the HPLC solvent, which could be achieved using a reverse-phase C18 cartridge. After a brief evaporation of MeCN from the HPLC solvent under vacuum, further dilution with additional water (10 mL) rendered the product retainable in the C18 cartridge. After drying the cartridge, the product could be eluted with a small volume of dry MeCN directly to the V-vial containing certinib. There was no loss of activity in the C18 cartridge during the concentration step.
Purification of the final product [18F]-2 by HPLC was not suitable due to the slow wash-out of ceritinib from the column. As a result, a small quantity of ceritinib remained in the final product, representing a major contaminant, which would compete with [18F]-2 during biological studies. Therefore, flash chromatography was used to provide the desired product [18F]-2, which was both chemically and radiochemically pure. An HPLC chromatogram of the radioactive product [18F]-2 co-injected with non-radioactive standard compound is presented in Figure 2.
Figure 2.
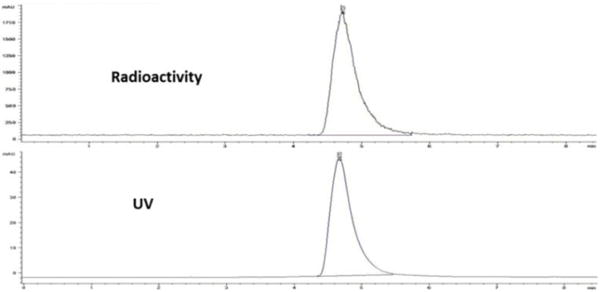
HPLC chromatogram of [18F]fluoroethyl-ceritinib, co-injected with the non-radioactive standard. Analytical column; 60% MeCN/H2O/0.5% TFA, flow 1 mL/min.
Although this method produced reasonably good yields and a clean product, the handling of radioactive material in multiple steps is not convenient and the method is difficult to automate for routine production. Therefore, we explored the synthesis of the compound using a simple alternative one-step method, fluorination followed by purification (Method 2).
Method 2
We initially sought to prepare tosylethyl-ceritinib, which could be fluorinated to afford FEC 2. We attempted the reaction of ceritinib with ethylene glycol ditosylate to obtain tosylethyl-ceritinib, but this reaction produced ethylene di-ceritinib, implicating that both tosylates of ethylene reacted with certinib. The compound was isolated in low yield and tentatively confirmed by 1H NMR spectrometry (data not shown). The mono-substituted product, tosylethyl-ceritinib could not be obtained from this reaction. Alternatively, we attempted to prepare tosylethyl-ceritinib via hydroxyethyl-ceritinib (Scheme 3). Compound 3 was prepared by the reaction of ceritinib with hydroxyethyl-tosylate in 88% yield (purified). The product was fully characterized by 1H NMR spectroscopy and the spectrum was consistent with that reported in the literature [13].
Scheme 3.
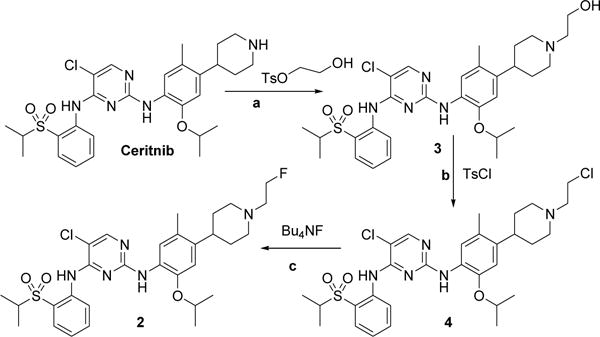
Reagents and conditions: (a) HOCH2CH2OTs (2.5 equiv.), Et3N (40 equiv.), CH3CN, 85°C, 3 h, 88% yield. (b) TsCl (4 eq.), Pyridine/DCM (3/1mL), 0°C 3 h, rt 15 h, 50% conversion, 40% yield, (c) Bu4NF (3 equiv.), CH3CN, 110°C, 2 h, 55% yield.
The reaction of compound 3 with either tosyl- or mesyl-chloride did not produce the desired sulfonylethyl-ceritinib, but instead produced compound 4 in a moderate yield (48%). The reaction of hydroxyethyl-ceritinib with mesyl chloride showed two spots on TLC, and the lower spot was gradually converted to the higher one with time at room temperature, which suggested that the mesylethyl-ceritinib was formed. However, it was difficult to isolate a pure product from the reaction mixture. Therefore, we allowed the reaction to run to completion at the higher spot, which resulted in chloroethyl-ceritinib (compound 4). Compound 4 was fully characterized by 1H and 13C NMR spectroscopy and HRMS. All data were consistent with the identity of the product. Fluorination of compound 4 produced the desired compound 2 in 55% yield. The spectral data of the product 2 by this method was identical to those obtained by Method 1.
