

Abstract
Hypertension is a risk factor for myocardial infarction, stroke, renal failure, heart failure, and peripheral vascular disease. One feature of hypertension is a hyperresponsiveness to contractile agents, and inhibition of vasoconstriction forms the basis of some of the treatments for hypertension. Hypertension is also associated with an increase in the growth and proliferation of vascular smooth muscle cells, which can lead to a thickening of the smooth muscle layer of the blood vessels and a reduction in lumen diameter. Targeting both the enhanced contractile responses, and the increased vascular smooth muscle cell growth could potentially be important pharmacological treatment of hypertension. Extracellular signal-regulated kinase (ERK) is a member of the mitogen-activated protein kinase family that is involved in both vasoconstriction and vascular smooth muscle cell growth and this, therefore, makes it attractive therapeutic target for treatment of hypertension. ERK activity is raised in vascular smooth muscle cells from animal models of hypertension, and inhibition of ERK activation reduces both vascular smooth muscle cell growth and vasoconstriction. This review discusses the potential for targeting ERK activity in the treatment of hypertension.
Keywords: ERK, hypertension, smooth muscle, vasoconstriction
Introduction
Hypertension is a risk factor for myocardial infarction, stroke, renal failure, heart failure, and peripheral vascular disease. It is associated with both enhanced responses to contractile agents, and an increase in the growth and proliferation of vascular smooth muscle cells. The protein kinase extracellular signal-regulated kinase (ERK) is involved in both vasoconstriction and vascular smooth muscle cell growth and this, therefore, makes it an attractive target for the treatment of hypertension.
The mitogen-activated protein kinase (MAPK) family of enzymes include the ERKs (ERK1, ERK2, ERK3/4, ERK5, ERK7), the c-Jun amino-terminal kinases, and p38 MAPKs.1 Common to all of the MAPK family of protein kinases is activation via a sequential phosphorylation cascade, made up of three protein kinases: MAPK kinase (MEK) kinase, MEK, and MAPK (Figure 1).2 ERK1 and ERK2 are activated through the Ras/Raf/MEK cascade. Activation of the heterotrimeric G protein Ras leads to activation of the MEK kinase Raf, which in turn activates MEK, which then phosphorylates ERK1 and ERK2 at threonine 202 and tyrosine 204 to activate the enzymes.2
Figure 1.
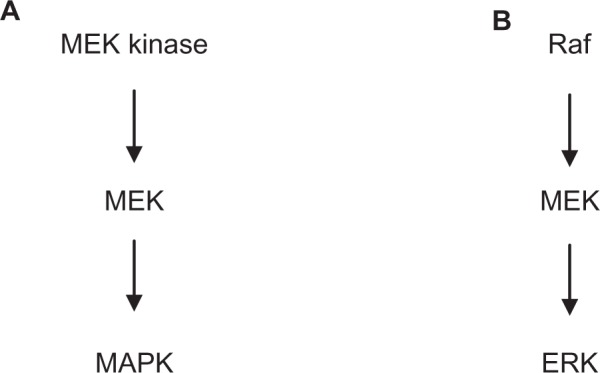
Schematic diagram showing (A) the general signaling pathway for activation of mitogen-activated protein kinases and (B) the specific pathway for activation of extracellular signal-regulated kinase.
Abbreviations: ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase.
Although other isozymes of ERK are known, ERK1 and ERK2 are the most studied. This review will concentrate on these two isozymes. Phosphorylated ERK1 and ERK2 enter the nucleus where they themselves phosphorylate transcription factors involved in regulation of the cell-cycle and tissue proliferation.3 In differentiated, contractile vascular smooth muscle, ERK1 and ERK2 in the cytoplasm are involved in the regulation of vascular smooth muscle contraction. As far as this review is concerned, ERK refers to ERK1 and ERK2.
ERK can be activated through stimulation of G protein-coupled receptors (GPCRs) and release of Gβγ subunits,4 or through activation of growth factor-stimulated tyrosine kinase receptors (Figure 2).5 As well as activation by Gβγ subunits after stimulation of GPCRs, ERK can also be activated through transactivation of growth factor tyrosine kinase receptors such as the epidermal growth factor (EGF) receptor, involving either tyrosine phosphorylation of the receptor, or the proteolytic cleavage of a membrane bound ligand, which then activates the receptor (Figure 2).6–9
Figure 2.

Schematic diagram summarizing the potential mechanisms of extracellular signal-regulated kinase activation.
