

Abstract
The phenotypic diversity of cancer results from genetic and nongenetic factors. Most studies of cancer heterogeneity have focused on DNA alterations, as technologies for proteomic measurements in clinical specimen are currently less advanced. Here, we used a multiplexed immunofluorescence staining platform to measure the expression of 27 proteins at the single-cell level in formalin-fixed and paraffin-embedded samples from treatment-naive stage II/III human breast cancer. Unsupervised clustering of protein expression data from 638,577 tumor cells in 26 breast cancers identified 8 clusters of protein coexpression. In about one-third of breast cancers, over 95% of all neoplastic cells expressed a single protein coexpression cluster. The remaining tumors harbored tumor cells representing multiple protein coexpression clusters, either in a regional distribution or intermingled throughout the tumor. Tumor uptake of the radiotracer 18F-fluorodeoxyglucose was associated with protein expression clusters characterized by hormone receptor loss, PTEN alteration, and HER2 gene amplification. Our study demonstrates an approach to generate cellular heterogeneity metrics in routinely collected solid tumor specimens and integrate them with in vivo cancer phenotypes.
An approach to quantifying regional heterogeneity in protein expression within tumors at the single-cell level is reported.
Introduction
Next-generation DNA sequencing has begun to uncover substantial heterogeneity within a single tumor biopsy, between different disease sites from a single cancer patient, and between tumors from different patients (1–8). A deeper understanding of tumor heterogeneity and its relationship to the phenotypic diversity of human cancer will likely require a broader investigation of cancer cell “states” and the interplay of the genome, transcriptome, and proteome (9, 10).
The most widely used method to evaluate in situ protein expression in clinical tumor samples is chromogenic IHC, which detects the presence of an antigen through the use of primary monoclonal antibodies, enzyme-linked secondary antibodies, and precipitation reactions resulting in chromogen deposition. Quantification of multiple antigens in the same tissue section is challenging with this technique due to its nonlinear dynamic range and inability to generate multiple individually identifiable signals. Several recent modifications to conventional IHC have improved the quantification of antigen-antibody interactions in tissue sections. In mass spectrometry IHC (MS-IHC), the primary antibody is conjugated to a lanthanide metal, which is subsequently detected by ion mass spectrometry (11, 12). Quantitative immunofluorescence (QIF) uses fluorescent reporters and can be linked with automated quantitative analysis (13–15).
While MS-IHC and QIF both broaden the dynamic range of chromogenic IHC, their ability to simultaneously assess multiple proteins in a single cell remains limited by the number of rare earth metals for antibody tagging and the overlapping photon emission spectra of fluorophores. One potential solution to achieve higher-level multiplexing of antibodies is the use of sequential rounds of fluorescent detection in situ (16). We recently described a method that allowed for the quantification of 61 protein antigens at single-cell resolution in a single unstained slide of routinely collected formalin-fixed and paraffin-embedded (FFPE) tumor tissue (17). In the current study, we used this platform to measure the expression of 27 proteins at the single-cell level in treatment-naive invasive ductal human breast cancer, derive spatial maps of protein colocalization, and determine protein expression patterns associated with in vivo tumor uptake of the PET radiotracer 18F-fluorodeoxyglucose (18F-FDG).
Results
Selection of antibodies and validation of staining.
Our image-based method to quantify protein expression in situ is based on sequential cycles of fluorescent staining, image acquisition, and chemical dye inactivation. It uses fluorescent dye-conjugated antibodies and a dye-cycling procedure that chemically inactivates the dyes and allows them to be reused on a new set of probes (17). This enables sequential staining of FFPE tissue sections (typically 3–5 μm) with many antibodies (Figure 1).
Figure 1. Experimental design.

Immunofluorescence approach. A single 3- to 5-μm unstained section from a routinely collected formalin-fixed and paraffin-embedded (FFPE) tumor tissue block was used from each tumor for the multiplex iterative imaging cycles (n = 20). Background autofluorescence (AF) tissue images were acquired before subsequent application of fluorescent dye-conjugated primary antibodies. Stained images were then acquired, followed by dye inactivation and restaining with new directly conjugated antibodies. New images were acquired, and the cycle was repeated until all target antigens were exhausted. Stained images were registered. Background AF was removed from each stained image. Images were segmented into epithelial and stromal regions using boundaries of cytokeratin staining, followed by identification of individual cells and corresponding plasma membrane (as determined by Na+K+ATPase staining), cytoplasm (S6 staining), and nuclear regions (DAPI). Biomarker pixel-level intensity data, which were subsequently queried in data analysis, were quantified at cell level. Three different metrics per marker (mean, standard deviation, and 90% hot spot) were used, amounting to about 155 million measurements. Data analysis included K-median clustering to groups of cells based on similar biomarker intensity levels. Each field of view (FOV) was manually reviewed. Only FOVs with >90% IDC cells on histopathological review were included in the analysis.
The selection of protein antigens and antibodies (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/jci.insight.87030DS1) for our current study was based on both biologic and technical considerations. We included (a) proteins with a documented role in human breast cancer and tumor metabolism, such as the estrogen receptor (ER), progesterone receptor (PR), the HER2 receptor tyrosine kinase; (b) members of the glycolysis and hypoxia pathways; (c) members of the phosphoinositide 3-kinase (PI3K)/mTOR signaling axis; and (d) proteins that could distinguish cellular and subcellular compartments, including cytokeratins (epithelial cells), Na+-K+-ATPase (cytoplasmic membranes), S6 ribosomal protein (cytoplasmic compartment), and DAPI (nuclear compartment).
