

Abstract
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and the leading single-gene cause of autism. It is caused by the lack of production of the Fragile X mental retardation protein (FMRP), resulting in cognitive deficits, hyperactivity, and autistic behaviors. Breakthrough advances in potential therapy for FXS followed the discovery that aberrant group 1 metabotropic glutamate receptor (mGluR) signaling is an important constituent of the pathophysiology of the syndrome. Research has indicated that upon neuronal stimulation, FMRP acts downstream of group 1 mGluRs (mGluRs1/5) to inhibit protein synthesis, long-term depression, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor internalization. To offset the deficits caused by the lack of FMRP, many pharmaceutical companies have designed medicinal drugs to target the unrestrained stimulation of mGluR5 signaling in FXS. Indeed, promising results from animal and clinical studies suggest that mGluR5 antagonists such as AFQ056 can successfully correct many of the deficits in FXS. In this review, we cover the animal studies performed to date that test the role of AFQ056 as a selective mGluR5 antagonist to alleviate the phenotypes of FXS.
Keywords: Fragile X syndrome, FMR1, Fragile X mental retardation protein, AFQ056, group 1 metabotropic glutamate receptor 5 antagonist, Fmr1 knockout mouse
Introduction
Fragile X syndrome (FXS) is the most common inherited cause of autism.1 It affects one in 5000 males and half as many females, possibly due to X chromosome inactivation in females.2 FXS is characterized by cognitive impairment, difficulties in learning and memory, developmental and/or language delays, hyperactivity, increased susceptibility to seizures, and autistic behaviours.1,3–5 Physical features may include a long, narrow face, prominent ears, and flat feet.5 Females with FXS tend to be at more risk for having emotional problems rather than cognitive deficits,5 and manifest milder symptoms overall. The monogenic nature of FXS makes it ideal for deconstructing the underlying pathology, and extending our understanding of intellectual disability in general. Furthermore, there is clinical overlap between FXS and autism spectrum disorders, and likely substantial overlap in the molecular pathology of these two disorders. Therefore, it is expected that many targeted treatments for FXS will be of benefit to individuals with autism spectrum disorders. Here, we review animal studies that have tested the therapeutic potential of AFQ056 in FXS.
Advances in the genetics of Fragile X syndrome
A number of milestones have been achieved since the discovery of FXS in 1943. Figure 1 lists the prominent events in genetics and neurobiology that have contributed to our understanding of FXS. Martin and Bell first attributed a familial form of intellectual disability afflicting males in two generations born to normal intelligent mothers to sex-linked recessive inheritance.6 Further research revealed a secondary constriction in the long arm of the X chromosome, which correlated with the sex-linked inheritance of intellectual disability.7 The ability to visualize such secondary constrictions consistently, or “fragile sites” as first termed by Hecht in Magenis et al,8 depended on the culture medium.9 However, as scientists studied more FXS pedigrees and performed cytogenetic analyses, it became clear that FXS does not follow simple Mendelian inheritance in what became known as the Sherman paradox.10,11 For instance, normal transmitting males carrying the Fragile X chromosome consistently had normal carrier mothers and daughters, and the siblings of such males were less likely to be impaired than the siblings of intellectually impaired males. Moreover, the penetrance of intellectual impairment in daughters of transmitting males was lower compared with that in daughters of transmitting females. Finally, the penetrance of intellectual impairment in brothers of transmitting males was lower than that in maternal grandsons of transmitting males.10,11 The Sherman paradox was soon resolved by the identification of the Fragile X mental retardation 1 (FMR1) gene,12 mutations in which are responsible for FXS. This discovery started a new era in FXS research by enabling scientists to create animal models and examine the pathophysiological basis of FXS, which in turn allowed for the development of new targeted treatments, such as AFQ056.
Figure 1.

Milestones in Fragile X research. Time scale map illustrating significant developments in the elucidation of Fragile X genetics, biology, and treatment. Numbers in superscript indicate references.
Abbreviations: FMR1, Fragile X mental retardation 1; FMRP, Fragile X mental retardation protein; RNA, ribonucleic acid; MPEP, 2-methyl-6-(phenylethynyl)-pyridine hydrochloride; mGluR, metabotropic glutamate receptor.
Animal models of FXS
The identification of FMR1 sparked the development of FXS animal models, such as the Fmr1 knockout (Fmr1-KO) mouse,13 which continues to be a very helpful tool for studying FXS. Animal models of FXS are particularly useful because Fmr1 is highly conserved across species.4 In fact, the Fmr1 mouse gene is 95% homologous to its human counterpart, and the resulting protein, Fragile X mental retardation protein (FMRP), has similar patterns of expression in terms of timing and tissue specificity in both species.3,14 Unlike humans, a CGG repeat expansion in Fmr1 does not lead to transcriptional silencing in mice;4 thus, the Fmr1-KO mouse model has the Fmr1 gene knocked out by the insertion of a neomycin cassette in exon 5.13 However, both the mouse model and the human are functionally similar in that no FMRP is produced.3 Fmr1-KO mice recapitulate many of the pathological features of FXS, such as learning deficits, macro-orchidism,15 hyperactivity,14 increased risk of seizures, abnormalities in synaptogenesis and synaptic structures,16 and an overabundance of immature dendritic spines.4 Additional strains of the FXS mouse model have been created, as well as other animal models, such as the Drosophila melanogaster and zebrafish models.17–20 The Drosophila model is widely used, and several targeted treatments have been tested in this model. The Drosophila Fmr1 homolog, dFmr1, shows high sequence homology relative to its human counterpart. In addition, its two K homology domains are 85% similar in amino acid sequence to the human FMR1 homolog, and the encoded protein has a similar function to that in humans.21
The molecular basis of FXS
Many of the phenotypes expressed in FXS are a result of the interplay between FMRP and glutamate signaling at the synapse.22 Scientists have made significant strides in understanding the mechanisms that underlie FXS, starting with FMR1 sequencing and elucidating the importance of FMRP in regulating protein synthesis-dependent plasticity at the synapse to the development of FXS-specific pharmacological treatments.