Notes: Extracellular signal-regulated kinase activation can occur through stimulation of either a G protein-coupled receptor or a growth factor receptor, followed by activation of the Ras, Raf, mitogen-activated protein kinase kinase pathway. Activation of extracellular signal-regulated kinase through G protein-coupled receptors could be through direct activation of the Ras, Raf, mitogen-activated protein kinase kinase pathway, or through transactivation of a growth factor receptor, such as the epidermal growth factor receptor. This can occur through activation of a matrix metalloprotease and subsequent cleavage of a membrane-bound ligand such as heparin-binding epidermal growth factor, leading to release of the ligand and activation of the receptor. Alternatively, activation of the G protein-coupled receptor could lead to tyrosine phosphorylation of the epidermal growth factor receptor.
Abbreviations: EGF, epidermal growth factor; ERK, extracellular signal-regulated kinase; HB-EGF, heparin-binding epidermal growth factor; GPCR, G protein-coupled receptor; MEK, mitogen-activated protein kinase kinase; MMP, matrix metalloprotease.
Role of ERK in regulation of vascular smooth muscle contraction
Hypertension is associated with an increase in the contraction of blood vessels in response to agonist stimulation, and impaired relaxation responses to both endothelium-dependent and -independent stimuli.10,11 Contraction of isolated blood vessels in response to activation of certain GPCRs is associated with an increase in ERK activity, and inhibition of ERK activation by inhibiting the upstream protein kinase MEK reduces contraction of blood vessels.12–14 Stretch-induced contractions are also associated with an increase in ERK activity.15 The relative role of ERK in the contractile response appears to depend upon the receptor activated and/or the blood vessel. For example, ERK appears to play a greater role in α2-adrenoceptor-mediated contractions in the porcine palmar lateral vein compared to α1-adrenoceptor-mediated contractions in the ferret aorta.12,14 Currently, the mechanism by which ERK causes contraction of the blood vessels is not clear. One hypothesis is that ERK phosphorylates the actin-binding protein caldesmon (Figure 3). Caldesmon can inhibit the adenosine triphosphatase activity of actomyosin,16 and it has been proposed that phosphorylation of caldesmon by ERK removes this inhibitory effect. Phosphorylation of caldesmon at serine 789 by ERK is thought to bring about a conformational change in caldesmon,17 resulting in decreased binding to actin and, hence, removal of the adenosine triphosphatase inhibition.18 Caldesmon phosphorylation that is inhibited by the MEK inhibitor PD98059 has been shown to be associated with vasoconstriction,12,19 although other studies have suggested that ERK only phosphorylates l-caldesmon, which is only present in dedifferentiated smooth muscle, and thus suggesting that ERK phosphorylation of caldesmon is involved in cell division rather than contraction.20 Phosphorylation of l-caldesmon in dedifferentiated cells may regulate migration of proliferating vascular smooth muscle cells and, therefore, may play a role in the development of smooth muscle hyperplasia in hypertension.21 Other studies have found no correlation between caldesmon phosphorylation by ERK and blood vessel contraction, further questioning the role of caldesmon in the ERK-mediated contractile response.22–25
Figure 3.
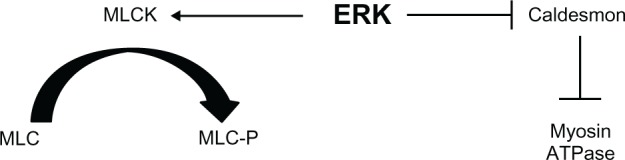
Schematic diagram summarizing the potential mechanisms by which extracellular signal-regulated kinase could regulate vasoconstriction.
Notes: Phosphorylation of caldesmon at serine 789 is thought to inhibit the activity of this protein, thus removing its inhibitory effect on myosin adenosine triphosphatase. Alternatively, extracellular signal-regulated kinase could activate myosin light-chain kinase leading to an increase in phosphorylation of the myosin light-chains.
Abbreviations: ATPase, adenosine triphosphatase; ERK, extracellular signal-regulated kinase; MLC, myosin light-chain; MLCK, myosin light-chain kinase; MLC-P, myosin light-chain phosphatase.
Alternative mechanisms by which ERK mediates vasoconstriction include activation of myosin light-chain kinase, leading to increased phosphorylation of myosin light-chains and hence contraction.26 This is supported by studies in the porcine palmar lateral vein showing that inhibition of ERK activation causes a reduction in myosin light-chain phosphorylation.27 Phosphorylated ERK has also been shown to be associated with the actin cytoskeleton,28 which could be related to phosphorylation of caldesmon or phosphorylation of myosin light-chains.
