

Abstract
Bicyclic nitroxyl derivatives, such as 2-azaadamantane N-oxyl (AZADO) and 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO), have emerged as highly effective alternatives to TEMPO-based catalysts for selective oxidation reactions (TEMPO = 2,2,6,6-tetramethyl-1-piperidine N-oxyl). Their efficacy is widely attributed to their smaller steric profile; however, electrocatalysis studies described herein show that the catalytic activity of nitroxyls is more strongly affected by the nitroxyl/oxoammonium redox potential than by steric effects. The inexpensive, high-potential TEMPO derivative, 4-acetamido-TEMPO (ACT), exhibits higher electrocatalytic activity than AZADO and ABNO for the oxidation of 1° and 2° alcohols. Mechanistic studies provide insights into the origin of these unexpected reactivity trends. The superior activity of ACT is especially noteworthy at high pH, where bicyclic nitroxyls are inhibited by formation of an oxoammonium-hydroxide adduct.
Graphical Abstract
Introduction
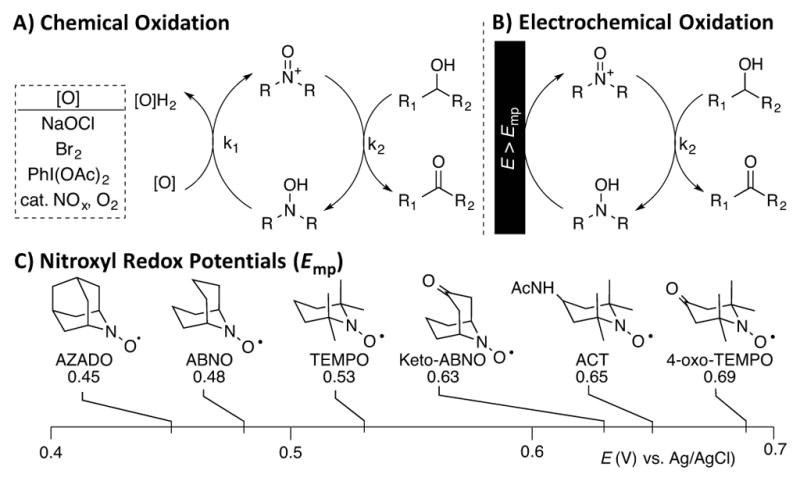
TEMPO (2,2,6,6-tetramethyl-1-piperidine N-oxyl) and related nitroxyl derivatives are important catalysts for alcohol oxidation,1 and they find widespread used in industrial2 and laboratory3 applications (Scheme 1). Diverse stoichiometric oxidants have been used to promote catalytic turnover, the most common of which include bleach (NaOCl), hypervalent iodine reagents, and O2 in combination with NOx co-catalysts (Scheme 1A).1–3 In addition to these chemical oxidation methods, nitroxyls have been widely studied as mediators for electrocatalytic alcohol oxidation (Scheme 1B).4,5 The latter approach has important implications for energy conversion applications, as demonstrated in recent studies of photoelectrochemical conversion of biomass4i and biofuel cells.4g,j
Scheme 1.
Chemical and Electrochemical Alcohol Oxidation Methods with Nitroxyls of Different Structures and Redox Potentials.
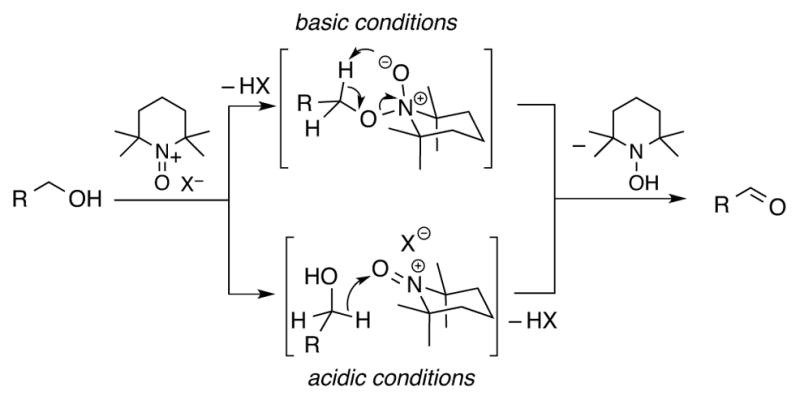
Under typical (basic) reaction conditions, TEMPO exhibits strong steric discrimination in its reaction with alcohols, including the ability to achieve chemoselective oxidation of 1° alcohols in the presence of unprotected 2° alcohols.6 This selectivity has been attributed to steric effects in the formation of an alkoxide adduct with the oxoammonium species (Scheme 2, top pathway).1b The selectivity changes when the reaction is performed under acidic conditions, wherein the mechanism is proposed to involve bimolecular hydride transfer, which favors more electron-rich substrates (i.e., 2° > 1° alcohols).7 The reactions under the acidic conditions are typically considerably slower than those under basic conditions, however.
Scheme 2.
Proposed Mechanisms for TEMPO+-Mediated Oxidation of Alcohols under Different Conditions.