Metabotropic glutamate receptors and plasticity
Glutamate is the major excitatory neurotransmitter in the brain. It signals through two types of receptors, ie, ionotropic and metabotropic glutamate receptors. The ionotropic glutamate receptors include the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), N-methyl-D-aspartate (NMDA), and kainate receptors, while the metabotropic glutamate receptors (mGluRs) are a family of eight receptors subdivided into three groups (1, 2, and 3) based on sequence homology, pharmacology, and signal transduction pathways.23,24 All mGluRs have seven α-helical transmembrane domains and belong to class C of G-protein coupled receptors.25 This class of receptors has a large, highly conserved extracellular amino-terminal domain to which the orthosteric ligands bind without much selectivity across the mGluR subtypes.25 For this reason, developing pharmacological drugs against the allosteric binding sites is more beneficial clinically in order to target mGluR subtypes differentially. Group 1 mGluRs couple to Gq-like proteins and are expressed postsynaptically.26,27 They are comprised of mGluR1 and mGluR5, which have complementary expression patterns. mGluR1 is more highly expressed in the cerebellum, whereas mGluR5 is more highly expressed in the forebrain.28 Group 1 mGluRs regulate protein synthesis primarily through the canonical phospholipase C signaling cascade, as well as through the phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (mTOR) and the extracellular signal-regulated kinase signaling cascades.23 In the hippocampus, mGluR-induced long-term depression (LTD), a form of synaptic plasticity, is controlled by the regulation of AMPA receptor trafficking and changes in the properties of AMPA receptors, which are comprised of the subunits GluR1 and GluR2. Research has shown that LTD triggered by mGluR activation (mGluR-LTD) requires new protein synthesis at the synapse. Moreover, the volume of a dendritic spine varies linearly with the surface area of the postsynaptic density, which in turn varies with the number of AMPA receptors at the synapse.29–32 These findings correlate with the abnormal dendritic spine structure and synaptic plasticity in FXS. Landmark evidence suggests that group 1 mGluRs are implicated in the pathophysiology of FXS and that rescuing the aberrant mGluR signaling corrects many of the phenotypes of FXS.
FMR1
FMR1, located at cytogenetic position Xq27.3, encodes FMRP, which is produced at lower levels or not at all in FXS. The majority of cases of FXS result from aberrant expansion of a CGG trinucleotide repeat upstream of the coding region of FMR1. In turn this leads to hypermethylation of the CGG repeats and an upstream CpG island, with consequential epigenetic transcriptional silencing of FMR1.33 The number of CGG repeats is polymorphic within the population.34 Normal trinucleotide repeat lengths range from six to 54, whereas a full mutation occurs when the number of repeats exceed 200, resulting in FXS.34 An intermediate number of repeats of up to 200 is a premutation that may lead to fragile X-related primary ovarian insufficiency in females and fragile X-associated tremor/ataxia syndrome, which more commonly affects males.4 Moreover, premutation lengths are unstable and prone to increase in successive meiosis events, eventually becoming full mutations. Interestingly, full mutations are only supported in female meiosis, which explains the Sherman paradox.34 Some individuals with full mutations may display mosaicism, with different cells having different CGG repeat lengths or incomplete methylation patterns of FMR1.34 In turn, incomplete methylation translates into the partial activation of FMR1, and this will result in milder symptoms, such as a borderline to low-normal intelligence quotient,5 with functional consequences on treatment choices.
Expression and function of FMRP
FMRP is widely expressed in all mammalian tissues, with the highest expression levels in the brain and testes.35 It is highly expressed in neurons throughout the brain,36 and is also expressed in astrocytes of the developing mouse brain.37 Sequence analysis shows that FMRP is a selective RNA-binding protein that contains an arginine-glycine-glycine (RGG) box and two K homology domains for binding RNA.38 In fact, FMRP binds up to 4% of the mRNAs in the brain.39 The importance of the RNA-binding motifs is underscored by the I304N (isoleucine to asparagine) missense mutation in the second K homology domain of FMRP, which results in a severe phenotype of FXS. This missense mutation prevents FMRP from associating with translating polyribosomes despite normal RNA binding and nucleocytoplasmic distribution.40 FMRP is mainly cytoplasmic, although it possesses nuclear localization and export signals,41 which allows it to shuttle between the cytoplasm and the nucleus, presumably to bind its mRNA targets.36 After binding to its mRNA targets, FMRP forms part of a messenger ribonucleoprotein complex and is involved in the transport and translation of mRNAs in the dendrites.42 Not all of the mRNA targets of FMRP require it for their proper localization.1,42,43 This is evident by the proper localization of postsynaptic density protein 95 (PSD-95), an FMRP target protein that is involved in the regulation of AMPA receptor internalization in the absence of FMRP.44
The important role of FMRP in synaptic plasticity is evident from its high expression levels in dendrites and spines.36,45 Research has shown that FMRP is localized in neurons in the form of granules, and is mobilized to dendrites upon mGluR1/5 activation.45 Spines are shaped by neural activity, resulting in synapse strengthening, weakening, or pruning,46 and are important for learning and memory. The cognitive deficits that are characteristic of FXS correlate with the abnormal spine morphology seen in FXS patients and Fmr1-KO mice. FXS is hallmarked by an abundance of spines with immature morphology, ie, long, thin, and tortuous, and a deficit in spines with mature morphology, ie, stubby and mushroom-shaped.42,46,47 Interestingly, the increase in spine density in FXS contrasts with that in other cognitive disorders, such as Down syndrome and Rett syndrome, which mostly show reduced spine density.46 This implies that there is a pruning deficit underlying the increased spine density in FXS.
