

Abstract
Complex mechanisms shape the genome of brain cells into transcriptional units, clusters of condensed chromatin, and many other features that distinguish between various cell types and developmental stages sharing the same genetic material. Only a few years ago, the field’s focus was almost entirely on a single mark, CpG methylation; the emerging complexity of neuronal and glial epigenomes now includes multiple types of DNA cytosine methylation, more than 100 residue-specific posttranslational histone modifications and histone variants, all of which superimposed by a dynamic and highly regulated three-dimensional organization of the chromosomal material inside the cell nucleus. Here, we provide an update on the most innovative approaches in neuroepigenetics and their potential contributions to approach cognitive functions and disorders unique to human. We propose that comprehensive, cell type-specific mappings of DNA and histone modifications, chromatin-associated RNAs, and chromosomal “loopings” and other determinants of three-dimensional genome organization will critically advance insight into the pathophysiology of the disease. For example, superimposing the epigenetic landscapes of neuronal and glial genomes onto genetic maps for complex disorders, ranging from Alzheimer’s disease to schizophrenia, could provide important clues about neurological function for some of the risk-associated noncoding sequences in the human genome.
1. INTRODUCTION
1.1. Chromatin and epigenetic regulation: General principles
The elementary unit of chromatin is the nucleosome, or 146 bp of genomic DNA wrapped around an octamer of core histones, connected by linker DNA and linker histones. As further described below, the collective set of DNA and histone modifications and variant histones provide the molecular substrates of the epigenome, here broadly defined as the epigenetic landscapes that define the functional architecture of the chromosomal material, including transcriptional and many other features of genome organization that are differentially regulated in different cell types and developmental stages of the organism.1,2
1.1.1 DNA (hydroxy)methylation
Two related but functionally very different types of DNA modifications, cytosine C5-methylation (5mC) and hydroxymethylation (5hmC) of cytosines in CpG dinucleotides, provide the bulk of the epigenetic modifications in vertebrate DNA.3 There are additional types of DNA modifications, including 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which are viewed as chemical breakdown products, from mC5 to hmC5 to 5fC to 5caC, and in addition may carry regulatory functions.4,5 While the majority of DNA (hydroxy)methylation is found at sites of CpG dinucleotides and, more generally, in the CpG-enriched sequences of the genome, a much larger fraction, or up to 25% of mC5, is found at non-CpG sites in the brain.6 The mC5 and hmC5 markings show a differential pattern of genomic occupancy, with the hmC5 mark concentrated toward the 5′ end of the genes and the proximal most portion of transcriptional units, broadly correlating with local gene expression levels,7–9 and a potential role in the regulation of intron/exon boundaries and splicing events of neuron-specific gene transcripts.10 On the other hand, less than 3% of methylcytosine (mC5) markings are positioned around the 5′ end of the genes.11
1.1.2 Histone modifications
There is evidence that far more than 100 amino acid residue-specific posttranslational modifications (PTMs) exist in the vertebrate cell,12 including monomethylation (me1), dimethylation (me2), and trimethylation (me3); acetylation and crotonylation; poly-ADP-ribosylation; and small protein (ubiquitin, SUMO) modification of specific lysine residues, as well as arginine (R) methylation and citrullination, serine (S) phosphorylation, tyrosine (T) hydroxylation, and several others.12–14 These site- and residue-specific PTMs often define chromatin structure and function, with an epigenetic histone code (a combinatorial set of histone PTMs) differentiating between promoters, gene bodies, enhancer and other regulatory sequences, and condensed heterochromatin.15 It is important to emphasize that histone PTMs rarely occur in isolation and instead multiple histone PTMs appear to be coregulated and, as a group, define the aforementioned chromatin states.16 Many active promoters, for example, are defined by high levels of histone H3 lysine 4 trimethylation in combination with various histone lysine acetylation markings, while repressive histone PTMs, including the trimethylated forms of H3K9, H3K27, and H4K20, potentially colocalize to some of the same loci in the genome and so forth.15 Proteins associated with the regulation of histone PTMs are sometimes referred to as “writers” or “erasers” or “readers,” essentially differentiating the process of establishing or removing a mark as opposed to its docking functions for chromatin remodeling complexes that regulate transcription or induce and maintain chromatin condensation.14,17,18
1.1.3 Histone variants, chromatin remodeling, and nucleosome positioning
In addition to the core histones H2A/H2B/H3/H4, a number of histone variants, with H3.3, H3.1, H3.2, H2A.Z, and H2A.X, are some of the best-studied examples. Variant histones, which differ from the canonical histone at few amino acid positions, could affect nucleosome stability and compaction.19
Chromatin remodeling complexes are composed of multiple subunits that, according to their classical definition, regulate sliding and mobility of nucleosomes, powered by ATP hydrolysis, thereby regulating gene expression and RNA polymerase II access at transcription start sites.20 Examples of well-known chromatin remodelers with a critical role in brain development include the BAF (SWI/SNF) complex and CHD family of proteins.20 Mutations in numerous members of the BAF complex and multiple CHD proteins have now been linked to psychiatric disease and developmental brain disorders, with additional investigations in mutant mice.20–22
1.1.4 Chromatin-associated RNAs
While the process of gene expression is obviously defined by nascent RNA emerging from genomic DNA packaged into chromatin, chromatin-associated RNAs (caRNAs) (or cbRNA for chromatin-bound RNA) could be reserved to RNA species as part of a chromatin structure, thereby regulating its functions. According to some estimates, up to 2–3% of the nucleic acid content in chromatin is contributed by polyadenylated RNAs.23 One of the best-known examples of a caRNA is provided by the X-chromosome inactive transcript that governs the silencing of the second X in female diploid cells.24,25 Some caRNAs, including a complex long noncoding RNA, composed of 148 exons and introns, at a neurodevelopmental risk locus on chromosome 15q11–13,26 produce “RNA clouds” in cis, thereby triggering lasting decondensation of the surrounding chromosomal material.27,28 In addition, some of the caRNAs expressed in the human brain, including a noncoding RNA at the DPP10 (chromosome 2q14.1) locus, show human-specific epigenetic regulation and could contribute to cognitive features and disease vulnerabilities not shared with other primate species.29
1.1.5 Higher order chromatin
DNA methylation, epigenetic decoration of nucleosomal (including variant) histones, and the various caRNAs/cbRNAs described to date still would fall short to adequately describe the epigenome and localized chromatin architectures at any given genomic locus. This is because the chromosomal arrangements in the interphase nucleus are not random. Specifically, loci at sites of active gene expression are more likely to be clustered together and situated toward a central position within the nucleus, while heterochromatin and silenced loci move more toward the nuclear periphery.30,31 Chromosomal loopings, in particular, are among the most highly regulated supranucleosomal structures and are associated with transcriptional regulation, by, for example, positioning distal regulatory enhancer or silencer elements that—in the linear genome—are positioned potentially many hundred kilobases apart from a gene, to interact directly with that specific promoter.32,33 Proper regulation of such types of higher order chromatin is certainly of critical importance for orderly brain development and function. For example, Cornelia de Lange syndrome (CdLS) with an estimated incidence of 1:10–30,000 live births among the more frequent genetic disorders (source: http://ghr.nlm.nih.gov) is associated with severe developmental delay and a range of neuropsychiatric symptoms.34 CdLS (including Online Mendelian Inheritance in Man (OMIM) 122470 and 300590) involves causative mutations in the cohesin complex, a multisubunit protein that includes, among others, nipped-B-like protein (NIPBL), structural maintenance of chromosomes 1A and 3 (SMC1A and SMC3) proteins, and histone deacetylase 8 (HDAC8).35,36 Cohesin is thought to form ring-like structures bringing together DNA segments from different locations, and by interaction with transcriptional coactivators, the complex could promote the physical interaction of promoters with enhancers separated by thousands of base pairs on the linear genome, thereby regulating cell type-specific gene expression programs.37
2. CHALLENGES FOR EPIGENETIC APPROACHES IN THE (HUMAN) BRAIN
2.1. Cellular specificity of epigenetic markings
Conventional chromatin assays designed to detect and quantify DNA methylation and histone modifications require an input material between 103 and 108 nuclei.38,39 Such types of assay typically lack cellular resolution, which poses a challenge given that brain tissue is composed of an extremely heterogeneous mixture of different cell types, including glia-to-neuron ratios that could show considerable fluctuations across normal development or in certain disease states, such as conditions associated with neurodegeneration or neuroinflammation. To date, many studies exploring epigenetic dysregulation of gene expression in major psychiatric disease examined DNA methylation and histone modifications in tissue homogenates, while typically, the gene(s) of interest often is expressed only in a select subpopulation of neurons or other cells.40–48 On the other hand, there is evidence that each cell type is defined by differential regulation of DNA methylation and histone modifications at hundreds or thousands of promoter and enhancer sequences, resulting in considerable “epigenetic distance” even between cortical neurons and their surrounding glia and other nonneuronal cells.49–51 Of note, statistical methodology has been developed to decompose neuronal and nonneuronal signals in DNA methylation assays on tissue homogenate.52,53 These in silico approaches could be extremely valuable not only for already existing datasets but also for future studies in which sorting is not feasible, including in projects in which the cell type-specific nuclear epitope is not reliably expressed in every postmortem specimen. In these cases, cell type-specific epigenomic maps could be constructed in a smaller set of brains, while the larger cohort undergoes deconvolution based on epigenomic maps in tissue homogenate.