Role of ERK in regulation of vascular smooth muscle growth
Abnormal growth and proliferation of vascular smooth muscle cells, leading to a thickening of the smooth muscle cell layer and a reduction of the lumen diameter, is a major feature of hypertension, and may underlie the increase in the contractility of blood vessels.29 Activation of ERK is associated with changes in gene transcription and cell proliferation (Figure 4).3 In vascular smooth muscle, ERK is activated by growth factors, such as EGF, to regulate gene transcription and cell proliferation.5,30–32 Transactivation of the EGF receptor leading to activation of ERK has been shown to be involved in the induction of vascular smooth muscle cell growth after activation of α1-adrenoceptors.33 A similar trans-activation of the EGF receptor is implicated in angiotensin II-induced vascular smooth muscle cell proliferation.34 These studies indicate the importance of ERK in the regulation of vascular smooth muscle cell proliferation. Indeed, modulation of primary vascular smooth muscle cells from a contractile to a proliferating phenotype is associated with prolonged ERK activation.35 This could be important in disease states such as hypertension and stenosis in which there are increases in vascular smooth muscle cell growth or remodeling (hypertrophy or hyperplasia).36,37
Figure 4.
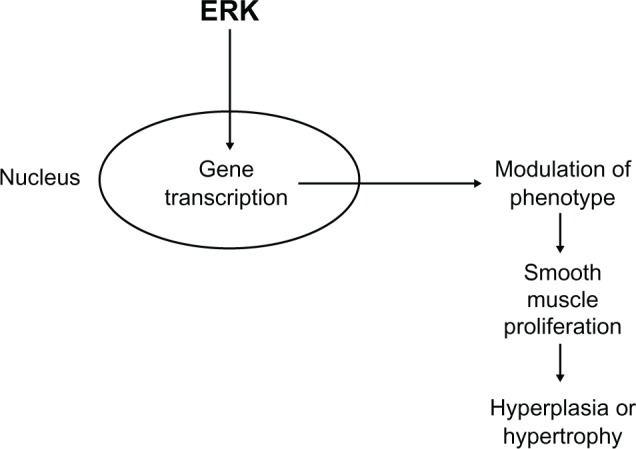
Schematic diagram summarizing the effect of extracellular signal-regulated kinase on vascular smooth muscle cell growth and proliferation through effects on gene transcription.
Abbreviation: ERK, extracellular signal-regulated kinase.
Vascular smooth muscle cells exist in two phenotypes: a fully differentiated, contractile phenotype and a dedifferentiated, proliferating phenotype. During hyperplasia of the smooth muscle layer, dedifferentiation from a contractile phenotype back into a proliferating phenotype causes a loss of contractile function. Altering the regulation of ERK activation may be a way of changing the phenotype of vascular smooth muscle cells back to the contractile type, thus reducing smooth muscle proliferation. For example, in an ex vivo organ culture model of smooth muscle hyperplasia, both platelet-derived growth factor and endothelin-1 induced smooth muscle cell growth were prevented by inhibiting ERK activation and caused a partial restoration of the contractile response,38 indicating that therapeutic inhibition of ERK activation could prevent smooth muscle hyperplasia.
Evidence for changes in ERK activity in hypertension
Basal levels of ERK are raised in a number of different animal models of hypertension, including spontaneously hypertensive rats (SHRs),39–41 and agonist-stimulated ERK activation is also enhanced in vascular smooth muscle cells from SHRs.42–44 Inhibition of ERK activation inhibits the enhanced contraction to agonists seen in hypertension. For example, angiotensin II-mediated contraction of smooth muscle cells from SHRs is increased compared to normotensive controls, and this is normalized by inhibition of ERK activation.45 On the other hand, treatment of SHRs with MEK inhibitor PD98059 for 24 hours had no effect on blood pressure.41 Similarly, chronic treatment with PD98059 (24 weeks) did not reverse the increase in blood pressure in SHRs.46 On the other hand, inhibition of ERK activity was associated with an improvement of endothelial function, and angiotensin II-mediated vasoconstriction was inhibited after 24-hour treatment with PD98059.40 Structure of the blood vessels was unaltered by treatment with PD98059 after 24 hours, but this would be as expected in such a short time frame. In comparison, chronic (24 week) treatment with PD98059 caused a partial reduction in the thickening of the vascular wall in renal arteries from SHRs.46 Although PD98059 had no effect on blood pressure in SHRs, in both deoxycorticosterone acetate salt-induced hypertension and angiotensin II-induced hypertension, treatment with PD98059 reduced blood pressure within 30–60 minutes,47,48 suggesting that the role of ERK in the regulation of blood pressure may depend on the animal model used.