The spine dysgenesis characteristic of FXS suggests abnormal regulation of translation at the synapse, especially given that FMRP regulates the translation of proteins important for synaptic plasticity and neuronal maturation and function.42 These proteins include cyclin-dependent kinase 4, glucocorticoid receptor α,48 PSD-95, the AMPA receptor subunits GluR1 and GluR2, and calcium/calmodulin-dependent kinase IIα, an inhibitor of mGluR-LTD.49 Direct evidence supports the role of FMRP as a translational repressor.50,51 Examples of synaptic proteins for which translation is repressed by FMRP include Arc, MAP1B, calcium/calmodulin-dependent kinase IIα, and SAPAP4.44 Thus, in the absence of FMRP, many proteins are elevated in the basal state, and their expression levels remain the same in response to group 1 mGluR activation, leading to a deficit in protein synthesis-dependent plasticity. One mechanism whereby FMRP regulates mGluR-LTD is through the control of mRNA stability of PSD-95 in hippocampal dendrites.44 In fact, Fmr1-KO mice show impaired stimulus-induced synthesis of calcium/calmodulin-dependent kinase IIα and PSD-95 in synaptoneurosomes,49 corroborating evidence that the lack of FMRP results in enhanced LTD and AMPA receptor internalization. Interestingly, FMRP also plays a role in glial function, whereby it regulates the mRNA stability of myelin basic protein.44 Another mechanism of FMRP-mediated translational repression is through ribosomal stalling in which FMRP prevents ribosomal translocation52 (for a review, please see Bassell and Warren).53
The mGluR theory of FXS
A primary pathway involved in FXS is the mGluR signal transduction cascade.23,54 The study of FMRP in mGluR-dependent LTD was first initiated after the discovery that activation of mGluRs stimulates the rapid translation of pre-existing mRNAs and promotes synthesis of FMRP in synaptoneurosomes.55,56 Since this finding, a growing number of studies have been carried out to support the role of group 1 mGluRs in the pathophysiology of FXS.57 Upon activation of excitatory postsynaptic group 1 mGluRs, translation rapidly ensues, due in part to the dissociation of FMRP from target mRNAs. One of the many consequences of the resultant protein synthesis is LTD and a reduction in synaptic transmission.58 Activation of mGluR by presynaptic glutamate release normally stimulates the synthesis of proteins involved in the stabilization of LTD. FMRP is also synthesized, creating a negative feedback mechanism for limiting mGluR-dependent LTD.59 mGluR-LTD requires rapid translation of pre-existing mRNAs in the post-synaptic dendrites.60 FMRP then functions to inhibit further protein synthesis (an example of end-product inhibition), serving as a brake to halt LTD.22 If unchecked, excessive LTD can lead to synapse elimination and weakened synapses.
The mGluR theory proposes that AMPA receptor internalization triggered by group 1 mGluR stimulation is exaggerated in FXS in the absence of the translational inhibitor FMRP. Increased translation of a subset of mRNAs perturbs the dynamics of receptor internalization, thereby amplifying internalization of AMPA-type glutamate receptors.61 A reduction in the number of AMPA receptors at the synaptic cleft results in enhanced mGluR-dependent LTD.62 Therefore, this model suggests that group 1 mGluRs and FMRP work in direct opposition to regulate mRNA translation at the synapse, and that in the absence of FMRP, dysregulated mGluR-dependent protein synthesis leads to the pathogenesis of FXS.22 Exaggerated LTD in FXS would, in turn, favor synaptic loss during critical periods of synaptogenesis, accounting for developmental delays and cognitive impairment. For comprehensive reviews, see Bhakar et al28 and Krueger and Bear.63
The therapeutic potential of mGluR5 antagonists
The mGluR theory provides a target to develop treatments for FXS, namely the group 1 mGluRs. Much of the focus has been specifically on mGluR5 since FXS phenotypes related to cognition, learning, and memory implies the involvement of the hippocampus, amygdala, and basal ganglia rather than the cerebellum. To support the notion that correcting the increased stimulation of mGluR5 rescues FXS phenotypes, scientists created Fmr1-mutant mice with reduced mGluR5 expression (Fmr1-CR).64 Indeed, the reduced expression of mGluR5 in Fmr1-CR rescued multiple phenotypes characteristic of FXS such as enhanced LTD, increased spine density, increased protein synthesis in the hippocampus, a hippocampus-dependent memory deficit, audiogenic seizures, and accelerated prepubescent growth. The only phenotype not rescued in this study was that of increased testicular weight in adult mice.64 Thus, this study lends support to the mGluR theory and shows that many of the phenotypes seen in FXS can be corrected by reducing the enhanced mGluR5 signaling.