Recently, significant advances in nitroxyl-catalyzed alcohol oxidation have been made through the use of bicyclic nitroxyls, such as 2-azaadamantane N-oxyl (AZADO) and 9-azabicyclo[3.3.1]nonane N-oxyl (ABNO) (cf. Scheme 1C).8 These nitroxyls exhibit excellent activity for the oxidation of both 1° and 2° alcohols, and their favorable catalytic properties are primarily attributed to the reduced steric hindrance around the active nitroxyl/oxoammonium site. Unfortunately, the cost and multi-step synthesis of these reagents constrain their use, especially for large-scale applications.9
Building on our studies of nitroxyl (co-)catalyzed aerobic oxidation reactions,10 we recently began exploring electrocatalytic reactions with nitroxyl mediators.11 Whereas TEMPO has been studied extensively as an electrocatalyst for alcohol oxidation,4,5 much less attention has been given to the bicyclic nitroxyls.4g,8c The mid-point potential (Emp) of the one-electron nitroxyl/oxoammonium couple varies, depending on the nitroxyl structure (i.e., mono- vs bicyclic) and/or substituents (Scheme 1C),12 but the effect of the nitroxyl redox potentials on catalytic activity and their implications for chemical and electrochemical oxidation methods has not been addressed. Here, we describe the electrocatalytic behavior of a series of bicyclic and TEMPO-based nitroxyls. Free-energy correlations show that nitroxyl-catalyzed alcohol oxidation rates under chemical vs. electrochemical conditions exhibit opposite trends with respect to the nitroxyl/oxoammonium redox potential. Mechanistic studies reveal the origin of this unexpected inversion of activity, and the results have important implications for practical applications of nitroxyl-based alcohol oxidation reactions. For example, the low-cost, readily accessible 4-acetamido-TEMPO (ACT) outperforms the more-expensive bicyclic nitroxyls under electrochemical conditions.
Results and Discussion
Kinetic analysis of electrocatalytic oxidation of alcohols with different nitroxyls
Cyclic voltammograms (CVs) of AZADO (a), ABNO (b), TEMPO (c), and ACT (d) reveal reversible electron transfer under conditions similar to those commonly used for alcohol oxidation (Figure 1A). In contrast, keto-ABNO and 4-oxo-TEMPO exhibit irreversible behavior under these basic conditions (cf. trace e in Figure 1A), reflecting the instability of the corresponding oxoammonium species. Keto-substituted oxoammonium species have been shown to be highly susceptible to base-promoted ring opening.13 ACT exhibits the highest mid-point potential (Emp = 0.65 V) among the four electrochemically reversible nitroxyls.
Figure 1.
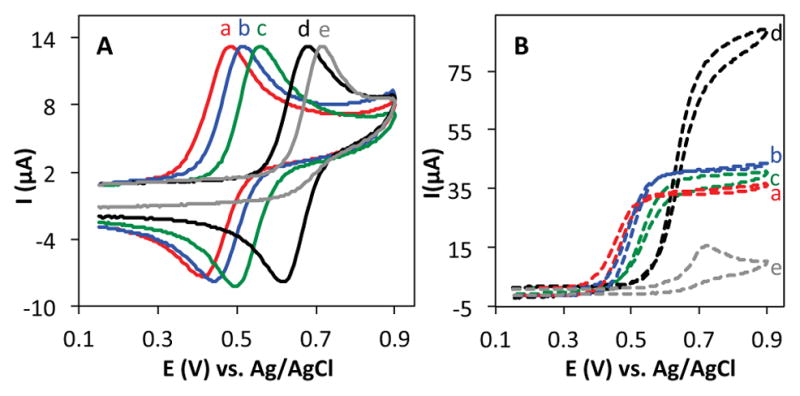
Cyclic voltammograms of 1 mM AZADO (a), ABNO (b), TEMPO (c), ACT (d) and 4-oxo-TEMPO (e) in the absence (left, A) and presence (right, B) of 50 mM 1-butanol in HCO3−/CO32− electrolyte (pH 10), scan rate 50 mVs−1.
CVs of the nitroxyls were then reacquired with 50 mM 1-butanol present in the solutions (Figure 1B). The S-shaped CVs, showing a significant increase in the oxidation peak and a disappearance of the reduction peak, are diagnostic of electrocatalysis, and the oxidation current is proportional to the catalytic turnovers that occur on the CV time scale. AZADO, ABNO and TEMPO perform similarly, while ACT exhibits more than 2-fold higher catalytic current than the other nitroxyls. No significant catalytic activity was observed for 4-oxo-TEMPO, reflecting the instability of the oxoammonium species.
To gain a more-quantitative comparison of the different nitroxyls, the electrocatalytic reactions were evaluated by additional electrochemical studies. Representative chronoamperograms (CAs) obtained with ABNO are shown in Figure 2. In the absence of alcohol, the CA exhibits an exponential decay (curve a), consistent with diffusion-controlled oxidation of the nitroxyl to the oxoammonium. The CA current increases upon addition of 1-butanol, and the steady-state current is proportional to the square root of the alcohol concentration (Figure 2 curves b-e).14 Similar studies were performed for each of the other nitroxyls (AZADO, TEMPO and ACT) in order to compare their catalytic activities and TOFs (see Figures S1–S4).
Figure 2.