Given the inherent problem of cellular heterogeneity in the brain, many groups have turned to fluorescence-activated cell sorting (FACS) for the isolation of more homogenous cell populations, including neurons, microglia, oligodendrocytes, astrocytes, and neural/glial progenitors at various stages of their lineage differentiation. Several useful markers exist to define these cell types in situ, but only few of them have shown the ability to label live cells for isolation (Table 8.1). A major obstacle to using most phenotypic markers for FACS is the necessity to target an extracellular cell-surface domain, particularly if downstream purification is needed.58,62 Several surface markers have been used to isolate oligodendrocyte progenitor cells (OPCs) A2B5,57 NG2,56 and CD140a-PDGFRα55; neural stem cells (NSCs) CD13360; microglia CD11b61; and most recently astrocyte-lineage cells from mice GLAST58 and from expanded human NSCs CD4459 (Table 8.1). While some of the abovementioned markers have sorted populations that are still quite heterogeneous,56,57,60 others have shown significant enrichment for specific cell types.55,58,61 The use of cell-surface antibodies to isolate cells is not ideal: cytoplasmic processes of more mature glia are often partially destroyed during enzymatic digestion and the rapidly flowing stream of FACS, leading to significant decrease in final cell yield. Cell sorting with the surface markers CD44 and GLAST (ASCA-1 antibody) has allowed the isolation of adult glia from postmortem subventricular zone tissue, providing sufficient yield of high-quality DNA for downstream genomic analysis (1000–100,000 cell events depending on cell population, brain age, and postmortem time) but insufficient for chromatin immunoprecipitation analysis (Tsankova lab, unpublished data). Furthermore, the use of cell-surface antibodies for FACS necessitates fresh tissue, due to the inevitable cell lysis of many cells during freezing and thawing. This adds another constraint: fresh autopsy tissue with short postmortem time or surgical brain tissue is not readily available and easily accessible outside of large neurosurgical centers or close collaboration with neuropathologists and neurosurgeons.
Table 8.1.
Common markers defining cell-type identity/lineage specificity of brain cells
| Marker | Cell type/lineage | Cellular localization | FACS fresh human tissue | Technique | Cell specificity |
|---|---|---|---|---|---|
| NeuN | Neuron | Nucleus | Y: Nuclei | Nuclei extraction>NeuN+FACS | Mature neurons |
| PDGFRα | Oligodendrocyte- lineage | Extracellular cell surface | Y: Whole cell | Papain dissociation>CD140a+FACS | OPCs |
| NG2 | Oligodendrocyte- lineage+ | Cell surface | Y: Whole cell (hESCs) | hESCs>NG2+FACS | OPCs, pericytes, endothelium |
| A2B5 | Oligodendrocyte-lineage+ | Cell surface | Y: Whole cell | Papain dissociation>A2B5+MACS | OPCs, astrocyte-lineage cells, immature neurons |
| Olig2 | Oligodendrocyte-lineage+ | Nucleus | N | OPCs to mature oligodendrocytes, few multipotent glial progenitors | |
| GLAST | Astrocyte-lineage | Extracellular cell surface | Y: Whole cell (mice) | Trypsin dissociation>ACSA-1+MACS | Astrocyte-lineage cells |
| CD44 | Astrocyte-lineage+ | Extracellular cell surface | Y: Whole cell (hNSC) | Expanded hNSC>CD44+CD184+FACS | Fibrous astrocytes (SVZ, WM), glial progenitors, inflammatory cells |
| GFAP | Astrocyte-lineage | Cytoplasm | N | Astrocytes, some neural stem cells/ radial glia | |
| ALDH1L1 | Astrocyte-lineage | Cytoplasm | N | Astrocyte-lineage cells | |
| CD133 | Neural stem cell+ | Extracellular cell surface | Y: Whole cell | Enzymatic dissociation>CD133+CD34−CD45−FACS | Neural stem cells, ependyma, endothelium |
| Sox2 | Neural stem cell | Nucleus | N | Embryonic and adult neural stem cells | |
| Nestin | Neural stem cell+ | Cytoplasm | N | Neural stem cells, ependyma | |
| CD11b | Microglia | Extracellular cell surface | Y: Whole cell | Enzymatic dissociation>CD11b+CD45dim MACS | Microglia, other monocytes |
Some of these markers have allowed isolation of cell populations using fresh human brain tissue by FACS, such as neurons (via NeuN54), oligodendrocyte-lineage cells (via PDGFRα,55 NG2,56 and A2B557), astrocyte lineage cells (via GLAST,58 in mice and CD4459 in expanded human NSCs), neural stem cells (via CD13360), and microglia (via CD11b61). +, more than one cell population isolated by FACS; MACS, magnetic-activated cell sorting; SVZ, subventricular zone; WM, white matter.
The limitation of using surface markers has caused many to turn to transgenic strategies for FACS in rodents, allowing the use of cytoplasmic or nuclear markers with good cell-type specificity, coexpressed with a fluorescent reporter; one example is the use of GFP–GFAP reporter mice for isolation of astrocytes.63 This approach, unfortunately, is not applicable for human tissue. An alternative, successful strategy to collect nuclei from human tissue has been the isolation of neuronal nuclei labeled with the specific antibody NeuN.64 This overcomes the caveats of heterogeneity, fragility of cytoplasmic processes, and availability of fresh tissue and offers satisfactory yield for downstream epigenetic studies. Protocols are available for efficient purification and immunotagging of nuclei from frozen postmortem brain specimens, which then could be processed by fluorescence-activated sorting. Thus, the separate collection of 107–108 neuronal and nonneuronal nuclei from a few hundred milligram of postmortem cerebral cortex in a single day is possible, thereby enabling separate processing of neuronal and nonneuronal chromatin.54,65,66 A similar neuronal marker for astrocytes has not yet been described, but the oligodendrocyte-lineage nuclear marker Olig2 could offer a comparable nuclear isolation solution for immature and maturing oligodendrocytes.66 Another exciting avenue to explore is the use of molecular beacons as mRNA-specific targets for FACS in human brain tissue, which has already shown feasibility in rodents.62
2.2. With focus on the candidate gene approach, only few high-resolution epigenomic mappings
With the exception of neurosurgical cases, the bulk of human brain studies, including those focused on neuropsychiatric disorders, rely on postmortem tissue. There is general consensus that nucleosomal arrays, histone modifications, and the activity of histone-modifying enzymes such as methyl- and acetyltransferases, DNA methylation markings, and even some of the chromosomal loop formations are preserved, at least partially, in postmortem brain tissue that typically is exposed to 5–30 or more hours of autolysis time before being stored in a −70 °C freezer.38,64,67–69 Thus, 20 years ago, epigenetic exploration of the diseased human brain started out with restriction enzyme-based DNA cytosine methylation mapping at a predetermined set of CpG dinucleotides surrounding the 5′ end of amyloid-beta precursor (APP)70 and FMR1 genes in single cases diagnosed with Alzheimer’s disease and fragile X mental retardation syndrome,71–73 followed 10 years later by histone methylation mappings at the site of NMDA glutamate receptor genes in developing cerebral and cerebellar cortices.74 In the case of fragile X, the expansion of CGG codon from (normally) 5 to 40 repeats to 50 to over 200 repeats triggers excessive DNA methylation at the promoter, effectively shutting down gene expression by silencing the surrounding chromatin.75 These highly reproducible molecular phenotypes in a monogenetic neurodevelopmental disorder then provided a road map for similar studies on disorders of a more heterogeneous and complex etiology, including Alzheimer’s dementia, depression, schizophrenia, and others.40–42,44,48,70 However, small cohort sizes, typically involving far less than 50–100 brains, in conjunction with the overall only very subtle differences between cases and controls45,46,48 and the considerable degree of sample heterogeneity in terms of etiology of disease, have thus far precluded a general consensus on which, if any, genomic locus shows a reproducible epigenetic defect in any of the aforementioned neuropsychiatric disorders.