Further evidence supporting the mGluR theory stems from the rescue of FXS phenotypes upon pharmacological downregulation of glutamate signaling. One such preliminary drug is 2-methyl-6-(phenylethynyl)-pyridine hydrochloride (MPEP).65,66 de Vrij et al examined the effect of acute MPEP treatment on the acoustic prepulse inhibition response, which is a test of sensorimotor processing, and on dendritic spine morphology in vitro.65 Their results showed 73% inhibition of the startle response in wild-type mice when preceded by an auditory prepulse stimulus compared with the response after the auditory stimulus alone. On the other hand, in Fmr1-KO mice, the startle response was inhibited only 30% of the time after a prepulse stimulus, which indicates a prepulse inhibitory response deficit. MPEP treatment 30 minutes before testing rescued the prepulse inhibitory response in Fmr1-KO mice to 70%, which is similar to wild-type levels without treatment. However, the prepulse inhibitory response was also increased in wild-type mice treated with MPEP. MPEP treatment lasting four hours also rescued the increased number of filopodia and lower ratio of spines to filopodia in Fmr1-KO neurons, without altering the number of protrusions in wild-type cultures.65 In addition, administration of MPEP rescued the increased audiogenic seizure phenotype as well as the increased time spent in the center field in the open field exploratory behavior of Fmr1-KO mice.66 Thus, MPEP treatment has a positive effect on some of the features of FXS. However, some studies have shown that chronic MPEP treatment changed the spine morphology into a more immature phenotype.67 Moreover, MPEP is readily metabolized, and is not mGluR-specific since increased dosages also affect NMDA receptors.24,68,69 Thus, many pharmaceutical companies are focusing on the development of MPEP analogs, such as the acetylenic analog, AFQ056 (Figure 2).24
Figure 2.
The structure of mGluR5 antagonists. Four common mGluR5 antagonists, ie, AFQ056, MPEP, CTEP, and fenobam.
Abbreviations: CTEP, 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy)phenyl)-1H-imidazol-4-yl)ethynyl)pyridine; MPEP, 2-methyl-6-(phenylethynyl)-pyridine hydrochloride; mGluR, metabotropic glutamate receptor.
AFQ056
Development
AFQ056 (mavoglurant) was developed by the Swiss pharmaceutical company, Novartis, as a selective negative allosteric modulator of mGluR5 with a noncompetitive inhibitory mode of action. Allosteric modulators of class C G-protein coupled receptors are thought to bind in the seven-transmembrane spanning region (Figure 3). AFQ056 was first tested for the treatment of anxiety and smoking cessation, but without a positive outcome.24 However, it showed positive results in treating the complications induced by chronic levodopa use, namely levodopa-induced dyskinesia in patients with Parkinson’s disease. It is currently under clinical development for this purpose.70 The first clinical trial using AFQ056 to treat patients with FXS showed promising results, primarily in patients with a fully methylated FMR1 promoter.71 It is unknown why subjects lacking FMR1 expression were the best responders to treatment with AFQ056. Full methylation at the FMR1 promoter site possibly modulates additional gene products that interact with AFQ056. A number of animal models have tested AFQ056, and it is currently in Phase II and III clinical trials as a potential targeted treatment for FXS.
Figure 3.
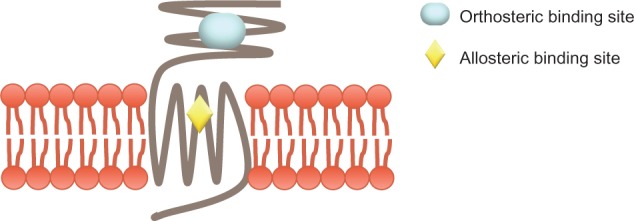
G-protein coupled receptors. A schematic of class C G-protein coupled receptors, to which mGluRs belong, showing the orthosteric and allosteric binding sites.
Preclinical trials in FXS
The first animal study to test the therapeutic potential of AFQ056 in FXS examined its effects on a sensorimotor-gating deficit, specifically the acoustic prepulse inhibition of the startle response, and also on the abnormal dendritic spine structure in adult male Fmr1-KO mice.72 Given that increased sensitivity to sensory stimuli is a hallmark of FXS, the researchers tested if a weaker acoustic stimulus would inhibit the eye blink response to a stronger startling acoustic stimulus (prepulse inhibitory response). As expected, Fmr1-KO mice manifested a deficit in the prepulse inhibitory response compared with their wild-type counterparts. An intraperitoneal injection of AFQ056 (3 mg/kg) rescued this deficit in Fmr1-KO mice without causing any side effects in wild-type mice. In addition, comparison of embryonic Fmr1-KO and wild-type mouse hippocampal neuronal cultures showed that Fmr1-KO neurons had significantly longer dendritic spines, with no difference in spine width or density between the genotypes. Three different concentrations of AFQ056, added to neuronal cultures after 14 days, successfully reduced the dendritic spine length in Fmr1-KO cultures in a concentration-dependent manner. However, treatment with AFQ056 resulted in a small but statistically significant decrease in dendritic spine width and a statistically significant increase in spine density in Fmr1-KO neuronal cultures. Such an increase in spine density following treatment is puzzling,42,72 because it suggests an aggravation of an FXS phenotype rather than an amelioration.
In a follow-up study, the same group analyzed dendritic spine morphology in vivo in the CA1 region of the hippocampus at different developmental stages, ie, in two-week-old, 10-week-old, and 25-week-old male mice.73 At two weeks, Fmr1 -KO2 mice74 had significantly shorter spines than their wild-type counterparts. This phenotype was reversed so that 10-week-old and 25-week-old Fmr1 -KO2 mice had significantly longer spines than wild-type mice of the same age. Surprisingly, there was no difference in spine density between Fmr1 -KO2 and wild-type mice at any of the ages tested. Comparing time points showed that there was a significant difference in spine density and length between each time point in Fmr1 -KO2 and wild-type mice. The scientists then examined the effect of chronic six-week AFQ056 treatment administered in food pellets starting in 19-week-old Fmr1 -KO2 and wild-type mice. Spines were significantly shorter in the Fmr1-KO2 mice after treatment, to the point that there was no significant difference in spine length between treated Fmr1 -KO2 and treated wild-type mice. Chronic AFQ056 treatment seemed to affect spine length in wild-type mice, which had significantly longer spines compared with their untreated wild-type counterparts. Treatment had no effect on spine density.73 Notably, the two studies72,73 differed in several aspects, including the choice of mouse model, experimental paradigm (in vitro versus in vivo), visualization method (transfection72 versus diOlistic labeling73), and software for visualizing confocal images, explaining the different results obtained.
