

Abstract
A cocatalyst system consisting of an alkylamine base and a bis(thiourea) featuring a linear alkane tether is shown to dramatically increase the rate of ring-opening polymerization (ROP) of L-lactide versus previously disclosed monothiourea H-bond donors. Rate acceleration occurs regardless of the identity of the alkylamine cocatalyst, and the ROP remains controlled yielding poly(lactide) with narrow molecular weight distributions, predictable molecular weights and high selectivity for monomer. This H-bond mediated ROP of L-lactide constitutes a rare, clear example of rate acceleration with bis(thiourea) H-bond donors versus monothioureas, and the bis(thiourea) is shown to remain highly active for ROP at fractional percent catalyst loadings. Activation at a single monomer ester by both thiourea moieties is implicated as the source of rate acceleration.
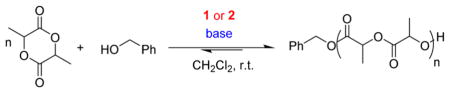
Graphical Abstract

INTRODUCTION
Thiourea (TU) H-bond donors1 have been a workhorse of organocatalytic transformations.2–5 This class of compounds features a wide array of functional motifs and geometries and has been employed in a multitude of reactions including Henry reactions, hydroaminations, conjugate additions and ring-opening polymerization (ROP).6–12 While the wealth of chemistry offered by H-bonding catalysts has attracted numerous research groups, these systems can require high catalyst loadings and/or long reaction times. A general means of producing rate-accelerated reactions with this widely used class of catalysts has been elusive. The thiourea 1 (Scheme 1), with a slate of base cocatalysts, has been widely applied to the synthesis of polyesters and polycarbonates via ROP.13,14 These systems are believed to effect “living” ROP via dual activation of monomer by 1 and of growing polymer chain by base, Scheme 1.15,16 Herein, we show a strategy for the rate enhancement of the ROP of L-lactide (L-LA) using bis(thiourea) H-bond donors with a variety of alkylamine cocatalysts, Figure 1. Such a rate acceleration is not usually observed upon switching from monothiourea to bis(thiourea) H-bond donating catalysts in small molecule systems.7,17–19
Scheme 1.

Second Order Behavior in 1 Inspires Tethered H-Bond Donor 2
Figure 1.

Alkylamine and thiourea cocatalysts evaluated for the ROP of L-LA.
RESULTS AND DISCUSSION
Our approach was inspired by the use of bis(thiourea) catalysts in small molecule transformations as well as our own investigations into the nature of 1/base catalyzed ROP.20 During the course of mechanistic studies into the 1/base catalyzed ROP of lactide initiated from benzyl alcohol, we observed that some 1/alkylamine combinations, like 1/Me6TREN in Scheme 1, exhibit second order kinetics in [1].21 This observation suggests that two 1 molecules are kinetically relevant in the rate-determining step. The kinetic orders of the previously studied ROP reactions are base dependent,21 which hints at the possibility of exploiting these differences for the development of advanced catalyst systems. We reasoned that tethering two thiourea moieties could enhance the rate of the 1/base cocatalyzed ROPs which exhibit second order dependence upon [1] and possibly enforce dual thiourea activation in the others, likewise enhancing rate.
Bis(thiourea) 2,7 combined with the alkyl amine base HMTETA (Figure 1), significantly accelerates the ROP of L-LA (1 M in CH2Cl2), initiated from benzyl alcohol ([M]o/[I]o = 100) versus the 1/HMTETA cocatalyzed ROP. Only the concentration of cocatalysts are varied between runs. The ROP of L-LA from benzyl alcohol achieves 90% conversion in 15 min when catalyzed by 2/HMTETA (2.5 mol % each), whereas the 1/HMTETA (5 mol % each) catalyzed reaction reaches 94% conversion in 90 min. This ROP is accelerated with 2 versus 1 when controlling for the concentrations of cocatalysts or the concentration of thiourea moiety present, Table 1. The reaction rate slows with a stoichiometric excess of HMTETA to 2 (Table 1, entries 3 and 4), which suggests that 1:1 stoichiometry of base:2 is optimal for ROP.
Table 1.