In a very different line of research, large, multi-institutional consortia, such as the Encyclopedia of DNA Elements (ENCODE), have harnessed powerful, next-generation sequencing-based technologies76,77 to generate comprehensive genome-scale maps, often at (near) base-pair resolution, for DNA methylation and histone modification landscapes, chromatin accessibility as measured by DNase I hypersensitivity, and transcription factor binding profiles in a variety of peripheral cell lines and tissues.78,79 In contrast, only very few epigenetic markings, including DNA methylation and hydroxymethylation,80 and a small number of histone methylation and acetylation markings that differentiate between active and inactive/repressed promoter and enhancer sequences81 have been charted with next-generation sequencing technology in the human brain and with a narrow focus mostly on epigenetic changes that occur during normal development of the cerebral cortex. Furthermore, with few exceptions,82 such types of studies were based on very small and even single-digit cohorts.81 In the nearby future, government-sponsored program such as PsychENCODE (www.grants.gov) and/or private- or industry-sponsored efforts should catalyze the generation of a much larger brain epigenomic dataset than the one currently available, with the expectation of a deeper understanding about the epigenetic mechanisms of normal development and aging and changes in chronic neuropsychiatric disease.
2.3. Higher order chromatin studies in the human brain
Despite the growing realization of the importance of chromosomal looping and other higher order chromatin structures for transcriptional regulation (discussed above), very little is known about their role in the nervous system, including only a handful of studies with human brain tissue.29,83,84 This is surprising given that chromosome conformation capture (commonly referred to as 3C85,86) as the standard approach to map chromosomal loopings, is applicable to brain tissue collected postmortem, at least for some the genomic loci that were examined so far.83 The 3C technique explores physical interactions between DNA fragments separated by Kb or Mb of interspersed sequence; cross-linked chromatin is digested with a specific restriction enzyme, religated and amplified using primer pairs for which forward and reverse primers match to different portions of the genomic locus of interest.83
One interesting example of 3C applied on human brain involves GAD1, encoding the 67Kda glutamic acid decarboxylase GABA synthesis enzyme.84 A loop, initially detected in the prefrontal cortex and then verified in neuronal cultures derived from pluripotent skin cells, connected sequences surrounding the GAD1 transcription start site with a region 50 kb further upstream. This loop showed a significant weakening in the prefrontal cortex of some subjects with schizophrenia that were affected by decreased GAD1 expression. There was evidence that the loop was conserved between the rodent and primate brain, and indeed, based on 3C studies in reporter mice, this loop appeared to be much stronger in cortical GABAergic interneurons (which express Gad1) compared with other cortical cells (which do not express Gad1). Such type of study could be viewed as “proof of principle” that higher order chromatin is (i) amenable to analyses in the human brain, (ii) potentially altered in common brain disorders, and (iii) potentially amenable to translational approaches and follow-up work in preclinical model systems, including human cell culture and animals.
Similar to DNA methylation and histone modification studies discussed above, there is a glaring vacuum of genome-scale and agnostic (not candidate gene-based) higher order chromatin studies in the human brain. This is not due to the lack of basic techniques. At the time this review was written, massively parallel sequencing has enabled the production of chromosome conformation libraries to detect interactions (i) between specific loci genome-wide using circular chromosome conformation capture (4C), (ii) between a large set of loci using carbon-copy chromosome conformation capture (5C), (iii) between all regions in the genome using HiC, and (iv) between all regions in the genome bound by a specific protein or histone mark using chromatin immunoprecipitation for using paired-end sequencing (ChIA-PET).87–90 The datasets that are grounded in these techniques have provided unique insights into the organizational complexities and nonrandomness of spatial genome architectures, with multiple types of looping interaction groups between highly transcribed genes, gene-rich active regions, nonactive centromere proximal clusters, coregulated genes, gene ontology groups, and so on.91–93 We predict that for the human brain too, a similar or perhaps even more complex organization will emerge, once neuroscientists and neuropsychiatrists embark on these types of studies.
HiC is the most high-throughput method, investigating interactions across the entire genome with a resolution of 5–10 kb using the best current algorithms starting with 300 million mappable reads or four lanes of Illumina sequencing.94,95 This technique, though powerful, is not always the most amendable method to detect higher order chromatin organization due to the depth of sequencing required. 4C, however, can be used to determine genome-wide interaction contacts for specific loci with a 1–8 kb resolution using only 500,000 mappable reads, allowing for the multiplexing of several experiments in one lane of Illumina sequencing.96,97 Both HiC and 4C are limited, however, due to the sheer number of interactions that exist at neighboring loci, which tend to result in a large majority of interaction read counts. 5C can overcome neighboring interaction artifacts from high-throughput sequencing by only designing neighboring primers on opposite strands and primers on the same strand apart by 15–30 kb. Thus, the determination of which “C” to use for individual experiments depends on the hypothesis in question.