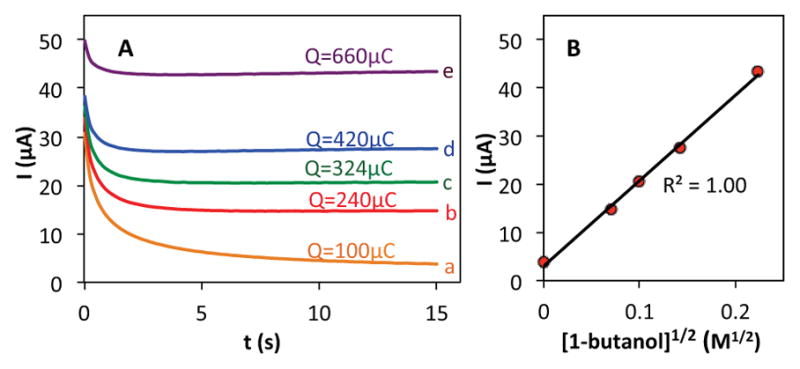
(A) Chronoamperograms of ABNO (1 mM) in the absence (a) and presence of 1-butanol at 5 mM (b), 10 mM (c), 20 mM (d) and 50 mM (e) concentrations in HCO3−/CO32− electrolyte (pH=10), and (B) a plot of current vs [1-butanol]1/2 with an applied potential 0.7 V vs. Ag/AgCl.
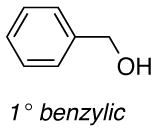
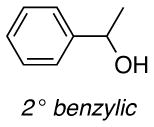
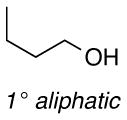
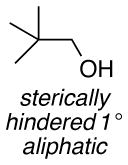
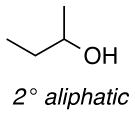
The difference between the current in the presence and absence of 1-butanol (i.e., curves b-e vs. curve a in Figure 2) is directly proportional to the nitroxyl turnover frequency (TOF). The TOFs for each of the four nitroxyls with a series of five different alcohols are presented in Table 1. The alcohol substrates were selected as representative examples of 1° and 2° benzylic, 1° and 2° aliphatic, and sterically hindered 1° aliphatic alcohols. The data show that ACT is nearly always the most active catalyst. The only exception was evident in the oxidation of 2-butanol, but even in this case, the activity of ACT is comparable to ABNO and AZADO. TEMPO exhibits the lowest activity in all cases, except for the oxidation of 1-butanol, for which AZADO is the least active. Most significantly, the activity of ACT is much higher than expected from recent literature reports that highlight the predominant role of steric effects.8
Table 1.
TOF (h−1) of different nitroxides for oxidation of alcohols
|
|
|
|
|
|
|---|---|---|---|---|---|
| ABNO | 1088 | 238 | 588 | 337 | 87 |
| AZADO | 1128 | 358 | 488 | 298 | 78 |
| TEMPO | 853 | 118 | 568 | 198 | 18 |
| ACT | 1228 | 378 | 708 | 388 | 73 |
Standard conditions for TOF determination: 50 mM alcohol, 1.0 mM nitroxyl, HCO3−/CO32− electrolyte (pH 10).
Analysis of pH effects on electrocatalytic oxidation of alcohols with different nitroxyls
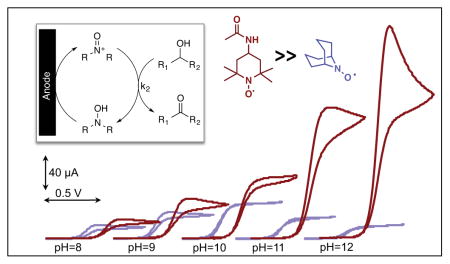
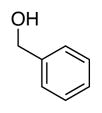
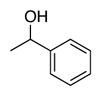
The advantageous performance of ACT relative to the bicyclic nitroxyls is even more evident when ACT and ABNO are compared at different pH (Figure 3A). CVs were obtained for the oxidation of 1-butanol by ACT reveal a significant increase in the catalytic current upon raising the pH from 8 to 12 (dashed CVs, Figure 3A). ABNO activity is lower than the ACT activity (solid CVs, Figure 3A), and while it increases upon raising the pH from 8 to 10, it then decreases at pH 11 and 12. A comparison of the ACT and ABNO TOFs for five different alcohols at pH 9 and 11 is shown in Figures 3B and 3C. These data demonstrate the strong impact of pH on catalyst activity, and show that increasing the pH significantly activates ACT, while it strongly suppresses ABNO activity. The pH trend for ACT continues beyond pH 12, and TOFs reach values above 1000 and 6000 h−1 for 2-butanol and 1-butanol, respectively, at pH 13. The effectiveness of ACT as an electrocatalyst was confirmed in bulk electrolysis experiments. Electrochemical oxidation of each of the five alcohols were performed on 1 mmol scale at pH 10 with 2 mol% ACT, and the oxidized products were obtained in 88–98% yields within 2 h at room temperature (Table 2).15 ABNO exhibits lower activity and is less stable under the same reaction conditions. For example, use of 2 mol% ABNO only achieves 47% conversion to benzaldehyde prior to complete loss of catalytic activity.
Figure 3.

(A) Cyclic voltammograms of ABNO (solid line) and ACT (dashed line) in the presence of 50 mM 1-butanol at various pH values, scan rate 50mVs−1, (B) TOFs (h−1) of ACT and ABNO for oxidation of various alcohols; 1 mM ACT or ABNO, 50 mM alcohol in aqueous carbonate buffers; 0.14 M NaHCO3 and 0.01 M Na2CO3 for pH 9, 0.02 M NaHCO3 and (C) 0.13 M Na2CO3 for pH 11.