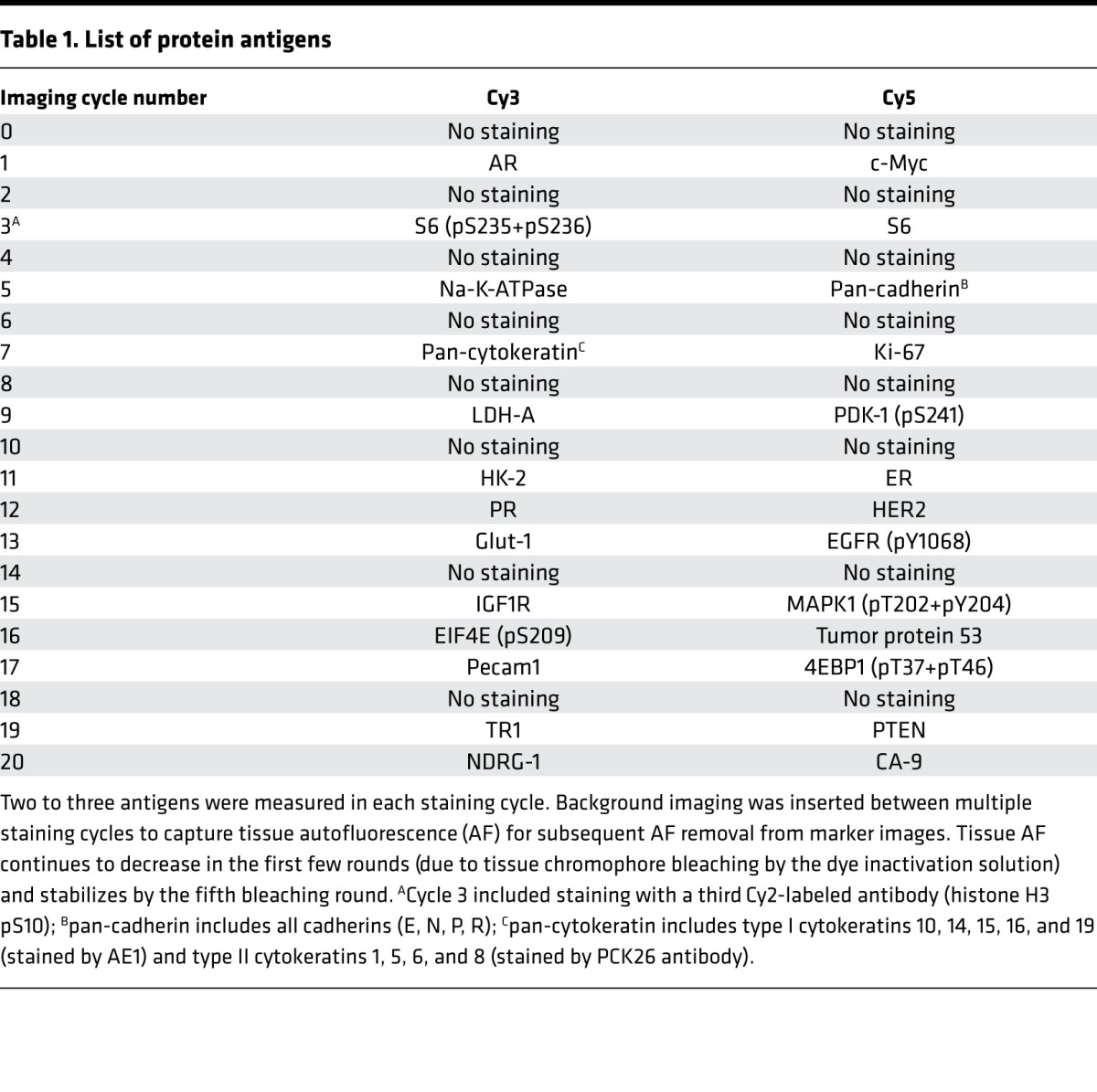
This combination of antibodies was multiplexed to be measured in a total of 20 imaging cycles (Table 1). Antigen sensitivity to the dye inactivation process was determined in preliminary experiments, and epitopes that appeared more sensitive to the effects of dye inactivation were quantified in earlier staining cycles. All samples were also stained with DAPI, a fluorescent stain that binds strongly to A-T–rich regions in DNA and labels nuclei. Images were registered using a previously described registration procedure using DAPI images from each staining round. Membrane, cytoplasm, and nuclear compartments in the tumor area were automatically segmented at the single-cell level using Na+-K+-ATPase, S6, DAPI, and cytokeratin.
Table 1. List of protein antigens.
To determine whether repeated staining and destaining might destabilize phosphoepitopes, we tested our multiplexed assay on tissue sections representing different degrees of PI3K pathway activity. These samples were generated by inoculating immunodeficient mice subcutaneously with HER2-amplified human BT-474 breast cancer cells and treating mice with a single dose of the dual PI-3K/mTOR inhibitor NVP-BEZ235 (18) 3 hours prior to tumor harvest. Three mice were treated with vehicle, three mice were treated with 10 mg/kg NVP-BEZ235, and three mice were treated with 40 mg/kg NVP-BEZ235. One FFPE section from each xenograft tumor was stained. Compared with tumors from vehicle-treated mice, tumors from mice treated with NVP-BEZ235 showed a dose-dependent decrease in phospho-eukaryotic translation initiation factor 4E-binding protein 1 (p-4EBP1, image cycle 17) and decreased staining for phospho S6 ribosomal protein (p-S6, image cycle 3). We observed no decrease in staining for total S6 ribosomal protein (Figure 2A).
Figure 2. Validation of staining approach.

(A) Quantification of phosphoepitopes. Nude mice (n = 3 per group) harboring subcutaneous BT-474 human breast cancer xenografts were treated with a single dose of the dual PI3K/mTOR kinase inhibitor NVP-BEZ235 (10 mg/kg or 40 mg/kg) or vehicle. Tumors were collected 3 hours later, routinely processed, and stained with our multiplexed immunofluorescence method. Top: representative immunofluorescence images from one tumor per cohort, p4EBP1 = red; pS6 = green; DAPI = blue. Bottom: quantification of staining results. *P < 0.05, **P < 0.01, t test, comparing each treatment group to the control group. Original magnification: ×20, zoom ×6. (B) Number of field of views (FOVs) examined for each tumor. Only FOVs with more than 90% IDC cells were included. (C) Tissue loss during successive imaging cycles shown for one representative tumor sample (tumor 072). The y axis depicts the fraction of tumor cells that remain attached to the slide in each FOV (FOVs 1–18) during each imaging cycle and can be coregistered through all cycles. Each line graph represents one FOV. (D) Tissue loss during successive imaging cycles shown for all 26 breast cancer samples. The y axis depicts the fraction of remaining cells (solid line) with a 95% confidence interval (between quantile 2.5% and 97.5%). 638,577 IDC cells that could be coregistered throughout all staining cycles were included in the subsequent proteomic analysis. (E) Correlation of multiplexed immunofluorescence staining results with clinical laboratory improvement amendments (CLIA) assays for estrogen receptor (ER) (left) and HER2 (right) from the same tumor. ER was determined by IHC, HER2 was determined by FISH. Green represents samples that were negative in the CLIA assay, and red represents samples that were positive in the CLIA assay. The cut-off for the ER CLIA assay was 1% of cells staining positive for ER. The cut-off used for CLIA HER2 FISH amplification was a HER2/CEP17 gene copy number ratio of 2.0 or greater.