3. EVIDENCE AND DEBATE
3.1. Epigenetic markings in the brain: State or trait?
The rationale of exploring certain types of epigenetic modifications in postmortem brain of subjects diagnosed with psychiatric disease is, as mentioned above, often based on the hypotheses that changes in RNA expression are associated with altered epigenetic decoration at the site of the corresponding gene promoter and related regulatory sequences. Quite often, the accompanying abnormalities in DNA methylation and histone modifications are then interpreted in terms of a stable and long-lasting epigenetic “lesion” in response to an environmental insult or some other pathogenic effect operating in early life, many years before the brain was obtained at autopsy. For example, different grades of maternal care in the early postnatal period lead to differential regulation of promoter-associated DNA methylation and histone acetylation at the aforementioned disease gene, Gad1, in the hippocampus of adult rats,98 and likewise, deficits in open chromatin-associated histone methylation at GAD1 in the prefrontal cortex of adult schizophrenics was discussed in the context of defective neurodevelopment in conjunction with a risk haplotype at the promoter.43 Furthermore, based on postmortem studies in adult suicide victims, there is evidence that the suffering of abuse in early childhood years leaves a lasting DNA methylation imprint on the stress-regulated glucocorticoid receptor NR3C1 promoter99 in the hippocampus and on DNA repeats encoding ribosomal RNAs.100 In addition, normal aging may be associated with widespread age-related changes in gene expression in the cerebral cortex, including downregulation of many neuronal genes.48,101–103
However, it is fair to admit that little is known about the stability and dynamic turnover of epigenetic markings in human brain, and therefore, it remains unclear whether any of the aforementioned epigenetic alterations in the brain of adult psychiatric subjects indeed reflect a (mal)adaptive “trait” stably maintained for years or, alternatively, whether disease-associated chromatin changes merely reflect the brain’s functional state at the time of death. The “trait” hypothesis appears very plausible in the context of monogenetic disorders associated with aberrant and excessive repressive DNA and histone methylation in cis (at the site of the mutation). Examples include the aforementioned CGG triplet expansion at the FMR1 (fragile X) gene promoter75 or the GAA triplet repeat expansion in the first intron of the FRATAXIN gene associated with Friedreich’s ataxia, an autosomal recessive neurodegenerative condition.104 In these cases, the epigenetic dysregulation is firmly linked to the pathophysiology of disease (resulting from silenced gene expression), and there can be little doubt that the observed changes in (postmortem) brain chromatin, like the impairments in neurological functions, most likely existed across the entire life span.73,104 Furthermore, there are many other examples strongly suggesting that the DNA sequence variation is a major driver for epigenetic differences between subjects. In addition to the abovementioned example gene, GAD1, many other single-nucleotide polymorphisms (SNPs) across the entire genome, including those that have been genetically implicated in the risk of major psychiatric disease (including bipolar disorder and schizophrenia), exhibit a robust effect on methylcytosine levels at the site of nearby genes.103,105 However, studies in mono- and dizygotic twins and related work in animals convincingly demonstrated that molecular mechanisms of heritability are unlikely due to DNA sequence differences alone.106
On the other hand, it will be difficult to confirm whether many of the reported epigenetic alterations observed in small cohorts of “sporadic” cases with schizophrenia, autism, depression, and other psychiatric diseases represent a type of molecular alteration stably related to the underlying disease. Given that most or perhaps all epigenetic markings studied to date are subject to bidirectional regulation in the cell culture system and animal model, it is reasonable to assume that the epigenetic decoration of human brain genomes is subject to similar types of dynamic regulation.
For example, DNA methylation at specific promoter sequences is subject to rapid up- or downregulation on the scale of minutes to hours.107,108 Hippocampal DNA methylation signatures are highly sensitive to acute depolarization109,110 and electroconvulsive seizures, affecting regulatory sequences regulating NMDA and GABA-A receptor genes, Notch signaling pathways, and other systems with a key regulatory role for synaptic signal and plasticity.111
Furthermore, changes in neuronal activity result in robust changes in expression and activity of multiple DNA methylation-associated proteins with an essential role for neuronal health and function, including the methyl-CpG-binding protein MeCP2 or Gadd45b that recruits cytidine deaminases and thymidine glycosylases at genomic sites subject to active DNA methylation.112–114 Furthermore, physiological activation of hippocampal circuitry during learning and memory is sufficient to elicit highly dynamic DNA methylation changes at PP1, REELIN, and other gene promoters regulating synaptic plasticity.115,116 The complex molecular machineries mediating demethylation of CpG dinucleotides or histone lysine residues are becoming increasingly understood, which is a remarkable progress given that these and other types of epigenetic modifications were not long ago considered to be potentially irreversible.117–120 These findings, taken together, would suggest that some of the epigenetic alterations reported in diseased postmortem brain are not necessarily stable for very long periods of time and, instead, are regulated by mechanisms that operate on a much shorter timescale, perhaps lasting only a few weeks or days or even less. Clinically relevant conditions that reportedly affect chromatin structure and function in the brain include ischemia,121 exposure to environmental toxins,122–124 abuse of nicotine,125,126 alcohol,127 psychostimulants,128–130 and antipsychotic and mood-stabilizing drugs.45,131–136
3.2. Mapping brain epigenomes from the culture dish?
While a significant portion of the epigenomic organization in brain nuclei is preserved in postmortem specimens stored at brain banks (with autolysis time typically in the range of 5–35 h), animal models suggest that some histone modification types and chromosomal loopings and other higher order chromatin structures show significant signal decay and other secondary effects already after 12–15 h of tissue autolysis and decay.38,68 Because access to fresh, neurosurgically obtained brain tissue is not an option for the vast majority of subjects diagnosed with neuropsychiatric disease, it is not surprising that many investigators in the field are harnessing pluripotent stem cells (iPS) from the skin or other cell types as the starting material to model brain tissue in the culture dish.
Once the generation of pluripotent stem cells by reprogramming somatic cells via retroviral transduction of four transcription factors (i.e., Oct4, Sox2, Klf4, and c-Myc)137 had been accomplished, the technique has been further advanced138 and applied by multiple groups to generate iPS-derived neuronal and glial cultures or even a three-dimensional organoid mimicking the cerebral cortex in a dish.139 These include neurodevelopmental syndromes140,141 and schizophrenia.142 Such type of studies undoubtedly will pave the way for broader future iPS-based approaches that most certainly will become a mainstay in the field of biological psychiatry. Cellular reprogramming with subsequent neural differentiation, including neural circuitry and active synapse formation in the dish, is likely to mimic many key steps of neurodevelopment and opens up the possibility of conducting electrophysiological recordings and other functional assays on nervous tissues of living subjects.142 Therefore, iPS technology provides an unprecedented opportunity to study the molecular and cellular biology of the nervous system from any patient (or at least from those who are able to give consent). A subset of psychiatric susceptibility genes may even, as in case of the MYLT1 transcription factor, which when mutated confers high risk for neurodevelopmental disability, promote the process of neuronal differentiation from stem cell preparations ex vivo.143,144
These recent advances in reprogramming technologies have also fueled general interest in the field to explore epigenetic regulation of the nervous system in patient- and control-derived iPS. For example, several studies explored chromatin structures and synaptic signaling in neuronal cultures of Rett syndrome (RTT) patients with MECP2 mutations and controls.141,145 The MECP2 gene product, methyl-CpG-binding protein 2, is highly expressed in the nervous system and occupies widespread territories of neuronal chromatin, dependent on the local density of methyl-CpG-dinucleotides.146 Loss-of-function mutations and other MECP2 structural variants have been linked to RTT, a disorder of early childhood associated with developmental and cognitive regression and a broad range of neurological symptoms.147,148 Furthermore, work on reprogrammed skin cells of Rett patients, which then was further confirmed in the Mecp2 mutant mouse brain, uncovered a genomic instability phenotype defined by disinhibition and increased mobility of retrotransposon and other parasitic DNA element activities due to altered DNA and histone methylation, in conjunction with changes in the global chromatin state.145,146 This work illustrates the promising potential of neural cultures, derived from skin fibroblasts, to study epigenetic (dys)regulation in specific disease cases and to gain knowledge about the molecular underpinnings of neurological disorders. Furthermore, the short-chain fatty acid derivative and anticonvulsant and mood-stabilizing drug valproic acid (VPA) induces pluripotency from skin fibroblasts when coadministered with Oct-4 and Sox-2 transcription factors.149 Furthermore, VPA promotes neuronal differentiation from progenitor stages150,151 and the drug induces, in cell culture and the brain, the upregulation of open chromatin-associated histone acetylation and methylation markings at promoters of genes with a key role in neurotransmission.43,152,153 It will be interesting to quantify these VPA-dependent effects on pluripotency and neuronal differentiation and to compare VPA treatment responders to non-responders. More broadly, pharmacoepigenomics, or a drug’s direct and indirect effects on chromatin structure and function, may perhaps in the future emerge as an interesting biomarker to predict treatment response and side effects or illuminate novel, hitherto unsuspected mechanisms of drug action.