Scheme 4 shows the radiosynthesis of [18F]-FEC following a direct fluorination of the precursor compound 4. The mean decay-corrected radiochemical yield of [18F]-2 using this method was 7% (range, 6–10%) in 4 runs. The product was >99% radiochemically pure and had an average specific activity of 1500 mCi/μmol. The synthesis time was 65–70 min from the end of bombardment.
Scheme 4.
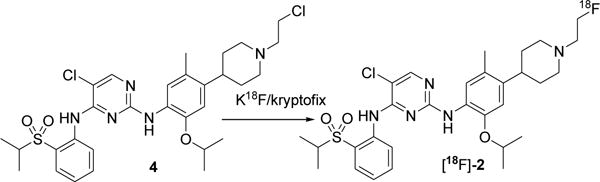
Reagents and conditions: K18F/kryptofix CH3CN, 110°C, 7% yield.
Although the fluorination of compound 4 with Bu4NF produced the nonradioactive product 2 in a 55% yield, the radiofluorination of 4 produced lower yields. This may be due to the limited availability of 18F-fluoride in the reaction mixture. The main drawback of Method 2 was the purification of the product [18F]-2. Analytical HPLC revealed that the crude product had a couple of unknown UV peaks, which eluted very close to the desired product; as a result, it was difficult to isolate a chemically pure product by HPLC. Alternatively, we used flash chromatography and separated the radioactive product, and HPLC of this radiochemically pure product also showed the unknown UV-active material. Although the second method seemed to be simple and straightforward, it produced lower yields, and the product contained chemical impurities that could potentially compete with the desired radioactive compound in biological studies. Nonetheless, further optimization of the procedure may improve the method. Of these two methods, we judged Method 1 to be the better choice for producing a pure compound for biological applications in vitro and in vivo. Further studies will be required to test the efficacy of the compound in PET imaging of specific types of solid tumors, including lung cancer.
Conclusion
We synthesized a novel [18F]-labeled analogue of ceritinib, [18F]-fluoroethyl-ceritinib, in good yields with high purity and specific activity. The compound is a potential PET imaging agent for the detection of ALK-overexpressing lung cancer and should be tested in vitro in cell culture and in vivo in tumor-bearing mice.
Acknowledgments
This work was supported by the National Institutes of Health through the Washington University-MD Anderson Cancer Center Inter-Institutional Molecular Imaging Center grant (NCI P50 CA94056) (DP-W) and by an MD Anderson Cancer Center Institutional Research Grant (MMA).
References
- 1.Grande E, Bolos MV, Arriola E. Targeting oncogenic ALK: A promising strategy for cancer treatment. Mol Cancer Ther. 2011;10:569–579. doi: 10.1158/1535-7163.MCT-10-0615. [DOI] [PubMed] [Google Scholar]
- 2.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 3.Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 4.Wang P, Cai J, Chen J, Ji M. Synthesis and anticancer activities of ceritinib analogs modified in the terminal piperidine ring. Eur J Med Chem. 2015;93:1–8. doi: 10.1016/j.ejmech.2015.01.056. [DOI] [PubMed] [Google Scholar]
- 5.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, Alk, to a nucleolar protein gene, Npm, in non-Hodgkins-lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara SI, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–563. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 7.Frampton JE. Crizotinib: a review of its use in the treatment of anaplastic lymphoma kinase-positive, advanced non-small cell lung cancer. Drugs. 2013;73:2031–2051. doi: 10.1007/s40265-013-0142-z. [DOI] [PubMed] [Google Scholar]
- 8.Shaw AT, Yasothan U, Kirkpatrick P. Crizotinib. Nat Rev Drug Discov. 2011;10:897–898. doi: 10.1038/nrd3600. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Wang F, Keats J, Zhu XT, Ning YY, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang YH, Narasimhan NI, Dalgarno D, Shakespeare WC, Miret JJ, Clackson T, Rivera VM. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib- resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11:122–132. doi: 10.1158/1541-7786.MCR-12-0569. [DOI] [PubMed] [Google Scholar]
- 11.Chen JY, Jiang C, Wang SM. LDK378: a promising anaplastic lymphoma kinase (ALK) inhibitor. J Med Chem. 2013;56:5673–5674. doi: 10.1021/jm401005u. [DOI] [PubMed] [Google Scholar]