We next applied the multiplexing method to FFPE tumor samples from 26 women who underwent primary surgery for stage II/III breast cancer at Memorial Sloan Kettering Cancer Center (MSKCC). All patients had locally advanced invasive ductal carcinoma (IDC) (Supplemental Table 2). A single 3- to 5-μm thick tissue section from each tumor was used, and 30 to 40 fields of view (FOVs) were placed throughout each specimen (Supplemental Figure 1). Each FOV was independently reviewed by a breast cancer pathologist (E. Brogi) to ensure the presence of invasive carcinoma. FOVs containing benign mammary glands or ductal carcinoma in situ, either in isolation or admixed with invasive carcinoma (Supplemental Figure 2), were excluded from further analysis. Eight to thirty FOVs per tumor (mean: 21.4; median: 22.5) contained at least 90% IDC cells and were included in our subsequent analysis (Figure 2B and Supplemental Table 3).
The quantification of protein coexpression patterns relies on the assumption that the majority of tumor cells complete all staining cycles and that the architecture of the tumor section remains sufficiently intact to allow image coregistration between different staining cycles. We therefore examined the extent of tissue loss/shift in each sample at the level of individual FOVs, shown for one representative sample (072, Figure 2C) and also for the entire data set (Figure 2D). Most FOVs (>80%) included the majority of cells (84.9%) after completion of all imaging cycles. About 10% of cells were removed from the analysis due to lack of image registration.
We next compared the staining intensity for ER (imaging cycle 11), PR (imaging cycle 12), and HER2 (imaging cycle 12) with the measurements of the same markers on an adjacent tissue section with validated assays used in routine clinical practice. Hormone receptor status was determined by IHC, and HER2 status was determined by IHC and FISH. These validation experiments were done without knowledge of the multiplexed staining results. For all 3 proteins, the staining distribution in our multiplexed immunofluorescence (IF) assay distinguished tumors that were considered “positive” versus “negative” (Figure 2E), as defined by established guidelines for hormone receptor and HER2 testing in breast cancer (19). Three of twenty-six samples showed divergent results for PR staining (Supplemental Figure 3). In two of these cases, the percentage of PR+ cells in the examined FOVs was 10% or fewer. Because the results from the clinical laboratory improvement amendments (CLIA) assay are based on the analysis of the entire tumor, regional heterogeneity in hormone receptor expression, which is well recognized in breast cancer (20), may have accounted for the discordant results.
Identification of protein coexpression patterns.
We next sought to identify patterns of protein coexpression across all breast cancer samples in our study. We pooled the intensity measurements for 18 proteins from all segmented IDC cells (n = 638,577) and performed K-medians clustering analysis to find common cell biomarker groupings. Segmentation markers (Na+-K+-ATPase, pan-cadherin, S6, pan-cytokeratin, CD31) and markers with weak focal or nonspecific staining (androgen receptor [AR], IGF-1 receptor [IGF1R], histone H3 pS10, and p53) were excluded from this analysis. We identified 8 distinct patterns of protein coexpression. Adding more clusters after this point did not add further resolution (Figure 3A and refs. 21, 22).
Figure 3. Protein coexpression clusters in human breast cancer.

(A) Consensus clustering was used to determine the optimum cluster set. The graph shows the relative change in area under cumulative distribution function for K = 2–15: when K increases, a positive change in the area under the curve decreases. After K = 8, the relative change is minimal and the curve flattens, suggesting that K = 8 is the best close-to-true partition. (B) Heatmap showing expression levels of each protein (log2) within the 8 clusters. IF, immunofluorescence. Rows were arranged following the lateral dendrogram. (C) Consensus hierarchical clustering of protein coexpression patterns in the TCGA breast cancer data set (747 human breast cancers). Protein expression was measured by reverse phase protein array (RPPA) using 187 antibodies. Pie charts indicate the association of each RPPA cluster with intrinsic breast cancer subtypes. Subtype annotation (PAM50) was available for 633 of 747 tumors. Numbers below each cluster indicate the number of human breast cancer samples belonging to each cluster. (D) Heatmap of the expression levels (RPPA) of the 10 proteins that were also represented in the multiplexed IF assay (see B). Genomic alterations in PTEN and HER2 are indicated above the RPPA cluster assignment.
Genome-wide RNA expression profiling of human breast cancer has characterized “intrinsic” disease subtypes (23). There are two predominantly ER+ intrinsic molecular subtypes (i.e., luminal A and luminal B) and two predominantly ER– intrinsic subtypes (i.e., HER2-enriched and basal-like) (24). Several of our protein expression clusters (Figure 3B) broadly aligned with these intrinsic breast cancer subtypes. Clusters 6 and 8, for example, were ER– and HER2– and resembled the basal-like subtype, with additional PTEN loss in cluster 8 compared with cluster 6. Cluster 7 was also hormone receptor– but showed the highest relative level of HER2 expression, consistent with the HER2-enriched subtype. Clusters 1 and 2 were hormone receptor+ and HER2– and showed low expression of the tumor cell proliferation antigen Ki-67, likely marking the luminal A subtype. The assignment of clusters 3 to 5 to a particular intrinsic breast cancer subtype was more ambiguous, perhaps reflecting the molecular heterogeneity of the luminal B breast cancer subtype (24).
To validate the patterns of protein coexpression identified in our analysis and their relationship to specific breast cancer subtypes, we turned to a data set of over 700 human breast cancer specimens that were previously analyzed by The Cancer Genome Atlas (TCGA) initiative (25). In addition to the genomic data, this data set contains reverse phase protein array (RPPA) data with 187 antibodies (26). Consensus hierarchical clustering of this group of tumors (n = 747) identified 8 clusters. Since each TCGA breast cancer sample has been assigned to one of the “intrinsic” breast cancer subtypes using a RNA-Seq–based 50-gene subtype predictor (PAM50) (27), we were able to examine the composition of each proteomic cluster by breast cancer subtype. Several clusters were enriched for a specific breast cancer subtype (Figure 3C). For example, most of the breast cancers expressing RPPA cluster 1 were basal-like cancers, whereas the majority of breast cancers expressing RPPA cluster 7 were of the HER2-enriched subtype. Luminal A and luminal B tumors appeared more heterogeneous in their patterns of protein coexpression, reminiscent of the findings from our IF-based analysis.
Ten of the eighteen protein antigens (50%) that we examined in our MultiOmyx analysis were also represented in the TCGA-RPPA analysis, including ER, PR, HER2, PTEN, phospho-EGFR (Y1068), phospho-PDK1 (S241), phospho-MAPK (T202/Y204), phospho-4EBP1 (T37/T46), c-Myc, and phospho-S6 ribosomal protein (S235/S236). We therefore examined the expression of these protein antigens in the 8 RPPA protein expression clusters (Figure 3D). Despite the differences in methodology, we identified consistent patterns. For example, RPPA cluster 1 (Figure 3D) resembled IF cluster 8 and was characterized by reduced expression of PTEN, ER, PR, HER2, phospho-EGFR (Y1068), and phospho-PDK1 (S241). RPPA cluster 7 resembled IF cluster 7 by showing elevated expression levels of HER2 and its phosphorylated coreceptor EGFR (Y1068) and reduced expression of ER and PR. In terms of genomic alterations, RPPA cluster 8 was enriched for clusters with PTEN deletion, whereas RPPA cluster 7 almost entirely consisted of breast cancers with HER2 gene amplification.