HMTETA and Bis(thiourea) cocatalyzed ROP of L-LA.a

| |||||||
|---|---|---|---|---|---|---|---|
| entry | TU | mol % (bis)TU | mol % base | convnb (%) | time (min) | Mnc (g/mol) | Mw/Mnc |
| 1 | 1 | 5 | 5 | 94 | 90 | 16 300 | 1.05 |
| 2 | 1 | 5 | 2.5 | 88 | 198 | 16 800 | 1.06 |
| 3 | 2 | 2.5 | 2.5 | 90 | 15 | 15 400 | 1.03 |
| 4 | 2 | 2.5 | 5 | 90 | 20 | 17 100 | 1.04 |
Reaction conditions: 1 M (0.7 mmol) L-LA, 0.007 mmol benzyl alcohol, CH2Cl2 (0.7 mL) and given amount of catalyst. Aliquots of the reaction were quenched with benzoic acid and characterized by GPC and 1H NMR.
Conversion to polymer obtained by 1H NMR.
Determined by GPC vs polystyrene standards.
The rate acceleration exhibited by 2 vs 1 is a general trend and is independent of the identity of the alkylamine cocatalyst being employed. Several commercially available alkylamines in combination with 1 have been shown previously to be effective cocatalysts for the ROP of lactide.21,22 The effects of base cocatalyst identity upon ROP have been explained computationally22 and experimentally21 in terms of chelating H-bonding interactions with alcohol or varied cocatalyst interactions, respectively. A selection of these cocatalysts were evaluated in the 2/base cocatalyzed ROP of L-LA (Table 2). For all base cocatalysts examined, the 2/base cocatalyzed ROP was faster than the comparative 1/base catalyzed ROP. This rate acceleration occurs regardless of base identity or the reaction order in [1] exhibited in the 1/base catalyzed ROP of L-LA.21 This H-bond mediated ROP of L-LA constitutes a rare, clear example of rate acceleration with bis(thiourea) H-bond donors versus monothioureas.
Table 2.
Comparison of Alkylamine and (bis)TU Cocatalyzed ROPs of L-Lactide.a
| |||||||
|---|---|---|---|---|---|---|---|
| entry | base | TU | [M]o/[I]o | convn (%) | time (min) | Mne (g/mol) | Mw/Mne |
| 1b | Me6TREN | 1 | 100 | 94 | 50 | 18 400 | 1.04 |
| 2c | Me6TREN | 2 | 100 | 94 | 10 | 17 500 | 1.03 |
| 3c | Me6TREN | 2 | 50 | 94 | 8 | 9700 | 1.05 |
| 4c | Me6TREN | 2 | 200 | 95 | 20 | 32 200 | 1.02 |
| 5b | TACN | 1 | 100 | 90 | 20 | 16 900 | 1.04 |
| 6c | TACN | 2 | 100 | 89 | 6 | 16 200 | 1.05 |
| 7b | PMDETA | 1 | 100 | 94 | 60 | 16 400 | 1.04 |
| 8c | PMDETA | 2 | 100 | 88 | 15 | 16 200 | 1.04 |
| 9b,d | TMEDA | 1 | 100 | 60 | 24 hf | 9200 | 1.07 |
| 10c,d | TMEDA | 2 | 100 | 81 | 24 hf | 14 600 | 1.04 |
| 11b,d | TEA | 1 | 100 | 40 | 24 hf | 6200 | 1.11 |
| 12d,g | TEA | 2 | 100 | 90 | 24 hf | 17 600 | 1.04 |
Reactions conducted in CH2Cl2 at 1 M (0.7 mmol) L-LA, except in the case in footnote d.
5 mol % each base and 1.
2.5 mol % each base and 2.
2 M L-LA.
Mn and Mw/Mn determined by GPC in CH2Cl2 vs polystyrene standards.
Reaction stopped at 24h.
2.5 mol % 2 and 5 mol % TEA.