- 12.Ou SHI, Azada M, Hsiang DJ, Herman JM, Kain TS, Siwak-Tapp C, Casey C, He J, Ali SM, Klempner SJ, Miller VA. Next-generation sequencing reveals a novel NSCLC ALK F1174V mutation and confirms ALK G1202R mutation confers high-level resistance to alectinib (CH5424802/RO5424802) in ALK rearranged NSCLC patients who progressed on Crizotinib. J Thorac Oncol. 2014;9:549–553. doi: 10.1097/JTO.0000000000000094. [DOI] [PubMed] [Google Scholar]
- 13.Marsilje TH, Pei W, Chen B, Lu WS, Uno T, Jin YH, Jiang T, Kim S, Li NX, Warmuth M, Sarkisova Y, Sun F, Steffy A, Pferdekamper AC, Li AG, Joseph SB, Kim Y, Liu B, Tuntland T, Cui XM, Gray NS, Steensma R, Wan YQ, Jiang JQ, Chopiuk G, Li J, Gordon WP, Richmond W, Johnson K, Chang J, Groessl T, He YQ, Phimister A, Aycinena A, Lee CC, Bursulaya B, Karanewsky DS, Seidel HM, Harris JL, Michellys PY. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem. 2013;56:5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 14.Kang GA, Lee M, Song D, Lee HK, Ahn S, Park CH, Lee CO, Yun CS, Jung H, Kim P, Ha JD, Cho SY, Kim HR, Hwang JY. Synthesis and evaluation of novel 2,4-diaminopyrimidines bearing bicyclic aminobenzazepines for anaplastic lymphoma kinase (ALK) inhibitor. Bioorg Med Chem Lett. 2015;25:3992–3998. doi: 10.1016/j.bmcl.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 15.Lovly1 CM, Heuckmann JM, Stanchina Ede, Chen H, Thomas RK, Liang C, Pao W. Therapeutics, Targets, and Chemical Biology: Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71(14):4920–31. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, Sakagami H, Furutani T, Doihara H, Kudoh M, Kuromitsu S. The Selective Anaplastic Lymphoma Receptor Tyrosine Kinase Inhibitor ASP3026 Induces Tumor Regression and Prolongs Survival in Non–Small Cell Lung Cancer Model Mice. Mol Cancer Ther. 2014;13(2):329–40. doi: 10.1158/1535-7163.MCT-13-0395. [DOI] [PubMed] [Google Scholar]
- 17.Ceccon M, Mologni L, Giudici G, Piazza R, Pirola A, Fontana D, Gambacorti-Passerini C. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor AP26113 in human NPM-ALK–positive anaplastic large cell lymphoma. Mol Cancer Res. 2014;13(4):775–83. doi: 10.1158/1541-7786.MCR-14-0157. [DOI] [PubMed] [Google Scholar]
- 18.Zou HY, Friboulet L, Kodack DP, Engstrom LD, Li Q, West M, Tang RW, Wang H, Tsaparikos K, Wang J, Timofeevski S, Katayama R, Dinh DM, Lam H, Lam JL, Yamazaki S, Hu W, Patel B, Bezwada D, Frias RL, Lifshits E, Mahmood S, Gainor JF, Affolter T, Lappin PB, Gukasyan H, Lee N, Deng S, Jain RK, Johnson TW, Shaw AT, Fantin VR, Smeal T. PF-06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell. 2015;28:70–81. doi: 10.1016/j.ccell.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Mittal V, Cang S, Liu D. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17–16. doi: 10.1186/s13045-015-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arkenau HT, Sachdev JC, Mita MM, Dziadziuszko R, Lin CC, Yang JCH, Infante JR, Anthony SP, Voskoboynik M, Su WC, De Castro J, Natale RB, Zhang ZY, Hughes L, Bobilev D, Weiss GJ. Phase (Ph) 1/2a study of TSR-011, a potent inhibitor of ALK and TRK, in advanced solid tumors including crizotinib-resistant ALK positive non-small cell lung cancer. J Clin Oncol. 2015 ASCO Annual Meeting (May 29 – June 2) Vol 33, No 15_suppl (May 20 Supplement), 2015, 8063. [Google Scholar]
- 21.News in American Association of cancer research. Ceritinib gains FDA approval for lung cancer. Cancer Discov. 2014;4:753–754. doi: 10.1158/2159-8290.CD-NB2014-074. [DOI] [PubMed] [Google Scholar]
- 22.Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, Liu Q, Zhao P, Zhao H, Lu D, Tang Z, Hakim AA, Boyd K, Keegan P, Justice R, Pazdur R. FDA Approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase–positive non–small cell lung cancer. Clin Cancer Res. 2015;21(11):2436–2439. doi: 10.1158/1078-0432.CCR-14-3157. [DOI] [PubMed] [Google Scholar]
- 23.Peng Z, Maxwell DS, Sun D, Prasad BAB, Pal A, Wang S, Balatoni J, Ghosh P, Lim ST, Volgin A, Shavrin A, Alauddin MM, Gelovani J, Bornmann WG. Imatinib analogs as potential agents for PET imaging of Bcr-Abl and c-KIT expression at a kinase level. Bioorg Med Chem. 2014;22:623–632. doi: 10.1016/j.bmc.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veach DR, Namavari M, Pillarsetty N, Santos EB, Beresten-Kochetkov T, Lambek C, Punzalan BJ, Antczak C, Smith-Jones PM, Djaballah H, Clarkson B, Larson SM. Synthesis and biological evaluation of a fluorine-18 derivative of dasatinib. J Med Chem. 2007;50:5853–5857. doi: 10.1021/jm070342g. [DOI] [PubMed] [Google Scholar]