Role of reactive oxygen species in ERK activation in hypertension
Hypertension is associated with an increase in the levels of reactive oxygen species in vascular smooth muscle cells,49,50 from both mitochondria and nicotinamide adenine dinucleotide phosphate oxidase.51,52 This increase in reactive oxygen species may be responsible for the increase in ERK activity in hypertension (Figure 5). For example, hydrogen-peroxide increases ERK activation, which is more prominent in SHRs compared to control animals.53 Furthermore, nicotinamide adenine dinucleotide phosphate oxidase activation and peroxide production can enhance vasoconstriction through activation of ERK.15 5-hydroxytryptamine stimulation of ERK in bovine pulmonary artery smooth muscle cells is mediated through production of reactive oxygen species,54 which may be related to the ability of 5-hydroxytryptamine to enhance smooth muscle proliferation.55 Interestingly, production of reactive oxygen species has been linked to the transactivation of the EGF receptor leading to ERK-mediated vascular smooth muscle cell growth.33 Protein kinase C and calcium/calmodulin-dependent pathways have also been suggested to be involved in hydrogen peroxide-mediated ERK activation in vascular smooth muscle cells.56 Although reactive oxygen species have been reported to activate Ras in vascular smooth muscle cells, this did not appear to lead to ERK activation.57 Together, these data indicate that the increase in reactive oxygen species in hypertension may produce responses through the ERK pathway and that reactive oxygen species-stimulated ERK activation may be involved in both vasoconstriction and increases in smooth muscle proliferation.
Figure 5.

Schematic diagram summarizing the role of extracellular signal-regulated kinase (ERK) in vascular smooth muscle and hypertension.
Notes: ERK can be activated through either stimulation of G protein-coupled receptors or growth factor tyrosine kinase receptors. As well as direct activation of ERK through G protein-coupled receptors, G protein-coupled receptor-stimulated transactivation of growth factor receptors can also lead to ERK activation. ERK can also be activated through production of reactive oxygen species from both nicotinamide adenine dinucleotide phosphate oxidase and mitochondria. Activated ERK mediates vascular smooth muscle contraction and could underlie the changes in vasoconstriction in hypertension. Activated ERK also causes changes in gene transcription leading to modulation of vascular smooth muscle phenotype and increases in vascular smooth muscle cell proliferation, which could underlie the hypertrophy/hyperplasia in hypertension.
Abbreviations: ERK, extracellular signal-regulated kinase; GPCR, G protein-coupled receptor; H2O2, hydrogen peroxide; MEK, mitogen-activated protein kinase kinase; NAPDH, nicotinamide adenine dinucleotide phosphate; O2, oxygen.
ERK as a therapeutic target in hypertension
The studies highlighted in this review demonstrate that ERK plays a role in mediating vasoconstriction and vascular smooth muscle cell growth, and that there is an increase in ERK activity in hypertension. The fact that ERK plays a role in both vasoconstriction and smooth muscle growth makes it an attractive target for treatment of hypertension as inhibition of ERK activity would lead to a reduced contraction of the vascular smooth muscle and would also prevent the increase in smooth muscle cell growth seen in vascular smooth muscle hyperplasia and hypertrophy. This has been demonstrated in animal models of hypertension in which there was a reduction in blood pressure and a reduction in the growth of the smooth muscle layer.46–48 Targeting the signaling pathway that is activated by multiple receptors, including GPCRs and growth factor receptors, would reduce the need to target each receptor independently. However, inhibition of ERK would have to be targeted specifically to the vascular smooth muscle in order to prevent nonselective inhibition of cell growth elsewhere in the body. Indirect inhibitors of ERK activation could be used, and some of these are already in use therapeutically. 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, or statins, for example, are used in the treatment of hypercholesterolemia. They inhibit the conversion of 3-hydroxy-3-methylglutaryl-coenzyme A to mevalonate, a precursor of cholesterol, which is also required for the production of the isoprenoids farnesyl pyrophosphate and geranylgeranyl pyrophosphate (Figure 6).58 Posttranslational modification of Ras by farnesyl pyrophosphate is required to enable its translocation to the cell membrane, where it is activated.58 For example, simvastatin inhibits angiotensin II-stimulated ERK activation,59 and inhibits proliferation of human vascular smooth muscle cells by preventing the farnesylation of Ras.60 A question mark, however, is whether high enough concentrations of statins could be obtained in vivo to replicate the effects seen in vitro.
Figure 6.
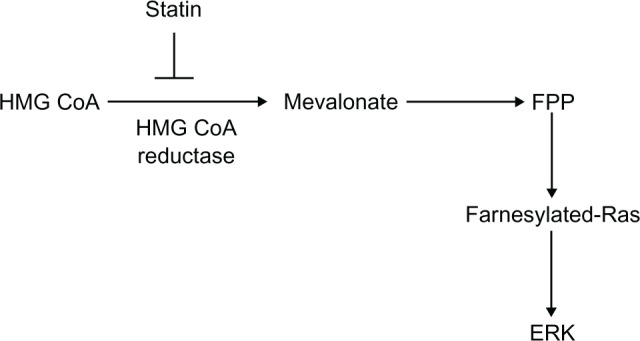
Schematic diagram showing the role of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway in the production of the isoprenoid farnesyl pyrophosphate and hence the farnesylation of Ras.