Table 2.
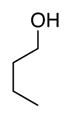
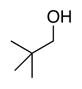
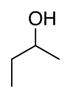
Bulk electrolysis data for ACT-mediated oxidation of alcoholsa
| Reactant: |
|
|
|
|
|
|
| |||||
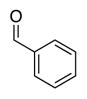
| Product: |
|
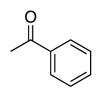
|
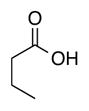
|
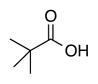
|
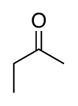
|
|
| |||||
| Time | 2 h | 2 h | 2 h | 2 h | 2 h |
| Yieldsb | 88% | 88% | 98% | 98% | 95% |
Conditions: 0.1 M alcohol in 10 mL aqueous carbonate buffer, 0.075 M NaHCO3, 0.075 M Na2CO3 (pH 10), 2 mol % ACT, electrolysis at 0.75 V vs Ag/AgCl.
NMR yields (int. std. = mesitylene).
Several lines of evidence suggest that the decrease in electrocatalytic activity of ABNO above pH 10 arises from its susceptibility to formation of an oxoammonium-hydroxide adduct, ABNOOH (eq 1). Adducts of this type have been described previously for TEMPO+ at pH > 12,16 and ring-strain considerations can account for the increased susceptibility of bicyclic oxoammonium species to such adduct formation. Insights into this adduct formation were obtain from CVs of ABNO at pH 9, 10, and 11, which show that the Emp shifts negative and the reduction peak decreases upon increasing the pH (Figure 4A). These data and CVs at different scan rates (Figure S5) indicate that ABNO+ undergoes a chemical reaction upon its electrochemical generation at pH > 9. In contrast, CVs of ACT are unaffected by changes to the solution pH (Figure 4B), indicating that ACT and ACT+ are stable under the voltammetry conditions. Steric effects, as well as reduced ring strain, probably contribute to the enhanced stability of ACT+ at high pH.
Figure 4.
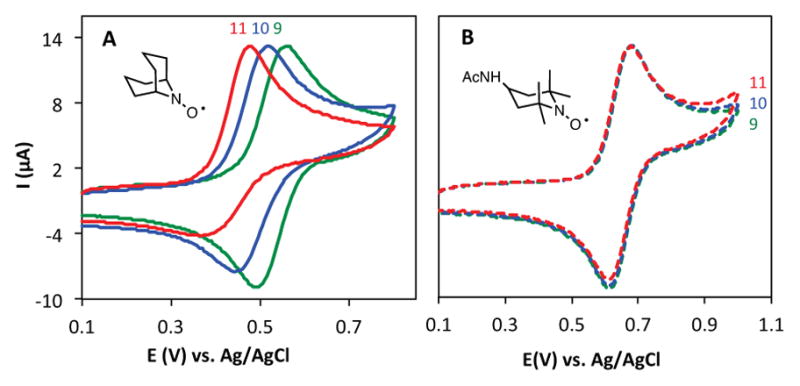
Cyclic voltammograms of ABNO (A) and ACT (B) from pH 9–11 (scan rate 50 mVs−1).
 |
(1) |
Spectroelectrochemical analysis of ABNO oxidation at different pH
UV-visible spectroelectrochemical studies complement the voltammetric data and support the hypothesis in eq 1. Well-behaved isosbestic behavior is observed for the redox interconversion of ABNO/ABNO+ at pH 8 (Figure 5). Near-isosbestic behavior is also observed at pH 10.2, but the product, which is different from ABNO+, corresponds to ABNOOH. Subsequent reduction of this species regenerates ABNO, consistent with reversible formation of the adduct (Figure 6). An equilibrium constant of 3.7 x 104 M−1 was derived for the formation of the ABNOOH from ABNO+ and OH− on the basis of the spectroelectrochemical data. At pH > 11, oxidation of ABNO is less reversible, possibly reflecting decomposition of ABNOOH under these conditions (Figure S6).
Figure 5.

Spectroelectrochemical UV-vis data collected during oxidation of ABNO (A) for 300 s (t = 0–300 s), immediately followed by reduction of the in situ generated ABNO+ (B) at for 300 s (t = 300–600 s). Insets: Concentration profiles of ABNO and ABNO+ during electrochemical oxidation (A) and reduction (B). Reaction conditions: 0.1 M HCO3− electrolyte (pH 8.3); oxidation performed at 0.7 V vs. Ag/AgCl and reduction at 0.3 V vs. Ag/AgCl; spectra recorded every 6 s.
Figure 6.

Spectroelectrochemical UV-vis data collected during oxidation of ABNO (A) in HCO3−/CO32− electrolyte (pH 10.2) at 0.65 V vs. Ag/AgCl for 300 s (time interval = 6 s), immediately followed by reduction of the in situ generated ABNO+/ABNOOH (B) at 0.25 V vs. Ag/AgCl for 300 s (time interval = 6 s). Insets: Concentration profiles of ABNO, ABNO+ and ABNOOH during electrochemical oxidation (A) and reduction (B).