Despite the increased rate, the ROPs cocatalyzed by 2 remain controlled and exhibit the characteristics of a “living” polymerization. In the 2/Me6TREN catalyzed ROP of L-LA, the Mn is predictable by [M]o/[I]o and Mw/Mn is narrow, < 1.05, Table 2, Entries 2–4. When initiated from pyrenebutanol, the RI and UV/vis signals overlap in the GPC trace of the resulting polymer which suggests end group fidelity, see Supporting Information. This conclusion is supported by MALDI–TOF analysis of a PLA sample which shows only the repeat pattern associated with PLA initiated from benzyl alcohol (see Supporting Information). Further, the sequential addition of LA monomer to a single polymerization solution results in quantitative chain-extension, see Supporting Information. These observations are consistent with those typically observed for the 1/base-catalyzed ROP of lactide.16,22 Previously, the best means of effecting higher rates of ROP were to employ stronger bases which typically result in the rapid post polymerization broadening of Mw/Mn.3,15 However, the higher rates of these 2/base-catalyzed ROPs are not associated with loss of selectivity for monomer; MALDI–TOF analysis confirms the remarkable selectivity of 2/base systems for monomer as multiples of 72 m/z which are associated with random chain scission are vanishingly small, see Supporting Information. The absence of these peaks in the MALDI–TOF suggests that near zero postpolymerization transesterification is occurring. Further, when the reaction solution was left to stir for 1 h after full conversion, the most active 2/base systems resulted in only modest erosion of Mw/Mn. After 1 h of stirring past full conversion, the initial Mw/Mn for the 2/HMTETA (Table 1, entry 3) and 2/Me6TREN (Table 2, Entry 2) experiments broadened only slightly to Mw/Mn = 1.06 for both samples. The 13C NMR spectrum of poly(L-lactide) shows only one resonance in the methine region, which suggests that the stereochemistry of the monomer is retained in the polymerization.
The bis(thiourea) (2) cocatalyst remains highly active at low concentrations which typically halt 1/alkylamine cocatalyzed ROP of lactide. For the ROP of L-lactide, the 2/Me6TREN (0.5 mol %, Table 3, entry 2) catalyzed reaction proceeded to 98% conversion in 45 min (Mn = 17 000; Mw/Mn = 1.05) whereas the 1/Me6TREN (1 mol %, Table 3, entry 1) catalyzed reaction only progressed to 3% conversion in 24 h. The same ROP with 2/Me6TREN cocatalysts (0.1 mol %, Table 3, entry 4) progressed to full conversion in 180 min. The development of highly selective catalysts for ROP which remain highly active at low catalyst loadings is vitally important to the increased applicability of these systems.23
Table 3.
Low Catalyst Loadings in the TU/Alkylamine Catalyzed ROP of L-LA.a

| ||||||
|---|---|---|---|---|---|---|
| entry | TU | mol % cats. (each)a | convnb (%) | time (min) | Mnc (g/mol) | Mw/Mnc |
| 1 | 1 | 1 | 3 | 24 hd | – | – |
| 2 | 2 | 0.5 | 98 | 45 | 17 000 | 1.05 |
| 3 | 2 | 0.25 | 93 | 80 | 20 000 | 1.02 |
| 4 | 2 | 0.10 | 98 | 180 | 17 900 | 1.03 |
Reactions conducted in CH2Cl2 at 2 M (0.7 mmol) L-LA with the given mol % (to LA) of each catalyst.
Conversion determined by 1H NMR.
Mn and Mw/Mn determined by GPC in CH2Cl2 vs polystyrene standards.
Reaction stopped at 24 h.
Tethered bis(thiourea)s, to our knowledge, have not been evaluated as ROP cocatalysts; however, such systems have been evaluated with mixed results as catalysts for small molecule transformations. Enhanced reaction rates have been observed when activation of two substrates is a possibility.17 However, rate acceleration with bis(thiourea)s is not general,7,18,19 although the introduction of chiral linkers facilitates increased enantioselectivity in some cases.6,19 The bis(thiourea) 2 does not feature a chiral linker and was not expected to alter the stereoselectivity of the ROP vis-à-vis monothiourea 1. The polymers resulting from the 1/Me6TREN and 2/Me6TREN catalyzed ROP of rac-LA from benzyl alcohol (conditions from Table 2, entries 1 and 2) were analyzed by 13C NMR (see Experimental Section). The 1H decoupled 13C NMR spectra suggested similar tacticities (Pm(1) = 0.69; Pm(2) = 0.66; where Pm is the probability of propagating with the retention of stereochemistry).16,24–26 This is consistent with previous suggestions that organocatalytic H-bonding catalysts display chain-end controlled stereochemistry.16