Note: The site of inhibition of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase pathway by statins is also indicated.
Abbreviations: ERK, extracellular signal-regulated kinase; FPP, farnesyl pyrophosphate; HMG CoA, 3-hydroxy-3-methylglutaryl-coenzyme A.
Conclusion
Hypertension is associated with increased levels of ERK activity in vascular smooth muscle. This increase in ERK activity appears to be involved in both the enhanced contractile responses and the increased smooth muscle proliferation seen in hypertension (Figure 2). This fact makes it an attractive target for treatment of hypertension as this would enable dual inhibition of both contractile responses and smooth muscle hyperplasia. A potential problem with targeting ERK would be selectively targeting the vascular smooth muscle to prevent inhibition of cell growth in other tissues in the body.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71(3–4):479–500. doi: 10.1016/s0079-6107(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 3.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 4.Hawes BE, Luttrell LM, van Biesen T, Lefkowitz RJ. Phosphati-dylinositol 3-kinase is an early intermediate in the Gβγ-mediated mitogen-activated protein kinase signaling pathway. J Biol Chem. 1996;271(21):12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 5.Yamanaka Y, Hayashi K, Komurasaki T, Morimoto S, Ogihara T, Sobue K. EGF family ligand-dependent phenotypic modulation of smooth muscle cells through EGF receptor. Biochem Biophys Res Commun. 2001;281(2):373–377. doi: 10.1006/bbrc.2001.4385. [DOI] [PubMed] [Google Scholar]
- 6.Eguchi S, Numaguchi K, Iwasaki H, et al. Calcium-dependent epidermal growth factor receptor transactivation mediates the angiotensin II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. J Biol Chem. 1998;273(15):8890–8896. doi: 10.1074/jbc.273.15.8890. [DOI] [PubMed] [Google Scholar]
- 7.Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of Erk and p38 MAPK but not for JNK. J Biol Chem. 2001;276(11):7957–7962. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- 8.Gao Y, Tang S, Zhou S, Ware A. The thromboxane A2 receptor activates mitogen-activated protein kinase via protein kinase C-dependent Gi coupling and Src-dependent phosphorylation of the epidermal growth factor receptor. J Pharmacol Exp Ther. 2001;296(2):426–433. [PubMed] [Google Scholar]
- 9.Zwick E, Hackel PO, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20(10):408–412. doi: 10.1016/s0165-6147(99)01373-5. [DOI] [PubMed] [Google Scholar]
- 10.Lais LT, Brody MJ. Mechanism of vascular hyperresponsiveness in the spontaneously hypertensive rat. Circ Res. 1975;36(6 Suppl 1):216–222. doi: 10.1161/01.res.36.6.216. [DOI] [PubMed] [Google Scholar]
- 11.Bruner CA. Vascular responsiveness in rats resistant to aldosterone-salt hypertension. Hypertension. 1992;20(1):59–66. doi: 10.1161/01.hyp.20.1.59. [DOI] [PubMed] [Google Scholar]
- 12.Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by α-adrenoceptor stimulation. Am J Physiol. 1998;275(4 Pt 1):C1081–C1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- 13.Banes A, Florian JA, Watts SW. Mechanisms of 5-hydroxytryptamine2A receptor activation of the mitogen-activated protein kinase pathway in vascular smooth muscle. J Pharmacol Exp Ther. 1999;291(3):1179–1187. [PubMed] [Google Scholar]
- 14.Roberts RE. Role of the extracellular signal-regulated kinase (ERK) signal transduction cascade in α2-adrenoceptor-mediated vasoconstriction in porcine palmar lateral vein. Br J Pharmacol. 2001;133(6):859–866. doi: 10.1038/sj.bjp.0704149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oeckler RA, Kaminski PM, Wolin MS. Stretch enhances contraction of bovine coronary arteries via an NADPH oxidase-mediated activation of the extracellular signal-regulated mitogen-activated protein kinase cascade. Circ Res. 2003;92(1):23–31. doi: 10.1161/01.res.0000051860.84509.ce. [DOI] [PubMed] [Google Scholar]
- 16.Ngai PK, Walsh MP. Inhibition of smooth muscle actin-activated myosin Mg2+ ATPase activity by caldesmon. J Biol Chem. 1984;259(22):13656–13659. [PubMed] [Google Scholar]