However, despite all these unprecedented perspectives of generating nervous tissue in the dish from skin cells, it is important to point out that the technique still faces hurdles and challenges, particularly in the context of epigenetic regulation. This is because the eraser and subsequent redecoration of epigenetic markings across the genome—a key mechanism for successful reprogramming—may be incomplete at some loci, resulting in carryover effects so that the reprogrammed cells (iPS) maintain DNA methylation signatures that define the original donor cell type (e.g., fibroblasts).154,155 Furthermore, while some of the chromosomal loopings and higher order chromatin structures that regulate neuronal gene expression in the human brain are tractable in stem cell-derived neuronal cultures, others are not.83,84
Given the considerable variability of epigenetic and cellular phenotypes after reprogramming,156,157 it will be difficult to faithfully “rebuild” in the culture dish the cortical neuronal networks with their constituents such as pyramidal neurons and their surrounding inhibitory cells. This is a challenge for any disease-related study, including many psychiatric disorders that are likely to show only subtle differences in cellular response patterns, as compared with controls. While purely speculative at this point in time, imagine the potential benefits of the iPS technology for pharmaco(epi)genomics and treatment response paradigm. For example, iPS-derived cells could be “challenged” with a compound and the epigenome signature measured to distinguish between treatment-resistant and treatment-responsive patients. In this context, one interesting biomarker appears to relate to histone methylation levels and acetylation levels in the peripheral blood cells of subjects exposed to the histone deacetylase inhibitor, valproate.158
3.3. Functional neuroepigenomics to inform disease-associated variants
Over the last decade, multiple genome-wide association (GWA) studies have produced strongly significant evidence that specific common DNA genetic variants among people influence their genetic susceptibility to a number of complex neuropsychiatric illnesses, including schizophrenia159 and bipolar disorder.160 The majority of common variant loci associated with genetic risk for these complex diseases reside within noncoding sequence of unknown function, and many are far from discovered genes. To mention just one example, consider the major histocompatibility complex (MHC) locus that has long been implicated in psychiatric disease, including three large genome-wide association studies (GWAS) published jointly in 2009.161–163 These studies identified up to 45 disease-associated SNPs in the 26–33 megabase region of the MHC locus on chromosome 6, but strikingly, fifty percent of these SNPs were not located near genes. Some of the SNPs with the statistically strongest disease association were approximately 30 kb away from the nearest gene.
Furthermore, the disease-associated genomic regions are frequently large and often contained multiple implicated genetic variants due to local linkage disequilibrium patterns. In order to be able to understand these associations mechanistically, it is critical to develop strategies for honing in on regions and genetic variants more likely to have functional effects. Thus, the elucidation of the function of noncoding disease-associated loci through neuroepigenomics is an important next step toward the development of testable hypotheses regarding biological processes involved in the pathogenesis of neuropsychiatric disorders.
Several lines of evidence suggest the involvement of a proportion of genome-wide associated variants in transcriptional regulatory mechanisms, including enrichment within expression quantitative trait loci (eQTL)164–167 and modulation of cis-regulatory elements (CREs).165,168–170 A CRE, such as a promoter, enhancer, or silencer, is a noncoding DNA sequence in, near, or distal to a gene that contains binding sites for regulatory factors and is required for proper spatiotemporal expression of the gene. The proposed mechanism is that disease-associated variants that lie within CREs affect the binding of regulatory proteins, such as transcription factors, leading to allele-specific differences in transcription and subsequent disease-related alterations in molecular pathways. The importance of neuroepigenomic annotations for informing the genetic influence on transcriptional regulation is supported by recent studies in human lymphoblastoid cell lines, where more than 50% of eQTLs were found to lie within CREs.166
Integration of neuroepigenomic annotations identifies a reduced set of functional SNPs to be tested for association with the disease in the GWAS datasets (Figs. 8.1 and 8.2). While still a young field with methods still under development, this approach bears significant promise because it would decrease the number of association tests from a couple of million variants (using imputed genotypes) to a couple of thousands. This approach leads to enhanced power by eliminating the excessive multiple testing corrections, (ii) identifying the true causal SNP out of nearby tag SNPs that are in linkage disequilibrium, and (iii) providing a plausible mechanistic and testable explanation for the effect of SNPs that can affect related functions, such as changes in gene expression, through allele-specific alterations in transcription factor binding site or alterations in the three-dimensional genome architecture associated with chromosomal loopings and transcriptional regulation in the brain.
Figure 8.1.

The epigenome, from nucleus to nucleosome. Schematic illustration of (green) gene poised for transcription by polymerase II (Pol II) initiation complex, with nucleosome-free interval at transcription start site (TSS); (blue) distal enhancer sequence that in loop-like structure moves in close proximity to active gene; and (red) marks a small subset of heterochromatic portions of the genome, including silenced gene and heterochromatic structures bordering the nuclear envelope and pore complex and also the nucleolar periphery. A small subset of representative histone variants and H3 site-specific lysine (K) residues at N-terminal tail (K4, K9, K27, and K36) or core fold domain of the (histone) octamer (K79) and the H4K20 residue are shown as indicated, together with panel of mono- and trimethyl or acetyl modifications that differentiate between active promoters, transcribed gene bodies, and repressive chromatin, as indicated. DNA cytosines that are hydroxymethylated at the C5 position are mostly found at active promoters, while methylated cytosines are positioned within the body of actively transcribed genes and around repressed promoters and in constitutive heterochromatin.
Figure 8.2.
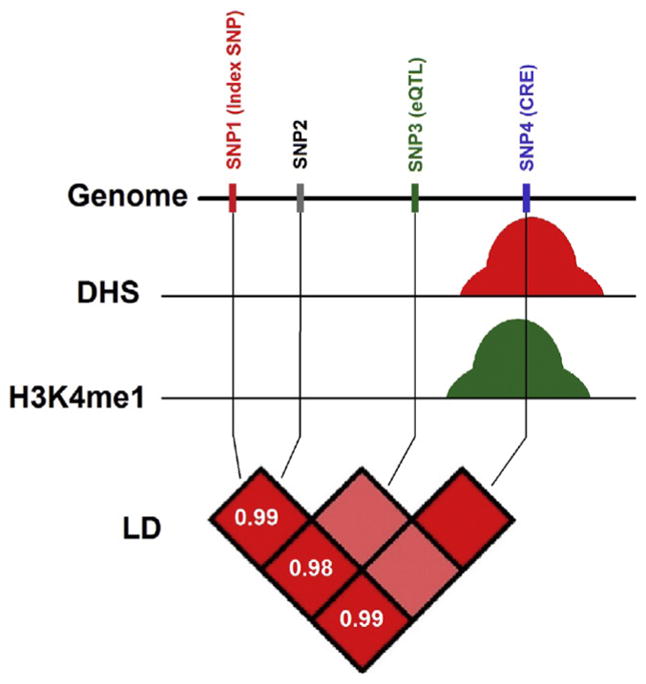
Neuroepigenomic annotations could prioritize variants with additional functional support. Example of four SNPs that are in strong LD (r2 ≈ 1). SNP1 is the associated variant with a phenotype in a GWAS. SNP2 is an eQTL associated with changes in gene expression in a different study. SNP4 overlaps a DNase I hypersensitivity peak (a measure for open chromatin and accessibility of the DNA by regulatory proteins)171,172 and a ChIP-seq peak for predicted enhancers (H3K4me1).173,174 Therefore, multiple sources of evidence support that SNP4 is in a regulatory region that regulates gene expression of specific transcript and is associated with the phenotype of interest.
4. SYNOPSIS AND OUTLOOK
Over the course of only a few years, we have witnessed a proliferation of epigenetic studies in the human brain, ranging from exploration of chromatin structures at a specific genomic locus to genome-wide epigenome mapping in defined cell types, generally with signal-to-noise ratios and signal quality comparable with those obtained in animal brains. Work from multiple groups, focusing mainly on human association cortex, points to large-scale remodeling of DNA and histone methylation landscapes during the late prenatal phase and early postnatal phase and early childhood, with comparatively less dramatic changes during subsequent stages of development and aging.51,80,103 Still, hundreds of promoters are subject to epigenetic changes that seemingly continue into old age, and these data, taken together, leave little doubt that chromatin structures undergo remodeling throughout the life span of the human brain,48,103,175 including neurons and other terminally differentiated cells.51 Based on postmortem brain work, epigenetic risk architectures are beginning to emerge for a number of common psychiatric conditions and disorders, including autism,176 schizophrenia,177 depression and bipolar disorder,105,178 and alcoholism.179 We predict that only very few, if any, loci will show replicated group-based differences when assayed in genome-wide epigenetic screens. Instead, we argue that epigenetic exploration of brain cells and tissue is ideally done in large cohorts, ideally hundreds, of phenotypically well-characterized subjects, in conjunction with next-generation sequencing-based epigenome and transcriptome profiling and high-coverage whole-genome sequencing of the same cases and tissues. Such type of datasets, when combined with the rapidly refining genetic risk maps for common neuropsychiatric diseases,159,180,181 will provide a powerful tool to gain deep and unprecedented insights into the genomic foundations of cognition and emotion, including disorders unique to human.