The source of the rate acceleration exhibited by bis(thiourea) 2 is proposed to be the activation at a single monomer ester by both thiourea moieties. While the possibility of 2 simultaneously binding base and monomer or simultaneous binding of monomer and polymer cannot be ruled out, the observed second order dependence upon [1] for some 1/alkylamine catalyzed ROPs of L-LA strongly indicates that both thiourea moieties of 2 are involved in the activation of a single ester moiety in the transition state.27 Presumably, the role of 2 is to enforce this favorable catalytic mode even in those 1/alkylamine systems which do not display second order dependence upon [1], Scheme 2. This suggestion is consistent with computational studies of a bis(thiourea) catalyzed Morita-Baylis-Hillman reaction wherein a bisTU-nitrate complex is believed to react with an uncomplexed aldehyde rather than bind both reagents prior to reaction.28,29 With the exception of the short-strong variety, H-bonds are electrostatic in nature and do not require orbital overlap,30 hence the mode of the 2-lactide activation could be due to direct, dual-thiourea activation of a single ester moiety or an activated-TU mechanism31 (Scheme 2). However, other unenvisioned processes are possible. Computational studies were conducted to differentiate between these mechanistic possibilities. Energies from geometry optimized structures (B3LYP/6-31G**) in CH2Cl2 solvent and the gas phase suggest that the C2 symmetric 2 structure leading to the activated-TU transition state is more stable than the CS structure required for a dual-thiourea activation mechanism by 5.7 or 9.4 kcal/mol, respectively, eq 1 (see Supporting Information). Further, computations suggest that LA activation via the activated-TU structure (Scheme 2, left) is lower in energy than the dual-thiourea activation structure (Scheme 2, right), see Supporting Information. Future studies will be aimed at experimentally determining the source of this increased activity.
Scheme 2.

Proposed Mechanism for the 2/Me6TREN Catalyzed ROP of L-Lactide
 |
(1) |
CONCLUSION
Achiral, bis(thiourea) H-bond donating molecules have been shown to be highly effective cocatalysts for the ROP of lactide. The rate accelerated 2/alkylamine systems retain ROP control, exhibiting the characteristics of a “living” polymerization, a high selectivity for monomer and marked activity at low catalyst loadings. The reaction rate enhancement is postulated to occur via an activated-TU mechanism, but ongoing mechanistic studies are expected to provide further insight into the source of the potency of the bis(thiourea) systems. The addition of a second thiourea moiety to these H-bond donating systems introduces the possibility of a multitude of structural variations, each of which could have dramatic ramifications on the course of the ROP.
EXPERIMENTAL SECTION
General Considerations
All manipulations were performed in an MBRAUN stainless steel glovebox equipped with a gas purification system under a nitrogen atmosphere. All chemicals were purchased from Fisher Scientific and used as received unless stated otherwise. Dichloromethane, toluene and THF (HPLC grade) were dried on an Innovative Technology solvent purification system with activated alumina columns. Thiourea catalysts were prepared as previously described.7,15 L-lactide and RAC-lactide from Acros Organics were recrystallized from dry toluene prior to use. Benzyl alcohol was distilled from CaH2 under high vacuum. Dialysis bags (MWCO = 3,000) were purchased from SpectraPor and stored in aqueous NaN3 solution. NMR experiments were performed on a Bruker Avance 300 MHz spectrometer except decoupled experiments which were performed on a Varian 500 MHz NMR spectrometer. Size exclusion chromatography (SEC) was performed at 30 °C in dichloromethane (DCM) at 1.0 mL/min using a Agilent Infinity GPC system equipped with three Agilent PLGel columns 7.5 mm ×300 mm (5 μm; pore sizes = 103, 104, and 105 Å) and multiwavelength detector (set to 254 nm) and refractive index detector connected in series. Molecular weight and Mw/Mn were determined versus PS standards (500 g/mol to 3150 kg/mol; Polymer Laboratories). MALDI–TOF data was acquired at the University of Akron Mass Spectrometry Center.
Example ROP of L-Lactide
In a typical polymerization, L-LA (100 mg, 0.7 mmol) was added to a 20 mL glass vial containing a stir bar, both of which were baked at 140 °C overnight. In another dried 20 mL glass vial with stir bar, 2 (17.5 μmol), Me6TREN (17.5 μmol) and benzyl alcohol (0.007 mmol) were added. Solvent (CH2Cl2, 1 M in L-LA) was added to both vials to bring the total volume of solvent to the desired level, approximately equal portions of solvent per vial. After stirring for 5 min, the L-LA solution was transferred via pipet to the vial containing catalysts and initiator. Aliquots were removed from the reaction with a micropipet at predetermined time points and quenched by the addition of benzoic acid (2 mol equivalents to base). The vial was removed from the glovebox, solvent removed under vacuum, conversion determined via 1H NMR, and the polymer was precipitated from CH2Cl2 by treatment with hexanes. The hexanes supernatant was decanted, and the polymer removed of volatiles under reduced pressure. Yield: 80%. Mw/Mn = 1.03; Mn(GPC) = 17 500. Comparative reactions were run side-by-side at room temperature.