- 17.Adam LP, Hathaway DR. Identification of mitogen-activated protein kinase phosphorylation sequences in mammalian h-caldesmon. FEBS Lett. 1993;322(1):56–60. doi: 10.1016/0014-5793(93)81110-l. [DOI] [PubMed] [Google Scholar]
- 18.Patchell VB, Vorotnikov AV, Gao Y, et al. Phosphorylation of the minimal inhibitory region at the C-terminus of caldesmon alters its structural and actin binding properties. Biochim Biophys Acta. 2002;1596(1):121–130. doi: 10.1016/s0167-4838(02)00210-8. [DOI] [PubMed] [Google Scholar]
- 19.Xiao DL, Pearce WJ, Longo LD, Zhang L. ERK-mediated uterine artery contraction: role of thick and thin filament regulatory pathways. Am J Physiol Heart Circ Physiol. 2004;286(5):H1615–H1622. doi: 10.1152/ajpheart.00981.2003. [DOI] [PubMed] [Google Scholar]
- 20.D’Angelo G, Graceffa P, Wang CA, Wrangle J, Adam LP. Mammal-specific, ERK-dependent, caldesmon phosphorylation in smooth muscle. Quantitation using novel anti-phosphopeptide antibodies. J Biol Chem. 1999;274(42):30115–30121. doi: 10.1074/jbc.274.42.30115. [DOI] [PubMed] [Google Scholar]
- 21.Yokouchi K, Numaguchi Y, Kubota R, et al. l-caldesmon regulates proliferation and migration of vascular smooth muscle cells and inhibits neointimal formation after angioplasty. Arterioscler Thromb Vasc Biol. 2006;26(10):2231–2237. doi: 10.1161/01.ATV.0000239441.29687.97. [DOI] [PubMed] [Google Scholar]
- 22.Gorenne I, Su X, Moreland RS. Inhibition of p42 and p44 MAP kinase does not alter smooth muscle contraction in swine carotid artery. Am J Physiol. 1998;275(1 Pt 2):H131–H138. doi: 10.1152/ajpheart.1998.275.1.H131. [DOI] [PubMed] [Google Scholar]
- 23.Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. Am J Physiol Cell Physiol. 2000;278(4):C718–C726. doi: 10.1152/ajpcell.2000.278.4.C718. [DOI] [PubMed] [Google Scholar]
- 24.Childs TJ, Watson MH, Sanghera JS, Campbell DL, Pelech SL, Mak AS. Phosphorylation of smooth muscle caldesmon by mitogen-activated protein (MAP) kinase and expression of MAP kinase in differentiated smooth muscle cells. J Biol Chem. 1992;267(32):22853–22859. [PubMed] [Google Scholar]
- 25.Nixon GF, Iizuka K, Haystead CM, Haystead TA, Somlyo AP, Somlyo AV. Phosphorylation of caldesmon by mitogen-activated protein kinase with no effect on Ca2+ sensitivity in rabbit smooth muscle. J Physiol. 1995;487(Pt 2):283–289. doi: 10.1113/jphysiol.1995.sp020879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137(2):481–492. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts RE. The role of Rho kinase and extracellular regulated kinase-mitogen-activated protein kinase in α2-adrenoceptor-mediated vasoconstriction in the porcine palmar lateral vein. J Pharmacol Exp Ther. 2004;311(2):742–747. doi: 10.1124/jpet.104.071100. [DOI] [PubMed] [Google Scholar]
- 28.Escano CS, Jr, Keever LB, Gutweiler AA, Andresen BT. Angiotensin II activates extracellular signal-regulated kinase independently of receptor tyrosine kinases in renal smooth muscle cells: implications for blood pressure regulation. J Pharmacol Exp Ther. 2008;324(1):34–42. doi: 10.1124/jpet.107.126300. [DOI] [PubMed] [Google Scholar]
- 29.Mulvany MJ, Hansen OK, Aalkjaer C. Direct evidence that the greater contractility of resistance vessels in spontaneously hypertensive rats is associated with a narrowed lumen, a thickened media, and an increased number of smooth muscle cell layers. Circ Res. 1978;43(6):854–864. doi: 10.1161/01.res.43.6.854. [DOI] [PubMed] [Google Scholar]
- 30.Mii S, Khalil RA, Morgan KG, Ware JA, Kent KC. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am J Physiol. 1996;270(1 Pt 2):H142–H150. doi: 10.1152/ajpheart.1996.270.1.H142. [DOI] [PubMed] [Google Scholar]
- 31.Wilden PA, Agazie YM, Kaufman R, Halenda SP. ATP-stimulated smooth muscle cell proliferation requires independent ERK and PI3K signaling pathways. Am J Physiol. 1998;275(4 Pt 2):H1209–H1215. doi: 10.1152/ajpheart.1998.275.4.H1209. [DOI] [PubMed] [Google Scholar]