Acknowledgments
Work conducted in the authors’ laboratories is sponsored by the National Institutes of Health, the Veterans Administration, Autism Speaks, and the Brain & Behavior Research Foundation.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17(3):330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 2.Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21(2):175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song CX, He C. Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci. 2013;38(10):480–484. doi: 10.1016/j.tibs.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song CX, Yi C, He C. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol. 2012;30(11):1107–1116. doi: 10.1038/nbt.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie W, Barr CL, Kim A, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148(4):816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39(12):5015–5024. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29(1):68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khare T, Pai S, Koncevicius K, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19(10):1037–1043. doi: 10.1038/nsmb.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466(7303):253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan M, Luo H, Lee S, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 14.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14(11):1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 16.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 17.Mosammaparast N, Shi Y. Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem. 2010;79:155–179. doi: 10.1146/annurev.biochem.78.070907.103946. [DOI] [PubMed] [Google Scholar]
- 18.Justin N, De Marco V, Aasland R, Gamblin SJ. Reading, writing and editing methylated lysines on histone tails: new insights from recent structural studies. Curr Opin Struct Biol. 2010;20(6):730–738. doi: 10.1016/j.sbi.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 19.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A. Z. Genes Dev. 2007;21(12):1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14(5):347–359. doi: 10.1038/nrg3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel-Ciernia A, Wood MA. Neuron-specific chromatin remodeling: a missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology. 2014;80:18–27. doi: 10.1016/j.neuropharm.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogel-Ciernia A, Matheos DP, Barrett RM, et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci. 2013;16(5):552–561. doi: 10.1038/nn.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez-Campos A, Azorin F. RNA is an integral component of chromatin that contributes to its structural organization. PLoS One. 2007;2(11):e1182. doi: 10.1371/journal.pone.0001182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brockdorff N. Noncoding RNA and polycomb recruitment. RNA. 2013;19(4):429–442. doi: 10.1261/rna.037598.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322(5902):750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Meur E, Watrin F, Landers M, Sturny R, Lalande M, Muscatelli F. Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev Biol. 2005;286(2):587–600. doi: 10.1016/j.ydbio.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 27.Leung KN, Chamberlain SJ, Lalande M, LaSalle JM. Neuronal chromatin dynamics of imprinting in development and disease. J Cell Biochem. 2011;112(2):365–373. doi: 10.1002/jcb.22958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xin Z, Allis CD, Wagstaff J. Parent-specific complementary patterns of histone H3 lysine 9 and H3 lysine 4 methylation at the Prader-Willi syndrome imprinting center. Am J Hum Genet. 2001;69(6):1389–1394. doi: 10.1086/324469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shulha HP, Crisci JL, Reshetov D, et al. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 2012;10(11):e1001427. doi: 10.1371/journal.pbio.1001427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cremer T, Cremer C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet. 2001;2(4):292–301. doi: 10.1038/35066075. [DOI] [PubMed] [Google Scholar]
- 31.Duan Z, Andronescu M, Schutz K, et al. A three-dimensional model of the yeast genome. Nature. 2010;465(7296):363–367. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood AJ, Severson AF, Meyer BJ. Condensin and cohesin complexity: the expanding repertoire of functions. Nat Rev Genet. 2010;11(6):391–404. doi: 10.1038/nrg2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7(9):703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- 34.Moss JF, Oliver C, Berg K, Kaur G, Jephcott L, Cornish K. Prevalence of autism spectrum phenomenology in Cornelia de Lange and Cri du Chat syndromes. Am J Ment Retard. 2008;113(4):278–291. doi: 10.1352/0895-8017(2008)113[278:POASPI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 35.Deardorff MA, Bando M, Nakato R, et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489(7415):313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gervasini C, Parenti I, Picinelli C, et al. Molecular characterization of a mosaic NIPBL deletion in a Cornelia de Lange patient with severe phenotype. Eur J Med Genet. 2013;56(3):138–143. doi: 10.1016/j.ejmg.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 37.Kagey MH, Newman JJ, Bilodeau S, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HS, Matevossian A, Jiang Y, Akbarian S. Chromatin immunoprecipitation in postmortem brain. J Neurosci Methods. 2006;156(1–2):284–292. doi: 10.1016/j.jneumeth.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 39.Adli M, Bernstein BE. Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat Protoc. 2011;6(10):1656–1668. doi: 10.1038/nprot.2011.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdolmaleky HM, Cheng KH, Faraone SV, et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15(21):3132–3145. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B(1):60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- 42.Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102(26):9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang HS, Matevossian A, Whittle C, et al. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27(42):11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iwamoto K, Bundo M, Yamada K, et al. DNA methylation status of SOX10 correlates with its downregulation and oligodendrocyte dysfunction in schizophrenia. J Neurosci. 2005;25(22):5376–5381. doi: 10.1523/JNEUROSCI.0766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mill J, Tang T, Kaminsky Z, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tochigi M, Iwamoto K, Bundo M, et al. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry. 2008;63(5):530–533. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Tamura Y, Kunugi H, Ohashi J, Hohjoh H. Epigenetic aberration of the human REELIN gene in psychiatric disorders. Mol Psychiatry. 2007;12(6):519, 593–600. doi: 10.1038/sj.mp.4002014. [DOI] [PubMed] [Google Scholar]
- 48.Siegmund KD, Connor CM, Campan M, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2(9):e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iwamoto K, Bundo M, Ueda J, et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21(5):688–696. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sugawara H, Iwamoto K, Bundo M, Ueda J, Ishigooka J, Kato T. Comprehensive DNA methylation analysis of human peripheral blood leukocytes and lymphoblastoid cell lines. Epigenetics. 2011;6(4):508–515. doi: 10.4161/epi.6.4.14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheung I, Shulha HP, Jiang Y, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci USA. 2010;107(19):8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montano CM, Irizarry RA, Kaufmann WE, et al. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013;14(8):R94. doi: 10.1186/gb-2013-14-8-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8(3):290–302. doi: 10.4161/epi.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. J Vis Exp. 2008;20:914. doi: 10.3791/914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sim FJ, McClain CR, Schanz SJ, Protack TL, Windrem MS, Goldman SA. CD140a identifies a population of highly myelinogenic, migration-competent and efficiently engrafting human oligodendrocyte progenitor cells. Nat Biotechnol. 2011;29(10):934–941. doi: 10.1038/nbt.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sundberg M, Skottman H, Suuronen R, Narkilahti S. Production and isolation of NG2+ oligodendrocyte precursors from human embryonic stem cells in defined serum-free medium. Stem Cell Res. 2010;5(2):91–103. doi: 10.1016/j.scr.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Nunes MC, Roy NS, Keyoung HM, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9(4):439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- 58.Jungblut M, Tiveron MC, Barral S, et al. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 2012;60(6):894–907. doi: 10.1002/glia.22322. [DOI] [PubMed] [Google Scholar]
- 59.Yuan SH, Martin J, Elia J, et al. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 2011;6(3):e17540. doi: 10.1371/journal.pone.0017540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uchida N, Buck DW, He D, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA. 2000;97(26):14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melief J, Koning N, Schuurman KG, et al. Phenotyping primary human microglia: tight regulation of LPS responsiveness. Glia. 2012;60(10):1506–1517. doi: 10.1002/glia.22370. [DOI] [PubMed] [Google Scholar]