Intertumoral and intratumoral distribution of protein coexpression clusters.
Our analysis of protein expression at the single-cell level allowed us to determine the distribution of cells representing each protein expression cluster within the entire patient cohort and also within each tumor. When viewed across all of 638,577 tumor cells included in our analysis, cells representing each of the protein expression clusters were fairly evenly distributed, ranging from 7.8% (cluster 6) to 18.5% (cluster 2) of all neoplastic cells (Figure 4A).
Figure 4. Distribution of protein coexpression clusters.

(A) Relative frequency of tumor cells representing each cluster within the complete data set (638,577 IDC cells from 26 human breast cancers). (B) Intertumoral distribution of cells representing each protein coexpression cluster. Each row in the table represents one tumor sample, and each column represents one protein coexpression cluster. Tumors were sorted by the extent of intratumoral heterogeneity, with the most homogenous tumor on top (99% of tumor cells expressing cluster C6) and the most heterogenous tumor at the bottom (coexistence of cancer cells representing 5 different protein coexpression clusters, with 3 clusters [C1, C2, and C4] contributing about one-third of the tumor cells each). The number in each cell indicates the percentage of neoplastic cells expressing a particular protein coexpression cluster. The percentage of cells expressing the most abundant cluster is shown in the area graph to the right and is 50% or greater (dashed line) in most tumors. (C) Intratumoral distribution of cells representing each protein coexpression cluster. Pie charts show the distribution of protein expression clusters in each field of view. Shown are 3 representative patterns. See Supplemental Figure 5 for a complete view of all breast cancer samples. (D) FDG tumor uptake in 26 patients with invasive ductal breast cancer. FDG uptake was measured by PET and quantified as standardized uptake values (SUVmax).
Breast cancers varied considerably in their extent of intratumoral heterogeneity. The majority of breast cancers (24 of 26) contained one protein expression cluster that was expressed in at least 50% of all tumor cells. In about one-third of tumors (9 of 26), this dominant cluster represented over 95% of all tumor cells. The remaining two-thirds of breast cancers were more heterogenous and consisted of admixtures of tumor cells representing multiple protein expression clusters (Figure 4B). There was a trend toward higher intratumoral heterogeneity in ER+ tumors, but this relationship was not significant when heterogeneity was defined as either the number of clusters expressed in more than 1% of neoplastic cells (P = 0.25) or the fraction of FOVs containing cells representing 3 of more different protein expression clusters (P = 0.06) (Supplemental Figure 4).
We also examined the distribution of cancer cells representing each protein coexpression cluster within the different regions of each tumor (Supplemental Figure 5). In some tumors, for example, tumor 673 (Figure 4C), heterogeneity appeared regional. Tumor 653 showed a similarly regional pattern of protein expression, with focal expression of clusters 6 and clusters 8. This combination of protein expression clusters within the same tumor is consistent with regional loss of PTEN (Supplemental Figure 6). Other breast cancers, for example, tumor 633 (Figure 4C), harbored an admixture of cells representing different protein expression clusters within many FOVs throughout the majority of the specimen.
Relationship between protein expression and in vivo tumor retention of FDG.
All patients in our study underwent PET imaging with the radiotracer 18F-fluorodeoxyglucose (FDG) within 4 weeks prior to their breast cancer surgery. As expected for a group of patients with treatment-naive breast cancer (28), we observed a wide range in FDG tumor uptake, quantified as standardized uptake value (SUVmax) (Figure 4D).
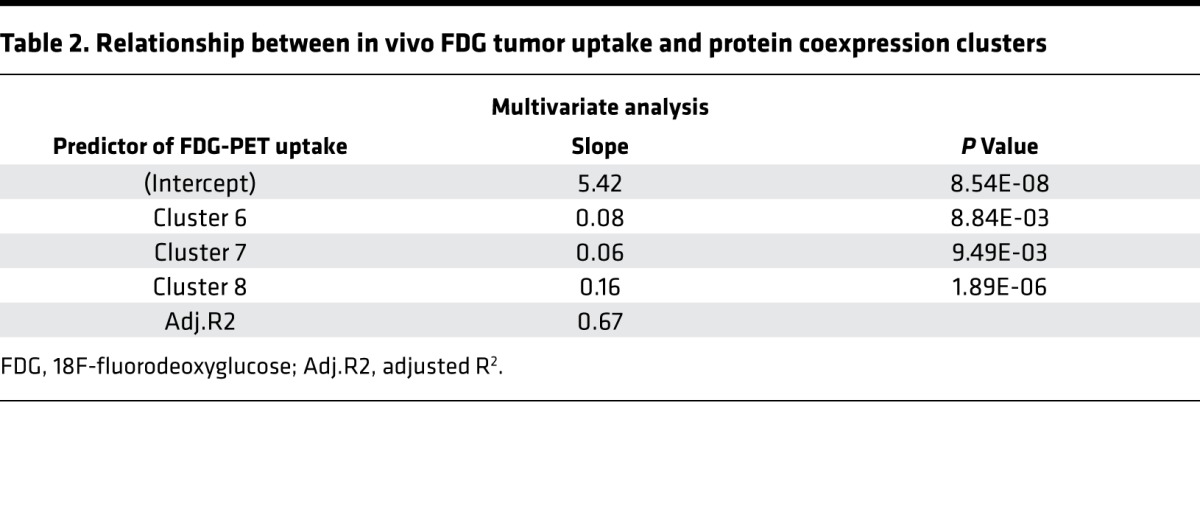
We next examined the relationship between in vivo FDG tumor uptake and the expression of each of the protein coexpression clusters identified by IF. In a multivariate analysis, expression of IF clusters 6, 7, and 8 was associated with high FDG uptake (Table 2). Perhaps surprisingly, tumors with the greatest extent of heterogeneity, defined as the fraction of FOVs containing 3 or more distinct protein expression clusters, did not show increased FDG uptake. In contrast, a linear regression model demonstrated that increased intratumoral heterogeneity correlated inversely with SUVmax (P = 0.04) in the subset of hormone receptor+ breast cancers (17 of 26) (Supplemental Figure 7).
Table 2. Relationship between in vivo FDG tumor uptake and protein coexpression clusters.