- 32.Yu SM, Hung LM, Lin CC. cGMP-elevating agents suppress proliferation of vascular smooth muscle cells by inhibiting the activation of epidermal growth factor signaling pathway. Circulation. 1997;95(5):1269–1277. doi: 10.1161/01.cir.95.5.1269. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Chalothorn D, Jackson LF, Lee DC, Faber JE. Transactivation of epidermal growth factor receptor mediates catecholamine-induced growth of vascular smooth muscle. Circ Res. 2004;95(10):989–997. doi: 10.1161/01.RES.0000147962.01036.bb. [DOI] [PubMed] [Google Scholar]
- 34.Liu G, Hitomi H, Hosomi N, et al. Mechanical stretch potentiates angio-tensin II-induced proliferation in spontaneously hypertensive rat vascular smooth muscle cells. Hypertens Res. 2010;33(12):1250–1257. doi: 10.1038/hr.2010.187. [DOI] [PubMed] [Google Scholar]
- 35.Roy J, Kazi M, Hedin U, Thyberg J. Phenotypic modulation of arterial smooth muscle cells is associated with prolonged activation of ERK1/2. Differentiation. 2001;67(1–2):50–58. doi: 10.1046/j.1432-0436.2001.067001050.x. [DOI] [PubMed] [Google Scholar]
- 36.Mulvany MJ, Baandrup U, Gundersen HJ. Evidence for hyperplasia in mesenteric resistance vessels of spontaneously hypertensive rats using a three-dimensional dissector. Circ Res. 1985;57(5):794–800. doi: 10.1161/01.res.57.5.794. [DOI] [PubMed] [Google Scholar]
- 37.Rizzoni D, Porteri E, Guefi D, et al. Cellular hypertrophy in subcutaneous small arteries of patients with renovascular hypertension. Hypertension. 2000;35(4):931–935. doi: 10.1161/01.hyp.35.4.931. [DOI] [PubMed] [Google Scholar]
- 38.Kida T, Chuma H, Murata T, et al. Chronic treatment with PDGF-BB and endothelin-1 synergistically induces vascular hyperplasia and loss of contractility in organ-cultured rat tail artery. Atherosclerosis. 2011;214(2):288–294. doi: 10.1016/j.atherosclerosis.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 39.Kim S, Murakami T, Izumi Y, et al. Extracellular signal-regulated kinase and c-Jun NH2-terminal kinase activities are continuously and differentially increased in aorta of hypertensive rats. Biochem Biophys Res Commun. 1997;236(1):199–204. doi: 10.1006/bbrc.1997.6926. [DOI] [PubMed] [Google Scholar]
- 40.Touyz RM, Deschepper C, Park JB, et al. Inhibition of mitogen-activated protein/extracellular signal-regulated kinase improves endothelial function and attenuates Ang II-induced contractility of mesenteric resistance arteries from spontaneously hypertensive rats. J Hypertens. 2002;20(6):1127–1134. doi: 10.1097/00004872-200206000-00024. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Lee YR, Lee CH, et al. Mitogen-activated protein kinase contributes to elevated basal tone in aortic smooth muscle from hypertensive rats. Eur J Pharmacol. 2005;514(2–3):209–215. doi: 10.1016/j.ejphar.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 42.Touyz RM, He G, El Mabrouk M, Diep Q, Mardigyan V, Schiffrin EL. Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. J Hypertens. 2001;19(3 Pt 2):553–559. doi: 10.1097/00004872-200103001-00006. [DOI] [PubMed] [Google Scholar]
- 43.Kubo T, Ibusuki T, Chiba S, Kambe T, Fukumori R. Altered mitogen-activated protein kinase activation in vascular smooth muscle cells from spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 2002;29(7):537–543. doi: 10.1046/j.1440-1681.2002.03694.x. [DOI] [PubMed] [Google Scholar]
- 44.Tabet F, Schiffrin EL, Callera GE, et al. Redox-sensitive signalling by angiotensin II involves oxidative inactivation and blunted phosphorylation of protein tyrosine phosphatase SHP-2 in vascular smooth muscle cells from SHR. Circ Res. 2008;103(2):149–158. doi: 10.1161/CIRCRESAHA.108.178608. [DOI] [PubMed] [Google Scholar]
- 45.Touyz RM, El Mabrouk M, He G, Wu XH, Schiffrin EL. Mitogen-activated protein extracellular signal-regulated kinase inhibition attenuates angiotensin II-mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ Res. 1999;84(5):505–515. doi: 10.1161/01.res.84.5.505. [DOI] [PubMed] [Google Scholar]