For Determination of Selectivity for Monomer
An aliquot of the reaction mixture was allowed to stir for 1 h past full conversion and the polymer was reanalyzed by GPC: Mw/Mn = 1.06; Mn(GPC) = 17,100.
For the Chain-Extension Experiment
The 2/Me6TREN (2.5 mol %) catalyzed ROP of LA (0.69 mmol, 1 equiv, 0.5 M in CH2Cl2) from benzyl alcohol (2 mol %) was stirred to full conversion (30 min) and an aliquot withdrawn. An additional 0.60 mmol of LA (to account for aliquot volume) was added to the reaction, and the process repeated at 60 min with a third addition of LA (0.49 mmol). Aliquot 1: Mn = 13 700 g/mol, Mw/Mn = 1.04. Aliquot 2: Mn = 29 000 g/mol, Mw/Mn = 1.02. Aliquot 3: Mn = 43 700 g/mol, Mw/Mn = 1.02.
Determination of Pm
The standard polymerization procedure was repeated but with rac-LA (100 mg, 0.7 mmol). The polymerization solution was stirred for enough time to achieve 90% conversion (to minimize postpolymerization reactivity). The reaction was quenched by the addition of benzoic acid and conversion determined by 1H NMR. The polymer was then dialyzed in methanol for 24 h to remove any trace of monomer impurity. The pure monomer was dissolved in chloroform-d and analyzed by 1H-decoupled 13C NMR at 70 °C. The procedure for determining Pm is thoroughly described elsewhere.16,24–26 Briefly, the experimental intensities of the five tetrads resulting from the ROP of rac-lactide were simulated using MNova software. The theoretical intensities of these resonances are determined from Markovian statistics from the Pm value. A calculated value of Pm was determined using Excel by systematically varying Pm subject to the minimization in the difference between the experimental and calculated tetrad intensities.
Computational Details
Computational experiments were performed in Spartan ’14 (Windows 7). Structures were geometry optimized at the DFT B3LYP/6-31G** level of theory in the gas phase. Energies in CH2Cl2 solvent were calculated as Single Point energies from the DFT-optimized structures. Energies, computed structures, and coordinates of optimized structures are given in the Supporting Information.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for partial support of this research. This research was partially supported by the NIH under RI-INBRE (8 P20 GM103430-12) and the University of Rhode Island. This material is based upon work conducted at a research facility at the University of Rhode Island supported in part by the National Science Foundation EPSCoR Cooperative Agreement #EPS-1004057.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macro-mol.5b01320.
Experimental details, kinetic plot, Mn vs conversion, GPC traces, computed structures, energies and coordinates (PDF)
References
- 1.Lippert KM, Hof K, Gerbig D, Ley D, Hausmann H, Guenther S, Schreiner PR. Eur J Org Chem. 2012;2012:5919–5927. [Google Scholar]
- 2.Thomas C, Bibal B. Green Chem. 2014;16:1687. [Google Scholar]
- 3.Kiesewetter MK, Shin EJ, Hedrick JL, Waymouth RM. Macromolecules. 2010;43:2093–2107. [Google Scholar]
- 4.Kamber NE, Jeong W, Waymouth RM, Pratt RC, Lohmeijer BGG, Hedrick JL. Chem Rev. 2007;107:5813–5840. doi: 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- 5.Dove AP. ACS Macro Lett. 2012;1:1409–1412. doi: 10.1021/mz3005956. [DOI] [PubMed] [Google Scholar]
- 6.Doyle AG, Jacobsen EN. Chem Rev. 2007;107:5713–43. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 7.Bertucci MA, Lee SJ, Gagne MR. Chem Commun. 2013;49:2055–2057. doi: 10.1039/c3cc00268c. [DOI] [PubMed] [Google Scholar]