It is important to characterize the social deficits inherent to FXS accurately in order to develop specific interventions that target the social disabilities in individuals with comorbid FXS and autism. For this reason, Gantois et al tested the prospect of chronic administration of AFQ056 to alleviate social deficits in adult male Fmr1-KO mice.75 Behavioral tasks included acclimation, sociability, and preference for social novelty in a three side-by-side chamber setup. In the sociability task, an unfamiliar (referred to as a “stranger”) mouse was randomly placed in a wire cup in one side (either the left or the right) chamber whereas the other side chamber contained an empty wire cup. Preferential interaction with the stranger mouse over the empty wire cup indicated sociability. The setup was modified in the preference for social novelty task by adding a second stranger mouse in the empty wire cup. The mouse tested was first placed in the central chamber, and its preference for interaction with the second stranger mouse was examined. Results showed that Fmr1-KO mice spent more time sniffing the stranger mouse in the sociability task, thereby showing increased social approach compared with wild-type mice. Chronic administration of AFQ056 for three weeks restored the abnormal social approach in Fmr1-KO mice. In addition, treatment with AFQ056 increased the preference of the Fmr1-KO mouse for the novel mouse over the familiar one in the social preference novelty task. It is noteworthy that chronic administration of AFQ056 had negative side effects in wild-type mice by reducing their speed and distance travelled during behavioral testing but not in Fmr1-KO mice.75
Other potential treatments
In addition to AFQ056, several other mGluR5 antagonists, such as 2-chloro-4-((2,5-dimethyl-1-(4-(trifluoromethoxy) phenyl)-1H-imidazol-4-yl)ethynyl)pyridine (CTEP), fenobam, STX107, and RO4917523, are being studied for the treatment of FXS. Research findings have indicated that acute administration of CTEP corrects the increased hippocampal LTD, protein synthesis, and audiogenic seizures in Fmr1-KO mice, whereas chronic administration rescues cognitive deficits, auditory hypersensitivity, dendritic spine density, overactive extracellular signal-regulated kinase and mTOR signaling, and partially corrects macro-orchidism.76 Likewise, fenobam alleviated some FXS symptoms, rescuing deficits in associative motor learning and avoidance behavior in Fmr1-KO mice; however, it had significant adverse effects on motor coordination in wild-type mice.77 A Phase I clinical trial will soon be launched to assess the safety of fenobam in healthy volunteers. Interestingly, a small, open-label, single-dose trial of fenobam had no significant adverse effects in patients with FXS and improved the prepulse inhibitory response in some subjects.78 STX107 and RO4917523 are currently in clinical trials for FXS. Figure 2 shows the structure of some mGluR5 antagonists.
γ-aminobutyric acid (GABA) agonists are emerging as contenders for treating FXS. GABA is the main inhibitory neurotransmitter in the adult central nervous system, and binds to two types of receptors, ie, the ionotropic GABAA receptor (GABAAR) and the metabotropic GABAB receptor (GABABR). In FXS, inhibitory neurotransmission involving GABA is insufficient, and treatments can target either type of receptor. Deficits in GABA inhibition underlie symptoms such as seizures, anxiety, and autistic-like behaviors in FXS.79 Recent findings indicate that Fmr1-KO mice express lower mRNA and protein levels of GABAAR subunits,80–82 which is not surprising given that mRNAs encoding the GABAAR subunits are targets of FMRP.48 Moreover, GABAAR agonists compensate for deficiencies in GABAAR subunits.83 Drugs that bind to GABAAR include diazepam, ganaxolone, and acamprosate. Administration of diazepam, a benzodiazepine, or ganaxolone has been found to rescue audiogenic seizures in Fmr1-KO mice.84 Open-label treatment with acamprosate, which also binds to NMDA receptors as well as GABAAR, remarkably improved communication in adult patients with FXS and comorbid autism,85 and improved social behavior, attention, and hyperactivity in youth without causing significant adverse effects or changes in vital signs.86 GABABR regulates cell excitability indirectly by affecting the release of glutamate.87,88 For instance, GABABR on presynaptic glutamatergic neurons inhibits the release of glutamate, and consequently, the signaling downstream of mGluR5.88 Therefore, the use of GABABR agonists can indirectly restore the aberrant basal protein levels in FXS. Arbaclofen (R-baclofen, R -4-amino-3-(4-chlorophenyl) butanoic acid or STX209) is a GABABR agonist. Its racemic mix, baclofen, has been used safely in the clinic for over 30 years, and the R-enantiomer shows promising results in treating FXS. Studies have shown that treatment with arbaclofen reduces mRNA translation and corrects the elevated basal protein synthesis in Fmr1-KO mice, and corrects increased spine density in juvenile Fmr1-KO mice.89 It also reduces AMPA receptor internalization in cultured Fmr1-KO neurons.89 A randomized, double-blind, placebo-controlled cross-over trial showed that arbaclofen had a positive effect on social function as tested by the ABC social avoidance scale in the whole study population and the Vineland-Socialization and ABC- social avoidance scales in subjects with more severe social impairments.90 Together, the mGluR theory and the GABA hypothesis suggest that the imbalance between excitation and inhibition favors overall excitation in the FXS brain and that targeting either mGluR5 or GABA receptors can treat FXS symptoms.