In terms of individual protein markers, high FDG uptake was associated with high expression of the proliferation marker Ki-67, low expression of ER and PR, and low expression of PTEN. High FDG uptake was also associated with intratumoral heterogeneity in expression (high standard deviation) of N-Myc downregulated gene 1 (NDRG1) (Supplemental Figure 8), perhaps representing regional differences in turnover of this tumor suppressor protein. A recent study reported that NRGD1 phosphorylation at threonine 346 targets NRGD1 for protein degradation and is associated with PTEN silencing in basal-like breast cancer (29). Consistent with this finding, we observed the comparatively lowest levels of NDRG1 protein expression (Figure 3B, IF cluster 8) and the comparatively highest levels of NDRG1 phosphorylation (threonine 346) (Figure 3D, RPPA cluster 1) in breast cancers with PTEN silencing.
Our univariate analysis also showed a positive correlation between GLUT1 expression and FDG-PET positivity, but this relationship was not statistically significant in our multivariate analysis (Supplemental Figure 8). The inconsistent relationship between GLUT1 protein expression and FDG-PET positivity has been well documented in the literature (30) and may be due to a variety of reasons, including the regulation of GLUT1 transport function through additional posttranslational mechanisms and the coexpression of other glucose transporters that mediate cellular import of the radiotracer.
Discussion
We present an approach to spatially map protein coexpression patterns in solid tumors, an unmet need in current tumor heterogeneity research (9). Several technologies have been developed to address this gap in knowledge. QIF with automated quantitative analysis provides a linear dynamic range and is clinically offered by Genoptix for prediction of recurrence risk in ER+ patients using a combination of ER, PR, Ki-67, and HER2 (31). QIF can simultaneously assess up to 10 proteins in a single cell with the use of multispectral fluorescence imaging (32). However, as the number of protein targets increases, overlapping dye emission spectra and the number of distinct animal species that are needed for each primary-secondary antibody combination become challenging. Labeled-mass spectrometry imaging (33) has the potential to provide simultaneous quantitative analysis of up to 100 proteins using lanthanide metal isotopes. These isotopes are not found in normal tissue, eliminating problems with tissue background signal and resulting in high sensitivity. However, this technology is limited by the availability of metal tags (only ~32) and validated antibody conjugates (34). Furthermore, the latter approach requires sample ablation for isotope release, rendering samples unsuitable for additional downstream analysis such as FISH. Advantages of MultiOmyx are that single-cell analysis of upward of 60 proteins is possible and tissue is kept intact.
Our examination of more than 600,000 breast cancer cells allowed us to identify distinct clusters of protein coexpression in breast cancer and examine their distribution within each tumor and between different tumors. For most proteins, the fold difference in expression between clusters was only modest (less than 2-fold). This might be explained by the limited dynamic range of antibody-based protein detection or could represent the true extent of differential protein expression. The latter conclusion seems more likely based on a recent proteomic comparison of different breast cancer subtypes using a modified stable isotope labeling with amino acids in cell culture approach (35). Together, these findings suggest that extensive protein measurements and robust bioinformatic approaches will be required to characterize the cancer proteome and its associated phenotypes in human cancer samples.
Our multiregion analysis suggests that a substantial fraction of treatment-naive human breast cancers show only modest intratumoral heterogeneity, with about one-third of all tumors expressing a dominant protein coexpression pattern in ≥95% of all malignant cells. In a prior study, 4 of 6 breast cancers showed remarkably similar genetic profiles across even morphologically distinct areas of each case (36). Another study reported a single clonal subpopulation in 7 of 16 examined human breast cancers (37). More recent multiregion sequencing failed to detect significant differences in mutations between different tumor regions in 23 of 50 (46%) breast cancers (5). Together, these findings suggest that at least a subset of treatment-naive human breast cancers may be considerably less heterogeneous than other human cancers submitted to similarly detailed analyses. In the subgroup of breast cancers with more extensive intratumoral heterogeneity, we identified two distinct spatial patterns. Some tumors showed geographically constrained expansion of subclones, whereas others showed intermingling of subclones in multiple areas, again consistent with recent genomic studies (5, 38). Differences in the extent and pattern of intratumoral heterogeneity might reflect differences in motility between distinct cancer cell populations (39), a hypothesis that remains to be tested. Taken together, the convergence of our proteomic results with recent genomic evaluations of intratumoral heterogeneity in human breast cancer supports the hypothesis (9) that there are fewer distinct cell states in a tumor than the degree of genetic, epigenetic, and transcriptional heterogeneity might suggest. Further studies are warranted to determine to what extent current breast cancer therapies alter the patterns of intratumoral heterogeneity and promote the acquisition of drug resistance (40).
Increased retention of the radiotracer FDG in breast cancer has been associated with loss of hormone receptor expression, increased HER2 expression, and the basal-like intrinsic breast cancer subtype (28, 41, 42). Silencing of the PTEN tumor suppressor has also been associated with increased glycolytic activity (43, 44) and is more common in basal-like breast cancer (45). The relationship between FDG-PET positivity and PI3K pathway activation in breast cancer warrants further study. Our current study showed that FDG-PET positivity is highest in tumors with hormone receptor loss and loss of PTEN, a negative regulator of the PI3K pathway. However, hormone receptor+ tumors, which harbor activating mutations in the catalytic domain of PI3K more often than other types of breast cancers, generally showed lower FDG-PET positivity, and we did not observe a significant correlation between FDG uptake and phosphorylation of the PI3K pathway members p-S6, p-eIF4E, or p-4EBP1. Our conclusion that protein clusters 6, 7, and 8 are markedly associated with increased FDG uptake emphasizes the prominence of glycolytic metabolism in ER– breast cancers. ER+ tumors show a wider range in FDG uptake, and high FDG uptake has been associated with worse clinical outcomes in this subgroup (28). The inverse relationship between intratumoral heterogeneity and FDG uptake in the subgroup of ER+ tumors suggests that heterogeneity per se may not be an indication of aggressive tumor biology, at least when measured at the level of the proteome. In contrast, it is intriguing to speculate that the coexistence of multiple tumor cell states and the absence of a dominant “clone” might be a reflection of less aggressive tumor growth.
While our methodology remains vulnerable to the general limitations of antibody-based protein quantification and observer-dependent selection of protein markers, the work presented here provides a template to interrogate the spatial distribution of cell populations and signaling networks in routinely collected tumor biopsies and link these findings with clinically relevant cancer phenotypes.