The above data provide a clear rationale for the enhanced activity of ACT relative to bicyclic nitroxyls above pH 10, but we also sought to understand the excellent performance of ACT below pH 10, where bicyclic oxoammonium-hydroxide adducts such as ABNOOH do not interfere. Our initial hypothesis was that the high activity of ACT+ arises from its high reduction potential (cf. Scheme 1C and Figure 1). Comparisons of TEMPO and bicyclic nitroxyl derivatives are typically performed with chemical oxidants, such as bleach (NaOCl).8 In these cases, high-potential nitroxyls (e.g., ACT) could undergo sluggish oxidation by the chemical oxidant, thereby masking their intrinsic alcohol-oxidation reactivity.17 Low-potential nitroxyls, such as AZADO and ABNO, will therefore experience a competitive advantage.
In situ monitoring of nitroxyl-catalyzed oxidation of 1-butanol by bleach
AZADO-, ABNO-, TEMPO- and ACT-catalyzed oxidation of 1-butanol were analyzed with bleach as the oxidant. It was possible to monitor the distribution of catalyst oxidation states during the reaction via cyclic chronoamperometry as an analytical technique. In these experiments, the potential at a rotating disk electrode (RDE) was cycled between 180 mV above and 180 mV below the midpoint potential (Emp) of the nitroxyl. The current measured at Emp + 180 mV is proportional to the [nitroxyl], while the current at Emp − 180 mV is proportional to the [oxoammonium] (see Figures 7 and S7).
Figure 7.
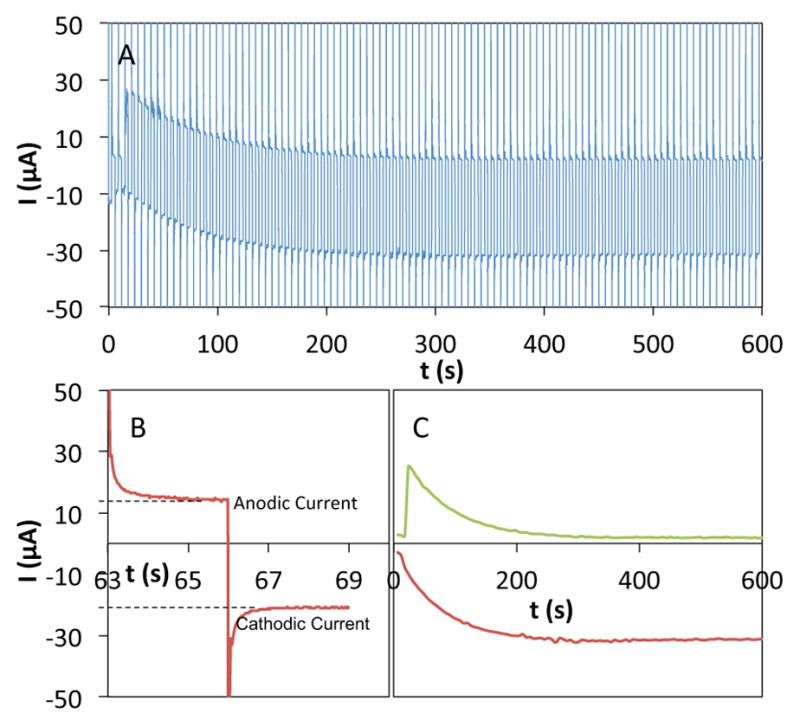
(A) Representative cyclic chronoamperometric data for the oxidation of TEMPO by bleach. (B) Expansion of the oxidation and reduction currents from a single cycle in plot A, obtained from potential steps at 0.71 and 0.25 V vs. Ag/AgCl. (C) Plots of the faradaic anodic (positive) and cathodic (negative) currents at each step in plot A (cf. plot B). (Pulse width = 3 s, RDE rotation rate = 2000 rpm; initial concentrations: TEMPO 5.0 mM, NaOCl 0.2 M, 0.12 M NaHCO3).
Before analyzing alcohol oxidation reactions, we probed the oxidation of the different nitroxyls by bleach via the cyclic chronoamperometric method described above. Representative data obtained during oxidation of TEMPO by bleach (in the absence of alcohol) is depicted in Figure 7. The data show progressive conversion of TEMPO to TEMPO+ over 600 s. The traces with positive current correspond to oxidation of TEMPO at 0.71 V vs. Ag/AgCl (Emp + 180 mV), and the traces with negative current correspond to reduction of TEMPO+ at 0.25 V vs. Ag/AgCl (Emp − 180 mV) [Emp (TEMPO) = 0.53 V]. Similar data were acquired for the four different nitroxyls, AZADO, ABNO, TEMPO, and ACT, and the data show that different nitroxyls undergo oxidation at significantly different rates (Figure 8). Their relative rates match the trend expected from their redox potentials: AZADO > ABNO > TEMPO > ACT. Oxidation of ACT is particularly slow, exhibiting a rate 60- and 7-fold slower than AZADO and ABNO, respectively, at pH 8.3.
Figure 8.