- 46.Jing L, Zhang J, Sun J, et al. Inhibition of extracellular signal-regulated kinases ameliorates hypertension-induced renal vascular remodeling in rat models. Int J Mol Sci. 2011;12(12):8333–8346. doi: 10.3390/ijms12128333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muthalif MM, Benter IF, Khandekar Z, et al. Contribution of Ras GTPase/MAP kinase and cytochrome P450 metabolites to deoxycor-ticosterone-salt-induced hypertension. Hypertension. 2000;35(1 Pt 2):457–463. doi: 10.1161/01.hyp.35.1.457. [DOI] [PubMed] [Google Scholar]
- 48.Muthalif MM, Karzoun NA, Gaber L, et al. Angiotensin II-induced hypertension: contribution of Ras GTPase/mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension. 2000;36(4):604–609. doi: 10.1161/01.hyp.36.4.604. [DOI] [PubMed] [Google Scholar]
- 49.Ren Y, D’Ambrosio MA, Liu R, Pagano PJ, Garvin JL, Carretero OA. Enhanced myogenic response in the afferent arteriole of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2010;298(6):H1769–H1775. doi: 10.1152/ajpheart.00537.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Budzyn K, Ravi RM, Miller AA, Sobey CG. Mechanisms of augmented vasoconstriction induced by 5-hydroxytryptamine in aortic rings from spontaneously hypertensive rats. Br J Pharmacol. 2008;155(2):210–216. doi: 10.1038/bjp.2008.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cruzado MC, Risler NR, Miatello RM, Yao G, Schiffrin EL, Touyz RM. Vascular smooth muscle cell NADPH oxidase activity during the development of hypertension: effect of angiotensin II and role of insulin-like growth factor-1 receptor transactivation. Am J Hypertens. 2005;18(1):81–87. doi: 10.1016/j.amjhyper.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 52.Zhang A, Jia Z, Wang N, Tidwell TJ, Yang T. Relative contribution of mitochondria and NADPH oxidase to deoxycorticosterone acetate-salt hypertension in mice. Kidney Int. 2011;80(1):51–60. doi: 10.1038/ki.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabet F, Schiffrin EL, Touyz RM. Mitogen-activated protein kinase activation by hydrogen peroxide is mediated through tyrosine kinase-dependent, protein kinase C-independent pathways in vascular smooth muscle cells: upregulation in spontaneously hypertensive rats. J Hypertens. 2005;23(11):2005–2012. doi: 10.1097/01.hjh.0000185715.60788.1b. [DOI] [PubMed] [Google Scholar]
- 54.Lee SL, Wang WW, Finlay GA, Fanburg BL. Serotonin stimulates mitogen-activated protein kinase activity through the formation of superoxide anion. Am J Physiol. 1999;277(2 Pt 1):L282–L291. doi: 10.1152/ajplung.1999.277.2.L282. [DOI] [PubMed] [Google Scholar]
- 55.Lee SL, Wang WW, Fanburg BL. Superoxide as an intermediate signal for serotonin-induced mitogenesis. Free Radic Biol Med. 1998;24(5):855–858. doi: 10.1016/s0891-5849(97)00359-6. [DOI] [PubMed] [Google Scholar]
- 56.Blanc A, Pandey NR, Srivastava AK. Distinct roles of Ca2+, calmodulin, and protein kinase C in H2O2-induced activation of ERK1/2, p38 MAPK, and protein kinase B signaling in vascular smooth muscle cells. Antioidx Redox Signal. 2004;6(2):353–366. doi: 10.1089/152308604322899422. [DOI] [PubMed] [Google Scholar]
- 57.Adachi T, Pimentel DR, Heibeck T, et al. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279(28):29857–29862. doi: 10.1074/jbc.M313320200. [DOI] [PubMed] [Google Scholar]
- 58.Lane KT, Beese LS. Thematic review series: lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J Lipid Res. 2006;47(4):681–699. doi: 10.1194/jlr.R600002-JLR200. [DOI] [PubMed] [Google Scholar]
- 59.Tristano AG, Castejon AM, Castro A, Cubeddu LX. Effects of statin treatment and withdrawal on angiotensin II-induced phosphorylation of p38 MAPK and ERK1/2 in cultured vascular smooth muscle cells. Biochem Biophys Res Commun. 2007;353(1):11–17. doi: 10.1016/j.bbrc.2006.11.044. [DOI] [PubMed] [Google Scholar]
- 60.Negre-Aminou P, van Vliet AK, van Erck M, van Thiel GC, van Leeuwen RE, Cohen LH. Inhibition of proliferation of human smooth muscle cells by various HMG-CoA reductase inhibitors; comparison with other human cell types. Biochim Biophys Acta. 1997;1345(3):259–268. doi: 10.1016/s0005-2760(96)00184-1. [DOI] [PubMed] [Google Scholar]