Methods
Patient selection and PET image acquisition
Our study included 26 patients who presented for operative management of primary breast carcinoma and were imaged with FDG-PET within 4 weeks prior to surgery. All patients had locally advanced disease, were treatment naive, and had a histopathological diagnosis of invasive ductal breast cancer (Supplemental Table 2). All patients fasted for at least 6 hours prior to 18F-FDG-PET imaging, and blood glucose levels were obtained before examination. Patients were injected with 370 to 555 MBq (10–15 mCi) pyrogen-free 18F-FDG, and imaging was performed 50 to 60 minutes later on an ADVANCE (General Electric Medical Systems) whole-body PET/CT scanner in accordance with the MSKCC PET protocol. A standard ROI analysis tool provided with the scanner was used to calculate the maximal FDG concentration within the primary tumor mass. SUVmax values were obtained by correcting for the injected dose and patient weight, again using the standard software tools. Only FDG uptake in the primary site was analyzed.
Tissue specimens
All tissue samples were obtained from surgically resected tissue and routinely processed and embedded in paraffin in the anatomic pathology laboratory at MSKCC. FFPE 3- to 5-micron-thick tumor tissue sections were stained with H&E and reviewed by a breast cancer pathologist (E. Brogi), and 25 to 30 FOVs were placed in each tumor for further analysis. Following the initial selection, FOVs containing predominantly benign breast parenchyma or ductal carcinoma in situ or showing invasive carcinoma intermingling with either were excluded from the analysis. Sections with >90% of IDC cells were subjected to analysis (Supplemental Figure 2 and Supplemental Table 3). A total of 8 to 30 FOVs (IDC only) per tumor were used for data analysis (Figure 2B). This represented on average about 35,000 neoplastic epithelial cells per carcinoma.
Reagents and cell lines
For each target antigen, we evaluated multiple clones of primary antibodies for sensitivity and specificity. Clones with the best performance characteristics were conjugated, compared with the unconjugated antibody, and then used for multiplex staining (Supplemental Table 1). The targeted epitope was also tested for stability following the signal inactivation as described previously (17) and summarized below. BT-474 cells were obtained from the ATCC. Subcutaneous human breast cancer xenografts were established by hind limb inoculation of nu/nu athymic mice with 1 × 106 BT-474 human breast cancer cells. When the mean tumor volume of the cohort reached 400 to 500 mm3, tumor-bearing mice (n = 3 per group) received a single dose of NVP-BEZ235 (10 and 30 mg/kg or vehicle) and were sacrificed 3 hours later. Tumors were excised, fixed in 4% paraformaldehyde, transferred to 70% ethanol, and stored at 4°C until embedding.
Image acquisition, registration, segmentation, and normalization
FFPE tissue sections were stained and imaged using the General Electric multiplex fluorescence microscopy platform. One tissue section was used for each tumor and underwent repeated cycles of staining, imaging, and signal removal. Prior to blocking and staining with antibody conjugates, samples were deparaffinized, hydrated, and processed through a 2-step antigen retrieval process (Michael J. Gerdes, Anup Sood, and Christopher J. Sevinsky, US patent 8067241 B2). Prior to multiplex staining and imaging, each antigen epitope was evaluated for its stability toward our dye inactivation solution. This evaluation involved exposure of serial sections to 1, 5, and 10 rounds of exposure to inactivation solution followed by staining of the target. The intensity and specificity of signal were compared with another serial section without exposure to the inactivation solution. This process identified both inactivation solution-sensitive and -insensitive antigens, with examples of both reported previously (17). For example, the expression level of S6 ribosomal protein showed a decrease in signal within increasing numbers of dye inactivation rounds, whereas phospho-4EBP1 (T37/T46) was not affected (Supplemental Figure 9).
Slides were imaged with Olympus IX-81 microscopes outfitted with internally developed software for repetitive imaging of the same FOV. DAPI was used for image alignment between different rounds of imaging prior to further processing for autofluorescence removal, image segmentation, and quantification of marker expression at subcellular level. Image alignment with DAPI minimized the effects of tissue movement and/or tissue loss between staining rounds. Only cells with perfect (100%) alignment with cells in round 0 were included in the analysis.
Staining quality was assessed manually as well as semiautomatically. Manual assessment was performed by visualizing staining patterns of individual markers across all samples. Segmentation markers (Na+-K+-ATPase, pan-cadherin, pan-cytokeratin, S6, CD31) and markers with technical issues (e.g., weak focal or nonspecific staining [AR, IGF1R, histone H3 pS10, p53]) were excluded from analysis. Semiautomatic analysis was used to identify high-intensity artifacts. An ImageJ (NIH) macro was developed to flag top 50 high-intensity objects in images of each marker. These objects were visually assessed for true or artifactual staining until the highest intensity object was positively identified as a true stain. If all 50 objects were found to be artifacts, the next 50 objects were selected for visual assessment.
Segmented images were visually assessed and compared with images of segmentation markers (e.g., pan-cytokeratin, S6, and Na+-K+-ATPase) and virtual H&E generated from a combination of pan-cytokeratin, tissue autofluorescence in the GFP channel, and DAPI. Images with failed segmentation due to weak staining or failed registration of one or more segmentation markers were excluded from the analysis. To minimize effects due to “oversegmentation” only cells with at least 10 pixels per compartment (membrane, cytoplasm, and nucleus) and no more than 2 nuclei were included.
Since illumination across the FOV is nonuniform and multiple microscopes were used, we included commercially available fluorescent beads and in-house prepared dye-impregnated gels to normalize intensity across microscopes and field flattening across the individual FOVs. Calibration standards were imaged every day prior to marker imaging to create calibration files that were used during image processing and quantification. All the slides for all the biomarkers were adjusted to a common exposure time per channel. Once the single-cell data was quality controlled, normalized, and transformed, each biomarker was scored for each patient (i.e., slide). Data analysis was limited to regions of the tumors representing IDC, representing on average about 35,000 epithelial carcinoma cells per tumor.
Statistics
Quantification of staining intensity.
Once the single-cell data was quality controlled, normalized, and transformed, each biomarker needed to be scored for each patient (slide). Three metrics of the cell distribution were used to score each protein marker in each patient. (a) The mean of the distribution metric (Fmk) represents the distribution of cell intensities for all the cells for biomarker m of patient k. The average cell intensity was used for patient k of biomarker m, which represents the central location of the distribution. (b) For the standard deviation of the distribution metric, a standard deviation represented the variability of a distribution. For each patient, each biomarker was scored using the sample standard deviation. (c) For the 90% hot spot metric, a threshold was chosen at the 90% quantile of all the cells from the entire cohort per biomarker. The proportion of cells exceeding this threshold was calculated for every patient per biomarker and was named the 90% hot spot metric. This metric describes the allocation of top 10% brightest cells in the whole cohort to each patient.
To document the concordance of our IF staining results with the determination of ER, PR, and HER2 using CLIA-certified laboratory assays on adjacent tissue sections, we graphed histograms of each marker (ER, PR, and HER2) on a single graph and color-coded it according to CLIA results for the same sample.