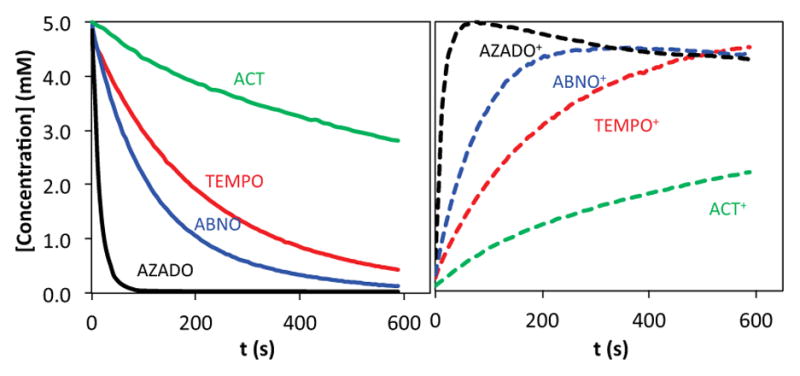
Concentration profiles of nitroxyl and oxoammonium species (solid and dashed lines, respectively) in the presence of bleach, derived from cyclic chronoamperometry. Reaction conditions: 5.0 mM nitroxyl, 120 mM NaOCl, 150 mM NaHCO3 (pH 8.3).
Oxidation of 1-butanol by bleach was then examined by 1H NMR spectroscopy using the same series of nitroxyls (Figure S8), and the results reveal that these reactions follow the same rate trends observed for oxidation of the nitroxyls by bleach: AZADO > ABNO > TEMPO > ACT. Cyclic chronoamperometry studies illuminate the origin of this trend. Data from the AZADO-, ABNO-, TEMPO- and ACT-catalyzed alcohol oxidation reactions, shown in Figure 9, reveal that AZADO and ABNO undergo rapid oxidation to AZADO+ and ABNO+ and that these oxoammonium species are the predominant form of the catalyst during steady-state turnover. In contrast, TEMPO and ACT convert more slowly to TEMPO+ and ACT+, and they reach steady-state concentrations of approximately 60% and 20%, respectively. A near-quantitative mass balance of the oxommonium and nitroxyl species is detected under the different reaction conditions, implying a negligible steady-state concentration of the hydroxylamine species. (The hydroxylamine formed upon oxidation of the alcohol could undergo rapid oxidation to the nitroxyl by direct reaction with the oxidant or via comproportionation with the oxoammonium species.) Collectively, these observations reveal that the poor catalytic activity of ACT with bleach as the oxidant arises from slow formation of the oxoammonium species (cf. Figure 8). In the electrocatalytic reactions, the electrode potential is adjusted to match the redox potential of the nitroxyl catalyst. Therefore, the catalyst reoxidation rate is normalized for different nitroxyl species and the catalytic rate reflects the intrinsic reactivity of the oxoammonium species with alcohol. The excellent electrocatalytic performance of ACT under basic conditions is attributed to the enhanced electrophilicity of the oxoammonium species ACT+, which should favor formation of the alkoxide adduct in the “basic” alcohol oxidation pathway (cf. Scheme 2).
Figure 9.
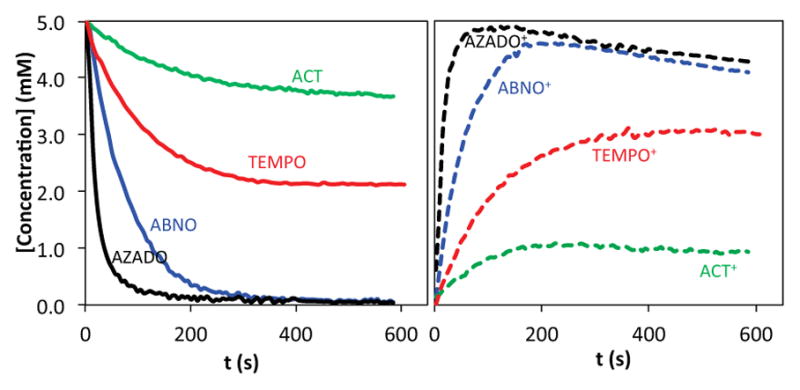
Concentration profiles of nitroxyl and oxoammonium species (solid and dashed lines, respectively) in the oxidation of 1-butanol by bleach, derived from cyclic chronoamperometry. Reaction conditions: 5.0 mM nitroxyl, 100 mM 1-butanol, 120 mM NaOCl, 150 mM NaHCO3 (pH 8.3).
Comparison of chemical and electrochemical nitroxyl-catalyzed alcohol oxidation
The very different catalyst activity trends observed for the bleach- vs electrode-driven alcohol oxidation reactions are depicted in the free-energy correlations in Figure 10. In addition to the four nitroxyls investigated systematically above, three additional nitroxyls 1-Me-AZADO, 4-MeO-TEMPO and 4-PhCO2-TEMPO were tested under the same conditions. The bleach-driven reactions (blue triangles) with the seven nitroxyl derivatives show a negative correlation with the nitroxyl Emp values. This trend is attributed to the influence of the catalyst reoxidation step on the overall rate: nitroxyls with lower Emp undergo more facile oxidation and are therefore more effective catalysts. The non-linearity of the trend is perhaps expected, considering the turnover-limiting step appears to change across this series of nitroxyls (cf. Figure 9) and/or because ring-strain or steric effects could impact the relative rates for the reactions of nitroxyls with bleach. On the other hand, the electrocatalytic reactions show a small, but positive, correlation with the nitroxyl Emp, The best catalytic rates are obtained with TEMPO derivatives bearing an electron-withdrawing substitutent in the 4-position, which have the highest Emp values among the nitroxyls studied. The trend clearly shows that the oxoammonium reduction potential is more significant than steric effects in controlling the reaction rate. The 4-benzoyl derivative exhibits the highest rate among the derivatives tested; however, the ester group is susceptible to hydrolysis under basic conditions. ACT is much less susceptible to hydrolysis and therefore represents a highly appealing low-cost electrocatalyst for alcohol oxidation.