- 62.Larsson HM, Lee ST, Roccio M, et al. Sorting live stem cells based on Sox2 mRNA expression. PLoS One. 2012;7(11):e49874. doi: 10.1371/journal.pone.0049874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Codega P, Silva-Vargas V, Paul A, et al. Prospective identification and purification of quiescent adult neural stem cells from their in vivo niche. Neuron. 2014;82(3):545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matevossian A, Akbarian S. A chromatin assay for human brain tissue. J Vis Exp. 2008;13:717. doi: 10.3791/717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hayashi Y, Nihonmatsu-Kikuchi N, Yu X, Ishimoto K, Hisanaga SI, Tatebayashi Y. A novel, rapid, quantitative cell-counting method reveals oligodendroglial reduction in the frontopolar cortex in major depressive disorder. Mol Psychiatry. 2011;16(12):1155–1158. doi: 10.1038/mp.2011.84. [DOI] [PubMed] [Google Scholar]
- 67.Ernst C, McGowan PO, Deleva V, Meaney MJ, Szyf M, Turecki G. The effects of pH on DNA methylation state: in vitro and post-mortem brain studies. J Neurosci Methods. 2008;174(1):123–125. doi: 10.1016/j.jneumeth.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 68.Mitchell AC, Bharadwaj R, Whittle C, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2013;75(12):961–969. doi: 10.1016/j.biopsych.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monoranu CM, Grunblatt E, Bartl J, et al. Methyl- and acetyltransferases are stable epigenetic markers postmortem. Cell Tissue Bank. 2011;12(4):289–297. doi: 10.1007/s10561-010-9199-z. [DOI] [PubMed] [Google Scholar]
- 70.Rogaev EI, Lukiw WJ, Lavrushina O, Rogaeva EA, St George-Hyslop PH. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics. 1994;22(2):340–347. doi: 10.1006/geno.1994.1393. [DOI] [PubMed] [Google Scholar]
- 71.West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci. 1995;6(2):141–146. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- 72.Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region −224 approximately −101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70(2):288–292. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 73.Tassone F, Hagerman RJ, Gane LW, Taylor AK. Strong similarities of the FMR1 mutation in multiple tissues: postmortem studies of a male with a full mutation and a male carrier of a premutation. Am J Med Genet. 1999;84(3):240–244. [PubMed] [Google Scholar]
- 74.Stadler F, Kolb G, Rubusch L, Baker SP, Jones EG, Akbarian S. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J Neurochem. 2005;94(2):324–336. doi: 10.1111/j.1471-4159.2005.03190.x. [DOI] [PubMed] [Google Scholar]
- 75.Oberle I, Rousseau F, Heitz D, et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252(5009):1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 76.Zhao J, Grant SF. Advances in whole genome sequencing technology. Curr Pharm Biotechnol. 2011;12(2):293–305. doi: 10.2174/138920111794295729. [DOI] [PubMed] [Google Scholar]
- 77.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10(10):669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thurman RE, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhu J, Adli M, Zou JY, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152(3):642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shulha HP, Cheung I, Guo Y, Akbarian S, Weng Z. Coordinated cell type-specific epigenetic remodeling in prefrontal cortex begins before birth and continues into early adulthood. PLoS Genet. 2013;9(4):e1003433. doi: 10.1371/journal.pgen.1003433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mitchell AC, Bharadwaj R, Whittle C, et al. The genome in three dimensions: a new frontier in human brain research. Biol Psychiatry. 2014;75(12):961–969. doi: 10.1016/j.biopsych.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bharadwaj R, Jiang Y, Mao W, et al. Conserved chromosome 2q31 conformations are associated with transcriptional regulation of GAD1 GABA synthesis enzyme and altered in prefrontal cortex of subjects with schizophrenia. J Neurosci. 2013;33(29):11839–11851. doi: 10.1523/JNEUROSCI.1252-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14(6):390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van Berkum NL, Dekker J. Determining spatial chromatin organization of large genomic regions using 5C technology. Methods Mol Biol. 2009;567:189–213. doi: 10.1007/978-1-60327-414-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simonis M, Klous P, Splinter E, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38(11):1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 88.Dostie J, Richmond TA, Arnaout RA, et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16(10):1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lieberman-Aiden E, van Berkum NL, Williams L, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li G, Fullwood MJ, Xu H, et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010;11(2):R22. doi: 10.1186/gb-2010-11-2-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gehlen LR, Gruenert G, Jones MB, Rodley CD, Langowski J, O’Sullivan JM. Chromosome positioning and the clustering of functionally related loci in yeast is driven by chromosomal interactions. Nucleus. 2012;3(4):370–383. doi: 10.4161/nucl.20971. [DOI] [PubMed] [Google Scholar]
- 92.Cagliero C, Grand RS, Jones MB, Jin DJ, O’Sullivan JM. Genome conformation capture reveals that the Escherichia coli chromosome is organized by replication and transcription. Nucleic Acids Res. 2013;41(12):6058–6071. doi: 10.1093/nar/gkt325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanizawa H, Iwasaki O, Tanaka A, et al. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010;38(22):8164–8177. doi: 10.1093/nar/gkq955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen Y, Yue F, McCleary DF, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488(7409):116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jin F, Li Y, Dixon JR, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gheldof N, Leleu M, Noordermeer D, Rougemont J, Reymond A. Detecting long-range chromatin interactions using the chromosome conformation capture sequencing (4C-seq) method. Methods Mol Biol. 2012;786:211–225. doi: 10.1007/978-1-61779-292-2_13. [DOI] [PubMed] [Google Scholar]
- 97.Stadhouders R, Kolovos P, Brouwer R, et al. Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat Protoc. 2013;8(3):509–524. doi: 10.1038/nprot.2013.018. [DOI] [PubMed] [Google Scholar]
- 98.Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci. 2010;30(39):13130–13137. doi: 10.1523/JNEUROSCI.1039-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McGowan PO, Sasaki A, Huang TC, et al. Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS One. 2008;3(5):e2085. doi: 10.1371/journal.pone.0002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Erraji-Benchekroun L, Underwood MD, Arango V, et al. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57(5):549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 102.Tang B, Chang WL, Lanigan CM, Dean B, Sutcliffe JG, Thomas EA. Normal human aging and early-stage schizophrenia share common molecular profiles. Aging Cell. 2009;8(3):339–342. doi: 10.1111/j.1474-9726.2009.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Numata S, Ye T, Hyde TM, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90(2):260–272. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Al-Mahdawi S, Pinto RM, Ismail O, et al. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17(5):735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- 105.Gamazon ER, Badner JA, Cheng L, et al. Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol Psychiatry. 2012;18(3):340–346. doi: 10.1038/mp.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaminsky ZA, Tang T, Wang SC, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41(2):240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 107.Kundakovic M, Chen Y, Costa E, Grayson DR. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol Pharmacol. 2007;71(3):644–653. doi: 10.1124/mol.106.030635. [DOI] [PubMed] [Google Scholar]
- 108.Levenson JM, Roth TL, Lubin FD, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281(23):15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 109.Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 110.Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28(2):395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guo JU, Ma DK, Mo H, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14(10):1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ma DK, Jang MH, Guo JU, et al. Neuronal activity-induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323(5917):1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cohen S, Gabel HW, Hemberg M, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72(1):72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14(8):1001–1008. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13(11):1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miller CA, Gavin CF, White JA, et al. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13(6):664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133(7):1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 118.Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- 119.Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci. 2011;36(1):7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 120.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 121.Endres M, Meisel A, Biniszkiewicz D, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20(9):3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bollati V, Baccarelli A, Hou L, et al. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Res. 2007;67(3):876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- 123.Desaulniers D, Xiao GH, Leingartner K, Chu I, Musicki B, Tsang BK. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-o-methyltransferase in prepubertal female Sprague–Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol Sci. 2005;86(1):175–184. doi: 10.1093/toxsci/kfi178. [DOI] [PubMed] [Google Scholar]