Conclusion and future directions
Since the identification of the FMR1 gene, many breakthrough discoveries in FXS have been made, such as elucidating the function of FMRP, proposing the mGluR theory, and developing new targeted treatments, including AFQ056. While AFQ056 seems promising in treating FXS based on animal studies, no conclusive evidence can be drawn at this time. More experiments need to be performed in order to validate the findings obtained to date, and examine the whole range of FXS phenotypes that AFQ056 can treat, such as repetitive behaviors, hyperactivity, and seizures. In addition, there are still unanswered questions about the impact of AFQ056 on spine density and morphology, especially in studies that tend to differ in spine definition and measurement techniques. There is a need to address the increase in spine density obtained in a neuronal culture upon acute treatment with AFQ056, which suggests aggravation of an FXS phenotype. This finding may not hold in vivo, in a chronic treatment administration paradigm, or by using different dosages of AFQ056. Studies can also directly test the role of AFQ056 in rescuing the abnormal protein synthesis-dependent plasticity in FXS. It is of utmost importance to address the safety of chronic AFQ056 use, especially given that ADX10059, an mGluR5 antagonist with a disubstituted alkyne functionality similar to that in AFQ056, was deemed unsafe for chronic use in Phase II clinical trials due to liver function abnormalities.25 Moreover, translational studies are necessary to validate the preclinical findings in the clinical population. AFQ056 may only be beneficial to a group of FXS patients, ie, those who have a completely methylated FMR1 promoter rather than those who display mosaicism or incomplete FMR1 methylation.71 Recently, it was suggested that the rescuing effect of AFQ056 is not due to epigenetic changes because AFQ056 does not impact the methylation status or translation of FMR1.91 It is anticipated that continued studies with AFQ056 and related drugs that target the mGluR5 pathway, if/when approved for FXS, will lead to human clinical trials for autism spectrum disorders.92
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Wang T, Bray SM, Warren ST. New perspectives on the biology of fragile X syndrome. Curr Opin Genet Dev. 2012;22(3):256–263. doi: 10.1016/j.gde.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coffee B, Keith K, Albizua I, et al. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009;85(4):503–514. doi: 10.1016/j.ajhg.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Donnell WT, Warren ST. A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci. 2002;25:315–338. doi: 10.1146/annurev.neuro.25.112701.142909. [DOI] [PubMed] [Google Scholar]
- 4.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of Fragile X syndrome: a twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 5.Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin JP, Bell J. A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry. 1943;6(3–4):154–157. doi: 10.1136/jnnp.6.3-4.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lubs HA. A marker X chromosome. Am J Hum Genet. 1969;21(3):231–244. [PMC free article] [PubMed] [Google Scholar]
- 8.Magenis RE, Hecht F, Lovrien EW. Heritable fragile site on chromosome 16: probable localization of haptoglobin locus in man. Science. 1970;170(3953):85–87. doi: 10.1126/science.170.3953.85. [DOI] [PubMed] [Google Scholar]
- 9.Sutherland GR. Fragile sites on human chromosomes: demonstration of their dependence on the type of tissue culture medium. Science. 1977;197(4300):265–266. doi: 10.1126/science.877551. [DOI] [PubMed] [Google Scholar]
- 10.Sherman SL, Morton NE, Jacobs PA, Turner G. The marker (X) syndrome: a cytogenetic and genetic analysis. Ann Hum Genet. 1984;48(Pt 1):21–37. doi: 10.1111/j.1469-1809.1984.tb00830.x. [DOI] [PubMed] [Google Scholar]
- 11.Sherman SL, Jacobs PA, Morton NE, et al. Further segregation analysis of the fragile X syndrome with special reference to transmitting males. Hum Genet. 1985;69(4):289–299. doi: 10.1007/BF00291644. [DOI] [PubMed] [Google Scholar]
- 12.Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 13.Bakker CE, Verheij C, Willemsen R, et al. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78(1):23–33. [PubMed] [Google Scholar]
- 14.Oostra BA, Hoogeveen AT. Animal model for fragile X syndrome. Ann Med. 1997;29(6):563–567. doi: 10.3109/07853899709007483. [DOI] [PubMed] [Google Scholar]
- 15.Tervonen TA, Louhivuori V, Sun X, et al. Aberrant differentiation of glutamatergic cells in neocortex of mouse model for fragile X syndrome. Neurobiol Dis. 2009;33(2):250–259. doi: 10.1016/j.nbd.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Castrén M, Tervonen T, Kärkkäinen V, et al. Altered differentiation of neural stem cells in fragile X syndrome. Proc Natl Acad Sci U S A. 2005;102(49):17834–17839. doi: 10.1073/pnas.0508995102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang YQ, Bailey AM, Matthies HJ, et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107(5):591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 18.Morales J, Hiesinger PR, Schroeder AJ, et al. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron. 2002;34(6):961–972. doi: 10.1016/s0896-6273(02)00731-6. [DOI] [PubMed] [Google Scholar]
- 19.Tucker B, Richards RI, Lardelli M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet. 2006;15(23):3446–3458. doi: 10.1093/hmg/ddl422. [DOI] [PubMed] [Google Scholar]
- 20.den Broeder MJ, van der Linde H, Brouwer JR, Oostra BA, Willemsen R, Ketting RF. Generation and characterization of FMR1 knockout zebrafish. PLoS One. 2009;4(11):e7910. doi: 10.1371/journal.pone.0007910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wan L, Dockendorff TC, Jongens TA, Dreyfuss G. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol Cell Biol. 2000;20(22):8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Dölen G, Carpenter RL, Ocain TD, Bear MF. Mechanism-based approaches to treating fragile X. Pharmacol Ther. 2010;127(1):78–93. doi: 10.1016/j.pharmthera.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Jaeschke G, Wettstein JG, Nordquist RE, Spooren W. mGlu5 receptor antagonists and their therapeutic potential. Expert Opin Ther Pat. 2008;18(2):123–142. [Google Scholar]