Figure 10.
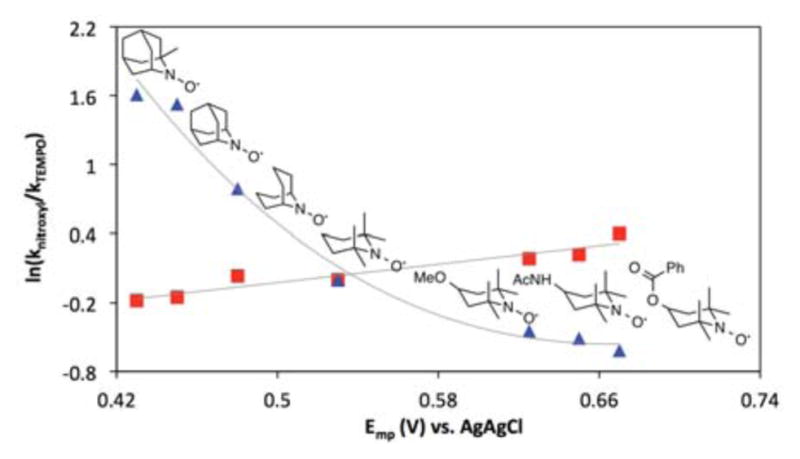
Linear-free-energy correlations for nitroxyl-catalyzed oxidation of 1-butanol with bleach (blue triangles) and under electrochemical conditions (red squares). See Figures S1–S4 and S8 for raw data.
Conclusion
The observations described herein provide extensive insights into the relative activity of TEMPO and bicyclic nitroxyl derivatives, and they have important fundamental and practical implications for this nitroxyl-catalyzed alcohol oxidation reactions. The higher activity of ACT relative to AZADO and ABNO was unexpected, but it shows that increased driving force can compensate for, and even overcome, steric effects in promoting the catalytic activity of nitroxyls. ACT is one of the least expensive nitroxyl derivatives available,2 and it exhibits excellent stability and activity over a broad pH range. The results described here bode well for expanded use of ACT in catalytic applications that will leverage the widespread use of ACT+ (commonly termed “Bobbitt’s salt”) as a stoichiometric reagent.18 The exceptional activity of ACT as an electrochemical mediator, particularly at high pH, also provides an important foundation for development of scalable electrocatalytic applications of ACT, for example, via continuous-flow electrolysis.
Supplementary Material
Acknowledgments
Financial support for this project was provided by the Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02-07ER64494).
Footnotes
Supporting Information: Experimental information, additional electrochemical, spectro-electrochemical, and kinetic data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For the first application of TEMPO-catalyzed alcohol oxidation and a comprehensive survey, see: Cella JA, Kelley JA, Kenehan EF. J Org Chem. 1975;40:1860.Bobbitt JM, Brückner C, Merbouh N. Org React. 2010;74:103.
- 2.For a review, see: Ciriminna R, Pagliaro M. Org Process Res Dev. 2010;14:245.
- 3.For reviews, see: de-Nooy AEJ, Besemer AC, van Bekkum H. Synthesis. 1996;10:1153.Sheldon RA, Arends IWCE. Adv Synth Catal. 2004;346:1051.Tebben L, Studer A. Angew Chem Int Ed. 2011;50:5034. doi: 10.1002/anie.201002547.Wertz S, Studer A. Green Chem. 2013;15:3116.Ryland BL, Stahl SS. Angew Chem, Int Ed. 2014;53:8824. doi: 10.1002/anie.201403110.Cao Q, Dornan LM, Rogan L, Hughes NL, Muldoon M. J Chem Commun. 2014;50:4524. doi: 10.1039/c3cc47081d.Seki Y, Oisaki K, Kanai M. Tetrahedron Lett. 2014;55:3738.Miles KC, Stahl SS. Aldrichimica Acta. 2015;48:8.
- 4.For leading references, see: Semmelhack MF, Chou CS, Cortes DA. J Am Chem Soc. 1983;105:4492.Osa T, Kashiwagi Y, Mukai K, Oshawa A, Bobbitt JM. Chem Lett. 1990;19:75.Kashiwagi Y, Kurashima F, Kikuchi C, Anzai J, Osa T, Bobbitt JM. Tetrahedron Lett. 1999;40:6469.Liaigre D, Breton T, Belgsir EM. Electrochem Commun. 2005;7:312.Hill-Cousins JT, Kuleshova J, Green RA, Birkin PR, Pletcher D, Underwood TJ, Leach SG, Brown RCD. ChemSusChem. 2012;5:326. doi: 10.1002/cssc.201100601.Green RA, Hill-Cousins JT, Brown RCD, Pletcher D, Leach SG. Electrochim Acta. 2013;113:550.Hickey DP, McCammant MS, Giroud F, Sigman MS, Minteer SD. J Am Chem Soc. 2014;136:15917. doi: 10.1021/ja5098379.Rafiee M, Karimi B, Alizadeh S. ChemElectroChem. 2014;1:455.Cha HG, Choi KS. Nat Chem. 2015;7:328. doi: 10.1038/nchem.2194.Hickey DP, Milton RD, Chen D, Sigman MS, Minteer SD. ACS Catal. 2015;5:5519.