Identification of protein expression clusters.
Unsupervised clustering was performed using 18 markers after exclusion of segmentation markers and markers with poor staining. A single metric, median cell intensity within the compartment of interest, was used for clustering. Since marker expression can vary significantly between markers, the median cell values were standardized by the overall marker mean and standard deviation. In total, 638,577 cells remained after quality control standards were applied. Cells were clustered into K groups based on the 18 dimensional marker space using K-medians clustering on all the cells. The stepFlexclust function of flexclust library (v. 1.3-3) for R (v. 2.15.0) was run with 20 replicates assuming K ranged between 2 and 15. The K-medians clustering algorithm uses Manhattan distances between cells and then partitions cells into K groups and calculates the median for each cluster to determine its centroid. For each K, the initial centroids are randomly chosen and the minimum within cluster distance solution is returned after 20 replicated runs.
For a given K, each tumor sample can be scored according to the proportion of cells belonging to one of the K clusters. For instance, if K = 3, 3 scores can be derived for each sample, the proportion of total cells belonging to cluster 1, to cluster 2, and to cluster 3. All the K-1 variables can then be associated with FDG uptake individually or collectively. 10-fold cross validation was utilized to assess the predictive accuracy of the K-1 variable model. The whole process was iterated for all the K values, ranging from 2 to 15, in order to find the best number of clusters for the data. Cluster sets K = 7 and 8 provided the highest correlation to FDG uptake, and the decision was made to use the 8 cluster set. Each of the 8 clusters was associated with FDG uptake using a univariate analysis approach to calculate the coefficients and the P values. All of the clusters were also fed together into the multivariate analysis pipeline.
Determine correlations with FDG uptake.
Several analyses were undertaken. (a) In the univariate analysis to correlate biomarker metrics with FDG uptake, FDG uptake was measured by SUVmax, which has a continuous range from 0 to 22.1 in our cohort. A linear regression (in R) was used to assess the association between each biomarker metric and FDG uptake individually. The coefficients and P values for the top metrics are reported in Supplemental Figure 9. (b) In the multivariate analysis, automatic least absolute shrinkage and selection operator (lasso) selection was conducted to build a multivariate model associated with FDG uptake. Only the metrics with P values of less than 0.05 from univariate analysis were considered in building up the multivariate models. Since multiple metrics for the same biomarker can appear highly correlated, only the top metric of a marker was selected into multivariate model building process. Lasso puts L1 constraints on the regression coefficients, which shrinks the coefficients toward 0, using the following formula: Σ |βj| < s, where β is the regression coefficient for each j and j is the jth predictor in the multivariate regression model. The bound s is a tuning parameter. When s is large enough, the constraint has no effect and the solution is the standard multiple linear least squares regression. However, for smaller values of s (s > 0) the solutions are shrunken versions of the least squares estimates. Often, some of the coefficients βj are 0. The predictors whose regression coefficient β is 0 will automatically be excluded. So choosing s is like choosing the number of predictors to use in a regression model, and cross-validation is a good tool for estimating the best value for s. When the tuning parameters vary, some of the solution coefficients were exactly 0. This makes lasso a model selection tool for achieving parsimony. The function glmnet in R was used to fit a lasso-regularized linear model. The built-in cross validation selects λ at 1 standard error away from minimum mean squared error. In this automatic selection process, we did not confine the number of markers in the model, but only fed the main effect of each metric into the selection pool. No interactions between markers were assessed. The coefficients for ER and PR stayed at non-zero after the optimal λ was applied. These markers were then fed into a linear regression model to produce the unbiased coefficient estimate and P values. A leave-one-out cross validation was then applied to the selected markers. Not surprisingly, the predicted SUVmax, using the (n – 1) samples, was highly correlated with the observed SUVmax. (c) Using correlation between FDG uptake and extent of tumor heterogeneity, the 8 clusters were not equally distributed among all the 26 tumor samples. In order to assess the degree of intratumoral heterogeneity of each tumor a score was determined. This was done by calculating the number of clusters that are present in each FOV (a 1% cut-off was applied so that a cluster was only counted as being present if >1% of total cells in the sample belonged to that cluster). Following this, tumors were scored according to the number of FOVs that were “homogenous” (<3 clusters) or “heterogeneous” (≥3 clusters). These counts were normalized by the number of FOVs sampled per tumor. Next, a linear regression model was applied to assess whether there was a correlation between heterogeneity and FDG uptake and evaluated for its correlation to ER positivity.
Study approval
The study was approved by the Institutional Review Board of MSKCC (protocol 06-107), and all participating patients signed informed consent prior to inclusion in the study. All animal experiments were approved by the MSKCC Institutional Animal Care and Use Committee (protocol 08-07-012).
Author contributions
A Sood, AMM, EB, FG, SML, and IKM designed the study. A Sood, YS, EM, ASP, ZP, SC, and QL performed research. A Sood, FG, YS, EM, A Stamper, CC, EP, TGG, and ZP contributed research material. A Sood, FG, JA, NS, and YS contributed to data analysis and interpretation. A Sood, AMM, EB, YS, FG, SML, and IKM wrote the paper.
Supplementary Material
Acknowledgments
We thank all members of the Mellinghoff laboratory for helpful suggestions. We would also like to acknowledge contributions of the following General Electric employees: Sireesha Kaanumalle, for coordinating antibody conjugation and validation efforts; Alexander Bordwell, Eric Williams, and Sarah Zhang, for antibody conjugation and validation; Vidya Kamath, for the development of visualization tools; and Christopher Sevinsky, for biological insights into tumor biology. This research was supported by grants from the NIH (1R01NS080944-01, P50-CA86438, P30-CA08748) and the National Brain Tumor Society (to I.K. Mellinghoff). Technical services provided by the Memorial Sloan Kettering Small-Animal Imaging Core Facility were supported in part by NIH grants R24-CA83084 and P50-CA92629. Parts of the study were supported by a research grant from GE Healthcare and GE Global Research Center (to S.M. Larson and I.K. Mellinghoff).
Footnotes
Elisa Port’s present address is: Department of Surgery, Mount Sinai Hospital, New York, New York, USA.
Conflict of interest: All authors affiliated with the GE Global Research Center are current employees of General Electric Company.
Reference information:JCI Insight. 2016;1(6):e87030. doi:10.1172/jci.insight.87030.
Contributor Information
Anup Sood, Email: anup.sood@ge.com.
Alexandra M. Miller, Email: millera2@mskcc.org.
Edi Brogi, Email: brogie@MSKCC.ORG.
Yunxia Sui, Email: suiy@ge.com.
Joshua Armenia, Email: armenia@cbio.mskcc.org.