- 25.Weiss JM, Jimenez HN, Li G, et al. 6-Aryl-3-pyrrolidinylpyridines as mGlu5 receptor negative allosteric modulators. Bioorg Med Chem Lett. 2011;21(16):4891–4899. doi: 10.1016/j.bmcl.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 26.Romano C, Sesma MA, McDonald CT, O’Malley K, Van den Pol AN, Olney JW. Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol. 1995;355(3):455–469. doi: 10.1002/cne.903550310. [DOI] [PubMed] [Google Scholar]
- 27.Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8(7):1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 28.Bhakar AL, Dölen G, Bear MF. The pathophysiology of Fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freire M. Effects of dark rearing on dendritic spines in layer IV of the mouse visual cortex. A quantitative electron microscopical study. J Anat. 1978;126(Pt 1):193–201. [PMC free article] [PubMed] [Google Scholar]
- 30.Spacek J, Hartmann M. Three-dimensional analysis of dendritic spines. I. Quantitative observations related to dendritic spine and synaptic morphology in cerebral and cerebellar cortices. Anat Embryol. 1983;167(2):289–310. doi: 10.1007/BF00298517. [DOI] [PubMed] [Google Scholar]
- 31.Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9(8):2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21(3):545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- 33.Oberlé I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252(5010):1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 34.Fu YH, Kuhl DP, Pizzuti A, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67(6):1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 35.Devys D, Lutz Y, Rouyer N, Bellocq JP, Mandel JL. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat Genet. 1993;4(4):335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- 36.Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM. Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci. 1997;17(5):1539–1547. doi: 10.1523/JNEUROSCI.17-05-01539.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pacey LKK, Doering LC. Developmental expression of FMRP in the astrocyte lineage: Implications for fragile X syndrome. Glia. 2007;55(15):1601–1609. doi: 10.1002/glia.20573. [DOI] [PubMed] [Google Scholar]
- 38.Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993;74(2):291–298. doi: 10.1016/0092-8674(93)90420-u. [DOI] [PubMed] [Google Scholar]
- 39.Ashley CT, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262(5133):563–566. doi: 10.1126/science.7692601. [DOI] [PubMed] [Google Scholar]
- 40.Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST. FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe fragile X syndrome abolishes this association. Mol Cell. 1997;1(1):109–118. doi: 10.1016/s1097-2765(00)80012-x. [DOI] [PubMed] [Google Scholar]
- 41.Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5(8):1083–1091. doi: 10.1093/hmg/5.8.1083. [DOI] [PubMed] [Google Scholar]
- 42.Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: the roots of fragile X syndrome. Nat Rev Neurosci. 2005;6(5):376–387. doi: 10.1038/nrn1667. [DOI] [PubMed] [Google Scholar]
- 43.Steward O, Bakker CE, Willems PJ, Oostra BA. No evidence for disruption of normal patterns of mRNA localization in dendrites or dendritic transport of recently synthesized mRNA in FMR1 knockout mice, a model for human fragile-X mental retardation syndrome. Neuroreport. 1998;9(3):477–481. doi: 10.1097/00001756-199802160-00022. [DOI] [PubMed] [Google Scholar]
- 44.Zalfa F, Eleuteri B, Dickson KS, et al. A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat Neurosci. 2007;10(5):578–587. doi: 10.1038/nn1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24(11):2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Irwin SA, Galvez R, Greenough WT. Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex. 2000;10(10):1038–1044. doi: 10.1093/cercor/10.10.1038. [DOI] [PubMed] [Google Scholar]
- 47.Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26(27):7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyashiro KY, Beckel-Mitchener A, Purk TP, et al. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37(3):417–431. doi: 10.1016/s0896-6273(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 49.Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27(20):5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10(4):329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- 51.Lu R, Wang H, Liang Z, et al. The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc Natl Acad Sci U S A. 2004;101(42):15201–15206. doi: 10.1073/pnas.0404995101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bassell GJ, Warren ST. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron. 2008;60(2):201–214. doi: 10.1016/j.neuron.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rooms L, Kooy RF. Advances in understanding fragile X syndrome and related disorders. Curr Opin Pediatr. 2011;23(6):601–606. doi: 10.1097/MOP.0b013e32834c7f1a. [DOI] [PubMed] [Google Scholar]
- 55.Weiler IJ, Greenough WT. Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci U S A. 1993;90(15):7168–7171. doi: 10.1073/pnas.90.15.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weiler IJ, Irwin SA, Klintsova AY, et al. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94(10):5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288(5469):1254–1256. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- 59.Politte LC, McDougle CJ. Phase II and III drugs for the treatment of fragile X syndrome. Expert Opinion on Orphan Drugs. 2013;1:47–65. [Google Scholar]