- 5.For a review, see Ciriminna R, Palmisano G, Pagliaro M. ChemCatChem. 2015;7:552.
- 6.Numerous examples are included in the reviews in ref. 3. See also: Siedlecka R, Skarzewski J, Młochowski J. Tetrahedron Lett. 1990;31:2177.de Nooy AEJ, Besemer AC, van Bekkum H. Tetrahedron. 1995;51:8023.Hoover JM, Stahl SS. J Am Chem Soc. 2011;133:16901. doi: 10.1021/ja206230h.
- 7.Bailey WF, Bobbitt JM, Wiberg KB. J Org Chem. 2007;72:4504. doi: 10.1021/jo0704614. [DOI] [PubMed] [Google Scholar]
- 8.For leading references, see refs. 3e and 3h and the following: Graetz B, Rychnovsky S, Leu WH, Farmer P, Lin R. Tetrahedron Asymm. 2005;16:3584.Shibuya M, Tomizawa M, Suzuki I, Iwabuchi Y. J Am Chem Soc. 2006;128:8412. doi: 10.1021/ja0620336.Demizu Y, Shiigi H, Oda T, Matsumura Y, Onomura O. Tetrahedron Lett. 2008;49:48.Kuang Y, Rokubuichi H, Nabae Y, Hayakawa T, Kakimoto M-a. Adv Synth Catal. 2010;352:2635.Shibuya M, Osada Y, Sasano Y, Tomizawa M, Iwabuchi Y. J Am Chem Soc. 2011;133:6497. doi: 10.1021/ja110940c.Steves JE, Stahl SS. J Am Chem Soc. 2013;135:15742. doi: 10.1021/ja409241h.Lauber MB, Stahl SS. ACS Catal. 2013;3:2612.Sasano Y, Nagasawa S, Yamazaki M, Shibuya M, Park J, Iwabuchi Y. Angew Chem, Int Ed. 2014;53:3236. doi: 10.1002/anie.201309634.
- 9.Prompted by a reviewer request, we provide the cost for 1 mmol of the commercially available nitroxyls considered in this study (prices reflect the lowest available cost from Sigma-Aldrich at http://www.sigmaaldrich.com/; accessed Sept. 11, 2015). TEMPO ($0.83), ACT ($0.96), 4-MeO-TEMPO ($5.69), 4-oxo-TEMPO ($7.14), 4-PhCO2-TEMPO ($16.81), ABNO ($32.76), AZADO ($155.64), 1-Me-AZADO ($328.68). The cost of individual nitroxyls will of course depend upon the scale of production.
- 10.See refs. 3d, 3g, 8f, 8g and the following: Xie X, Stahl SS. J Am Chem Soc. 2015;137:3767. doi: 10.1021/jacs.5b01036.
- 11.Gerken JB, Stahl SS. ACS Cent Sci. 2015;1:234. doi: 10.1021/acscentsci.5b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For redox potentials of diverse nitroxyls, see ref. 8g and the following: Shchukin GI, Ryabinin VA, Grigorev IA, Volodarskii LB. J Gen Chem USSR. 1985;56:753.Bobbitt JM, Flores MCL. Heterocycles. 1988;27:509.Shibuya M, Pichierri F, Tomizawa M, Nagasawa S, Suzuki I, Iwabuchi Y. Tetrahedron Lett. 2012;53:2070.
- 13.Golubev VA, Sen’ VD. Russ J Org Chem. 2011;47:869. [Google Scholar]
-
14.The rate of the catalytic reaction can be derived from the following equation.
Icat represents the catalytic current, CN and CA are the bulk concentrations of nitroxide and alcohol, and n, F, and A denote the number of electrons, Faraday constant and the electrode surface area respectively. The amount of 5.0×10−6 cm2s−1 has been considered as the diffusion coefficient (DA) of all alcohols. See ref. 4h and the following: Costentin C, Drouet S, Robert M, Savéant J-M. J Am Chem Soc. 2012;134:11235. doi: 10.1021/ja303560c. - 15.The results in Table 2 are included to demonstrate that ACT can serve as an effective electrocatalytic mediator under bulk electrolysis conditions. Ongoing studies are focused on optimizing these conditions for synthetic applications, including consideration of the preferred electrochemical apparatus and the substrate scope and functional group compatibility.
- 16.(a) Golubev VA, Sen VD, Rozantsev ÉG. Bull Acad Sci USSR. 1979;28:1927. [Google Scholar]; (b) Fish JR, Swarts SG, Sevilla MD, Malinski T. J Phys Chem. 1988;92:3745. [Google Scholar]
- 17.For relevant examples, see ref. 12c and the following: Hamada S, Furuta T, Wada Y, Kawabata T. Angew Chem, Int Ed. 2013;52:8093. doi: 10.1002/anie.201302261.
- 18.Mercadante MA, Kelly CB, Bobbitt JM, Tilley LJ, Leadbeater NE. Nat Protoc. 2013;8:666. doi: 10.1038/nprot.2013.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.