Elizabeth McDonough, Email: elizabeth.mcdonough@ge.com.
Alberto Santamaria-Pang, Email: santamar@ge.com.
Sean Carlin, Email: carlins@MSKCC.ORG.
Aleksandra Stamper, Email: astamper@smith.edu.
Carl Campos, Email: camposc@mskcc.org.
Zhengyu Pang, Email: pang@ge.com.
Qing Li, Email: liq@ge.com.
Elisa Port, Email: Elisa.port@mountsinai.org.
Nikolaus Schultz, Email: schultzn@mskcc.org.
Fiona Ginty, Email: ginty@research.ge.com.
Ingo K. Mellinghoff, Email: mellingi@mskcc.org.
References
- 1.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 2.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerlinger M, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46(3):225–233. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014;346(6206):256–259. doi: 10.1126/science.1256930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yates LR, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boutros PC, et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat Genet. 2015;47(7):736–745. doi: 10.1038/ng.3315. [DOI] [PubMed] [Google Scholar]
- 7.Izumchenko E, et al. Targeted sequencing reveals clonal genetic changes in the progression of early lung neoplasms and paired circulating DNA. Nat Commun. 2015;6:e87030. doi: 10.1038/ncomms9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murugaesu N, et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015;5(8):821–831. doi: 10.1158/2159-8290.CD-15-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alizadeh AA, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8):846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabassum DP, Polyak K. Tumorigenesis: it takes a village. Nat Rev Cancer. 2015;15(8):473–483. doi: 10.1038/nrc3971. [DOI] [PubMed] [Google Scholar]
- 11.Angelo M, et al. Multiplexed ion beam imaging of human breast tumors. Nat Med. 2014;20(4):436–442. doi: 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giesen C, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11(4):417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 13.Wang HA, et al. Fast chemical imaging at high spatial resolution by laser ablation inductively coupled plasma mass spectrometry. Anal Chem. 2013;85(21):10107–10116. doi: 10.1021/ac400996x. [DOI] [PubMed] [Google Scholar]
- 14.Rimm DL. Next-gen immunohistochemistry. Nat Methods. 2014;11(4):381–383. doi: 10.1038/nmeth.2896. [DOI] [PubMed] [Google Scholar]
- 15.Carvajal-Hausdorf DE, Schalper KA, Neumeister VM, Rimm DL. Quantitative measurement of cancer tissue biomarkers in the lab and in the clinic. Lab Invest. 2015;95(4):385–396. doi: 10.1038/labinvest.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schubert W, et al. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat Biotechnol. 2006;24(10):1270–1278. doi: 10.1038/nbt1250. [DOI] [PubMed] [Google Scholar]
- 17.Gerdes MJ, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A. 2013;110(29):11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maira SM, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 19.Hammond ME, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer (unabridged version) Arch Pathol Lab Med. 2010;134(7):e48–e72. doi: 10.5858/134.7.e48. [DOI] [PubMed] [Google Scholar]
- 20.Zardavas D, Irrthum A, Swanton C, Piccart M. Clinical management of breast cancer heterogeneity. Nat Rev Clin Oncol. 2015;12(7):381–394. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 21.Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach Learn. 2003;52(1):91–118. [Google Scholar]
- 22.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 24.Anderson WF, Rosenberg PS, Prat A, Perou CM, Sherman ME. How many etiological subtypes of breast cancer: two, three, four, or more? J Natl Cancer Inst. 2014;106(8):e87030. doi: 10.1093/jnci/dju165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parker JS, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009;27(8):1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn SG, et al. Standardized uptake value of (1)(8)F-fluorodeoxyglucose positron emission tomography for prediction of tumor recurrence in breast cancer beyond tumor burden. Breast Cancer Res. 2014;16(6):e87030. doi: 10.1186/s13058-014-0502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell. 2014;56(4):595–607. doi: 10.1016/j.molcel.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avril N. GLUT1 expression in tissue and (18)F-FDG uptake. J Nucl Med. 2004;45(6):930–932. [PubMed] [Google Scholar]
- 31.Cuzick J, et al. Prognostic value of a combined estrogen receptor, progesterone receptor, Ki-67, and human epidermal growth factor receptor 2 immunohistochemical score and comparison with the Genomic Health recurrence score in early breast cancer. J Clin Oncol. 2011;29(32):4273–4278. doi: 10.1200/JCO.2010.31.2835. [DOI] [PubMed] [Google Scholar]
- 32.Levenson RM, Mansfield JR. Multispectral imaging in biology and medicine: slices of life. Cytometry A. 2006;69(8):748–758. doi: 10.1002/cyto.a.20319. [DOI] [PubMed] [Google Scholar]
- 33.Camp RL, Chung GG, Rimm DL. Automated subcellular localization and quantification of protein expression in tissue microarrays. Nat Med. 2002;8(11):1323–1327. doi: 10.1038/nm791. [DOI] [PubMed] [Google Scholar]
- 34.Levenson RM, Borowsky AD, Angelo M. Immunohistochemistry and mass spectrometry for highly multiplexed cellular molecular imaging. Lab Invest. 2015;95(4):397–405. doi: 10.1038/labinvest.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyanova S, Albrechtsen R, Kronqvist P, Cox J, Mann M, Geiger T. Proteomic maps of breast cancer subtypes. Nat Commun. 2016;7:e87030. doi: 10.1038/ncomms10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geyer FC, et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J Pathol. 2010;220(5):562–573. doi: 10.1002/path.2675. [DOI] [PubMed] [Google Scholar]
- 37.Navin N, et al. Inferring tumor progression from genomic heterogeneity. Genome Res. 2010;20(1):68–80. doi: 10.1101/gr.099622.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Navin N, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472(7341):90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525(7568):261–264. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janiszewska M, et al. In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet. 2015;47(10):1212–1219. doi: 10.1038/ng.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basu S, et al. Comparison of triple-negative and estrogen receptor-positive/progesterone receptor-positive/HER2-negative breast carcinoma using quantitative fluorine-18 fluorodeoxyglucose/positron emission tomography imaging parameters: a potentially useful method for disease characterization. Cancer. 2008;112(5):995–1000. doi: 10.1002/cncr.23226. [DOI] [PubMed] [Google Scholar]
- 42.Palaskas N, et al. 18F-fluorodeoxy-glucose positron emission tomography marks MYC-overexpressing human basal-like breast cancers. Cancer Res. 2011;71(15):5164–5174. doi: 10.1158/0008-5472.CAN-10-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gregorian C, et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci U S A. 2009;106(46):19479–19484. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia-Cao I, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149(1):49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saal LH, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat Genet. 2008;40(1):102–107. doi: 10.1038/ng.2007.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.