- 60.Steward O, Schuman EM. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2001;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- 61.Nakamoto M, Nalavadi V, Epstein MP, Narayanan U, Bassell GJ, Warren ST. Fragile X mental retardation protein deficiency leads to excessive mGluR5-dependent internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2007;104(39):15537–15542. doi: 10.1073/pnas.0707484104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4(11):1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- 63.Krueger DD, Bear MF. Toward fulfilling the promise of molecular medicine in Fragile X syndrome. Annu Rev Med. 2011;62(1):411–429. doi: 10.1146/annurev-med-061109-134644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dölen G, Osterweil E, Rao BSS, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56(6):955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Vrij FM, Levenga J, van der Linde HC, et al. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31(1):127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49(7):1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 67.Catania MV, Bellomo M, Di Giorgi-Gerevini V, et al. Endogenous activation of group-I metabotropic glutamate receptors is required for differentiation and survival of cerebellar Purkinje cells. J Neurosci. 2001;21(19):7664–7673. doi: 10.1523/JNEUROSCI.21-19-07664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Popoli P, Pintor A, Tebano MT, et al. Neuroprotective effects of the mGlu5R antagonist MPEP towards quinolinic acid-induced striatal toxicity: involvement of pre- and post-synaptic mechanisms and lack of direct NMDA blocking activity. J Neurochem. 2004;89(6):1479–1489. doi: 10.1111/j.1471-4159.2004.02448.x. [DOI] [PubMed] [Google Scholar]
- 69.Lea PM, Movsesyan VA, Faden AI. Neuroprotective activity of the mGluR5 antagonists MPEP and MTEP against acute excitotoxicity differs and does not reflect actions at mGluR5 receptors. Br J Pharmacol. 2005;145(4):527–534. doi: 10.1038/sj.bjp.0706219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berg D, Godau J, Trenkwalder C, et al. AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov Disord. 2011;26(7):1243–1250. doi: 10.1002/mds.23616. [DOI] [PubMed] [Google Scholar]
- 71.Jacquemont S, Curie A, Portes des V, et al. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 2011;3(64):64ral. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]
- 72.Levenga J, Hayashi S, de Vrij FM, et al. AFQ056, a new mGluR5 antagonist for treatment of fragile X syndrome. Neurobiol Dis. 2011;42(3):311–317. doi: 10.1016/j.nbd.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 73.Pop AS, Levenga J, de Esch CE, et al. Rescue of dendritic spine phenotype in Fmr1 KO mice with the mGluR5 antagonist AFQ056/mavoglurant. Psychopharmacology (Berl) 2012 doi: 10.1007/s00213-012-2947-y.. [DOI] [PubMed] [Google Scholar]
- 74.Mientjes EJ, Nieuwenhuizen I, Kirkpatrick L, et al. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiol Dis. 2006;21(3):549–555. doi: 10.1016/j.nbd.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 75.Gantois I, Pop AS, de Esch CE, et al. Chronic administration of AFQ056/Mavoglurant restores social behaviour in Fmr1 knockout mice. Behav Brain Res. 2013;239:72–79. doi: 10.1016/j.bbr.2012.10.059. [DOI] [PubMed] [Google Scholar]
- 76.Michalon A, Sidorov M, Ballard TM, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74(1):49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vinueza Veloz MF, Buijsen RA, Willemsen R, et al. The effect of an mGluR5 inhibitor on procedural memory and avoidance discrimination impairments in Fmr1 KO mice. Genes Brain Behav. 2012;11(3):325–331. doi: 10.1111/j.1601-183X.2011.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berry-Kravis E, Hessl D, Coffey S, et al. A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J Med Genet. 2009;46(4):266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hagerman R, Hoem G, Hagerman P. Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol Autism. 2010;1(1):12. doi: 10.1186/2040-2392-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gantois I, Vandesompele J, Speleman F, et al. Expression profiling suggests underexpression of the GABA(A) receptor subunit delta in the fragile X knockout mouse model. Neurobiol Dis. 2006;21(2):346–357. doi: 10.1016/j.nbd.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 81.D’Hulst C, De Geest N, Reeve SP, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121(1):238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 82.Curia G, Papouin T, Séguéla P, Avoli M. Downregulation of tonic GABAergic inhibition in a mouse model of fragile X syndrome. Cereb Cortex. 2009;19(7):1515–1520. doi: 10.1093/cercor/bhn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagerman R, Lauterborn J, Au J, Berry-Kravis E. Fragile X syndrome and targeted treatment trials. Results Probl Cell Differ. 2012;54:297–335. doi: 10.1007/978-3-642-21649-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heulens I, D’Hulst C, Van Dam D, De Deyn PP, Kooy RF. Pharmacological treatment of fragile X syndrome with GABAergic drugs in a knockout mouse model. Behav Brain Res. 2012;229(1):244–249. doi: 10.1016/j.bbr.2012.01.031. [DOI] [PubMed] [Google Scholar]
- 85.Erickson CA, Mullett JE, McDougle CJ. Brief report: acamprosate in fragile X syndrome. J Autism Dev Disord. 2010;40(11):1412–1416. doi: 10.1007/s10803-010-0988-9. [DOI] [PubMed] [Google Scholar]
- 86.Erickson CA, Wink LK, Ray B, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology (Berl) 2013;228(1):75–84. doi: 10.1007/s00213-013-3022-z. [DOI] [PubMed] [Google Scholar]
- 87.Sohn J-W, Lee D, Cho H, et al. Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J Physiol (Lond) 2007;580(Pt 2):411–422. doi: 10.1113/jphysiol.2006.125914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Isaacson JS, Hille B. GABA(B)-mediated presynaptic inhibition of excitatory transmission and synaptic vesicle dynamics in cultured hippocampal neurons. Neuron. 1997;18(1):143–152. doi: 10.1016/s0896-6273(01)80053-2. [DOI] [PubMed] [Google Scholar]
- 89.Henderson C, Wijetunge L, Kinoshita MN, et al. Reversal of disease-related pathologies in the Fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012;4(152):152ra128. doi: 10.1126/scitranslmed.3004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berry-Kravis EM, Hessl D, Rathmell B, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with Fragile X syndrome: a randomized, controlled, phase 2 trial. Sci Transl Med. 2012;4(152):152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- 91.Tabolacci E, Pirozzi F, Gomez-Mancilla B, Gasparini F, Neri G. The mGluR5 antagonist AFQ056 does not affect methylation and transcription of the mutant FMR1 gene in vitro. BMC Med Genet. 2012;13:13. doi: 10.1186/1471-2350-13-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spooren W, Lindemann L, Ghosh A, Santarelli L. Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharmacol Sci. 2012;33(12):669–684. doi: 10.1016/j.tips.2012.09.004. [DOI] [PubMed] [Google Scholar]