- 8.Nakayama Y, Gotanda T, Ito K. Tetrahedron Lett. 2011;52:6234–6237. [Google Scholar]
- 9.Klauber EG, De CK, Shah TK, Seidel D. J Am Chem Soc. 2010;132:13624–13626. doi: 10.1021/ja105337h. [DOI] [PubMed] [Google Scholar]
- 10.Berkessel A, Roland K, Neudörfl JM. Org Lett. 2006;8:4195–4198. doi: 10.1021/ol061298m. [DOI] [PubMed] [Google Scholar]
- 11.Li BJ, Jiang L, Liu M, Chen YC, Ding LS, Wu Y. Synlett. 2005:603–606. [Google Scholar]
- 12.Knowles RR, Lin S, Jacobsen EN. J Am Chem Soc. 2010;132:5030–5032. doi: 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Becker JM, Tempelaar S, Stanford MJ, Pounder RJ, Covington JA, Dove AP. Chem Eur J. 2010;16:6099–6105. doi: 10.1002/chem.200902518. [DOI] [PubMed] [Google Scholar]
- 14.Koeller S, Kadota J, Peruch F, Deffieux A, Pinaud N, Pianet I, Massip S, Léger JM, Desvergne JP, Bibal B. Chem -Eur J. 2010;16:4196–4205. doi: 10.1002/chem.200902912. [DOI] [PubMed] [Google Scholar]
- 15.Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, Nederberg F, Choi J, Wade C, Waymouth RM, Hedrick JL. Macromolecules. 2006;39:8574–8583. [Google Scholar]
- 16.Pratt RC, Lohmeijer BGG, Long DA, Lundberg PNP, Dove AP, Li H, Wade CG, Waymouth RM, Hedrick JL. Macromolecules. 2006;39:7863–7871. [Google Scholar]
- 17.Sohtome Y, Tanatani A, Hashimoto Y, Nagasawa K. Tetrahedron Lett. 2004;45:5589–5592. [Google Scholar]
- 18.Li X, Luo S, Cheng JP. Eur J Org Chem. 2008;2008:4350–4356. [Google Scholar]
- 19.Shi Y, Lin A, Mao H, Mao Z, Li W, Hu H, Zhu C, Cheng Y. Chem Eur J. 2013;19:1914–1918. doi: 10.1002/chem.201202937. [DOI] [PubMed] [Google Scholar]
- 20.Kazakov OI, Datta PP, Isajani M, Kiesewetter ET, Kiesewetter MK. Macromolecules. 2014;47:7463–7468. doi: 10.1021/ma501847x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kazakov OI, Kiesewetter MK. Macromolecules. Submitted for publication. [Google Scholar]
- 22.Coady DJ, Engler AC, Horn HW, Bajjuri KM, Fukushima K, Jones GO, Nelson A, Rice JE, Hedrick JL. ACS Macro Lett. 2012;1:19–22. doi: 10.1021/mz2000048. [DOI] [PubMed] [Google Scholar]
- 23.Helou M, Miserque O, Brusson JM, Carpentier JF, Guillaume SM. Chem Eur J. 2010;16:13805–13813. doi: 10.1002/chem.201001111. [DOI] [PubMed] [Google Scholar]
- 24.Chamberlain BM, Cheng M, Moore DR, Ovitt TM, Lobkovsky EB, Coates GW, VCU, York N, Recei V. J Am Chem Soc. 2001;123:3229–3238. doi: 10.1021/ja003851f. [DOI] [PubMed] [Google Scholar]
- 25.Thakur KM, Kean RT, Hall ES, Kolstad JJ, Lindgren TA, Doscotch MA, Siepmann JI, Munson EJ. Macromolecules. 1997;30:2422–2428. [Google Scholar]
- 26.Ovitt TM, Coates GW. J Am Chem Soc. 2002;124:1316–1326. doi: 10.1021/ja012052+. [DOI] [PubMed] [Google Scholar]
- 27.The simultaneous activation of both ester moieties of lactide by 2 is a geometric possibility, but such dual-ester activation would not be expected to display the observed rate dependence in [1].
- 28.Breugst M, Houk KN. J Org Chem. 2014;79:6302–6309. doi: 10.1021/jo501227m. [DOI] [PubMed] [Google Scholar]
- 29.Sohtome Y, Nagasawa K. Synlett. 2010;2010:1–22. [Google Scholar]
- 30.Anslyn E, Dougherty DA. Modern Physical Organic Chemistry. University Science Books; Mill Valley, CA: 2006145206. [Google Scholar]
- 31.Jones CR, Dan Pantos G, Morrison AJ, Smith MD. Angew Chem, Int Ed. 2009;48:7391–7394. doi: 10.1002/anie.200903063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.