- 124.Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 2008;21(1):28–44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Satta R, Maloku E, Zhubi A, et al. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic inter-neurons. Proc Natl Acad Sci USA. 2008;105(42):16356–16361. doi: 10.1073/pnas.0808699105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Satta R, Maloku E, Costa E, Guidotti A. Stimulation of brain nicotinic acetylcholine receptors (nAChRs) decreases DNA methyltransferase 1 (DNMT1) expression in cortical and hippocampal GABAergic neurons of Swiss albino mice. Soc Neurosci Abstr. 2007 [Google Scholar]
- 127.Marutha Ravindran CR, Ticku MK. Changes in methylation pattern of NMDA receptor NR2B gene in cortical neurons after chronic ethanol treatment in mice. Brain Res Mol Brain Res. 2004;121(1–2):19–27. doi: 10.1016/j.molbrainres.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 128.Numachi Y, Shen H, Yoshida S, et al. Methamphetamine alters expression of DNA methyltransferase 1 mRNA in rat brain. Neurosci Lett. 2007;414(3):213–217. doi: 10.1016/j.neulet.2006.12.052. [DOI] [PubMed] [Google Scholar]
- 129.Numachi Y, Yoshida S, Yamashita M, et al. Psychostimulant alters expression of DNA methyltransferase mRNA in the rat brain. Ann N Y Acad Sci. 2004;1025:102–109. doi: 10.1196/annals.1316.013. [DOI] [PubMed] [Google Scholar]
- 130.LaPlant Q, Vialou V, Covington HE, 3rd, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13(9):1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cheng MC, Liao DL, Hsiung CA, Chen CY, Liao YC, Chen CH. Chronic treatment with aripiprazole induces differential gene expression in the rat frontal cortex. Int J Neuropsychopharmacol. 2008;11(2):207–216. doi: 10.1017/S1461145707008048. [DOI] [PubMed] [Google Scholar]
- 132.Shimabukuro M, Jinno Y, Fuke C, Okazaki Y. Haloperidol treatment induces tissue-and sex-specific changes in DNA methylation: a control study using rats. Behav Brain Funct. 2006;2:37. doi: 10.1186/1744-9081-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li J, Guo Y, Schroeder FA, et al. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90(5):1117–1131. doi: 10.1111/j.1471-4159.2004.02569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci USA. 2008;105(36):13614–13619. doi: 10.1073/pnas.0805493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kwon B, Houpt TA. Phospho-acetylation of histone H3 in the amygdala after acute lithium chloride. Brain Res. 2010;1333:36–47. doi: 10.1016/j.brainres.2010.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14(4):268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 138.Kim KS. Induced pluripotent stem (iPS) cells and their future in psychiatry. Neuropsychopharmacology. 2010;35(1):346–348. doi: 10.1038/npp.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shcheglovitov A, Shcheglovitova O, Yazawa M, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503(7475):267–271. doi: 10.1038/nature12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Farra N, Zhang WB, Pasceri P, Eubanks JH, Salter MW, Ellis J. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 2012;17(12):1261–1271. doi: 10.1038/mp.2011.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Brennand KJ, Simone A, Jou J, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 2011;473(7346):221–225. doi: 10.1038/nature09915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Pang ZP, Yang N, Vierbuchen T, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476(7359):220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stevens SJ, van Ravenswaaij-Arts CM, Janssen JW, et al. MYT1L is a candidate gene for intellectual disability in patients with 2p25.3 (2pter) deletions. Am J Med Genet A. 2011;155A(11):2739–2745. doi: 10.1002/ajmg.a.34274. [DOI] [PubMed] [Google Scholar]
- 145.Muotri AR, Marchetto MC, Coufal NG, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468(7322):443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Skene PJ, Illingworth RS, Webb S, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37(4):457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 148.Chouery E, Ghoch JA, Corbani S, et al. A novel deletion in ZBTB24 in a Lebanese family with Immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin Genet. 2012;82(5):489–493. doi: 10.1111/j.1399-0004.2011.01783.x. [DOI] [PubMed] [Google Scholar]
- 149.Huangfu D, Osafune K, Maehr R, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26(11):1269–1275. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 150.Yu IT, Park JY, Kim SH, Lee JS, Kim YS, Son H. Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology. 2009;56(2):473–480. doi: 10.1016/j.neuropharm.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 151.Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci USA. 2004;101(47):16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Guidotti A, Dong E, Kundakovic M, Satta R, Grayson DR, Costa E. Characterization of the action of antipsychotic subtypes on valproate-induced chromatin remodeling. Trends Pharmacol Sci. 2009;30(2):55–60. doi: 10.1016/j.tips.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 153.Gavin DP, Kartan S, Chase K, Jayaraman S, Sharma RP. Histone deacetylase inhibitors and candidate gene expression: an in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J Psychiatr Res. 2009;43(9):870–876. doi: 10.1016/j.jpsychires.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 154.Ohi Y, Qin H, Hong C, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat Cell Biol. 2011;13(5):541–549. doi: 10.1038/ncb2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nishino K, Toyoda M, Yamazaki-Inoue M, et al. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011;7(5):e1002085. doi: 10.1371/journal.pgen.1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tobin SC, Kim K. Generating pluripotent stem cells: differential epigenetic changes during cellular reprogramming. FEBS Lett. 2012;586(18):2874–2881. doi: 10.1016/j.febslet.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tobe BT, Brandel MG, Nye JS, Snyder EY. Implications and limitations of cellular reprogramming for psychiatric drug development. Exp Mol Med. 2013;45:e59. doi: 10.1038/emm.2013.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gavin DP, Kartan S, Chase K, Grayson DR, Sharma RP. Reduced baseline acetylated histone 3 levels, and a blunted response to HDAC inhibition in lymphocyte cultures from schizophrenia subjects. Schizophr Res. 2008;103(1–3):330–332. doi: 10.1016/j.schres.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ripke S, O’Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sklar P, Ripke S, Scott LJ, et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Shi J, Levinson DF, Duan J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460(7256):753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Stefansson H, Ophoff RA, Steinberg S, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhong H, Beaulaurier J, Lum PY, et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:e1000932. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nat Rev Genet. 2009;10(3):184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Degner JF, Pai AA, Pique-Regi R, et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature. 2012;482(7385):390–394. doi: 10.1038/nature10808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6(4):e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Musunuru K, Strong A, Frank-Kamenetsky M, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466(7307):714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Harismendy O, Notani D, Song X, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-gamma signalling response. Nature. 2011;470(7333):264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Karnik R, Meissner A. Browsing (Epi)genomes: a guide to data resources and epigenome browsers for stem cell researchers. Cell Stem Cell. 2013;13(1):14–21. doi: 10.1016/j.stem.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ballare C, Zaurin R, Vicent GP, Beato M. More help than hindrance: nucleosomes aid transcriptional regulation. Nucleus. 2013;4(3):189–194. doi: 10.4161/nucl.25108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Aday AW, Zhu LJ, Lakshmanan A, Wang J, Lawson ND. Identification of cis regulatory features in the embryonic zebrafish genome through large-scale profiling of H3K4me1 and H3K4me3 binding sites. Dev Biol. 2011;357(2):450–462. doi: 10.1016/j.ydbio.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hernandez DG, Nalls MA, Gibbs JR, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shulha HP, Cheung I, Whittle C, et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2011;69(3):314–324. doi: 10.1001/archgenpsychiatry.2011.151. [DOI] [PubMed] [Google Scholar]
- 177.Akbarian S. Epigenetics of schizophrenia. Curr Top Behav Neurosci. 2010;4:611–628. doi: 10.1007/7854_2010_38. [DOI] [PubMed] [Google Scholar]
- 178.Tang B, Dean B, Thomas EA. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry. 2011;1:e64. doi: 10.1038/tp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Taqi MM, Bazov I, Watanabe H, et al. Prodynorphin CpG-SNPs associated with alcohol dependence: elevated methylation in the brain of human alcoholics. Addict Biol. 2011;16(3):499–509. doi: 10.1111/j.1369-1600.2011.00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fromer M, Pocklington AJ, Kavanagh DH, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506(7487):179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]