

Abstract:
Endothelium-dependent relaxations are predominantly regulated by nitric oxide (NO) in large conduit arteries and by endothelium-dependent hyperpolarization (EDH) in small resistance vessels. Although the nature of EDH factors varies depending on species and vascular beds, we have previously demonstrated that endothelial NO synthases (eNOS)-derived hydrogen peroxide (H2O2) is an EDH factor in animals and humans. This vessel size-dependent contribution of NO and EDH is, at least in part, attributable to the diverse roles of endothelial NOSs system; in large conduit arteries, eNOS mainly serves as a NO-generating system to elicit soluble guanylate cyclase–cyclic guanosine monophosphate-mediated relaxations, whereas in small resistance vessels, it serves as a superoxide-generating system to cause EDH/H2O2-mediated relaxations. Endothelial caveolin-1 may play an important role for the diverse roles of NOSs. Although reactive oxygen species are generally regarded harmful, the physiological roles of H2O2 have attracted much attention as accumulating evidence has shown that endothelium-derived H2O2 contributes to cardiovascular homeostasis. The diverse functions of endothelial NOSs system with NO and EDH/H2O2 could account for a compensatory mechanism in the setting of endothelial dysfunction. In this review, we will briefly summarize the current knowledge on the diverse functions of endothelial NOSs system: NO and EDH/H2O2.
Key Words: endothelium-derived relaxing factor, endothelium-dependent hyperpolarization, hydrogen peroxide, nitric oxide, nitric oxide synthase
INTRODUCTION
The endothelium plays a crucial role in modulating vascular tone by synthesizing and releasing endothelium-derived relaxing factors, including vasodilator prostaglandins (PGs), nitric oxide (NO), and endothelium-dependent hyperpolarization (EDH) factors and also endothelium-derived contracting factors (Fig. 1).1–4 In 1988, Feletou and Vanhoutte5 and Chen et al6 independently demonstrated that a diffusible substance released by the endothelium causes hyperpolarization and relaxation of underlying vascular smooth muscle cells (VSMC), attributing to the existence of putative EDH factors. Since then, a quarter century has passed and now several candidates have been proposed for the nature of EDH factors (Fig. 1).7 It is widely accepted that the nature of EDH factors varies depending on species and vascular beds examined, including epoxyeicosatrienoic acids (EETs), metabolites of arachidonic P450 epoxygenase pathway,8,9 electrical communication through gap junctions,10 K+ ions,11 hydrogen sulfide (H2S),12 and as we have previously identified, hydrogen peroxide (H2O2) (Fig. 1).13 EETs mainly take part in EDH-mediated relaxations in bovine, porcine, canine and human large coronary arteries, gap junctions in rodents, rabbit, and human mesenteric arteries, K+ ions in rat hepatic and mesenteric arteries, porcine and canine coronary arteries and human arteries, H2S in mouse mesenteric arteries, and H2O2 in various vascular beds as described below.2 Intriguingly, the contribution of endothelium-derived relaxing factors (vasodilator PGs, NO, and EDH) to endothelium-dependent vasodilatation markedly varies depending on blood vessel size with the physiological balance between NO and EDH; vasodilator PGs play a small but constant role, and NO predominantly modulates the tone of large conduit vessels, and the contribution of NO decreases as the vessel size decreases, whereas that of EDH increases as the vessel size decreases, which phenomenon is well preserved from rodents to humans.14–16 Thus, EDH rather than NO plays a dominant role in small resistance vessels where blood pressure and organ perfusion are finely regulated. Indeed, accumulating evidence has demonstrated the critical roles of EDH in modulating blood pressure17 and vascular metabolic functions18 in general, and coronary autoregulation19 and coronary metabolic dilatation20 in particular. We have previously demonstrated the diverse roles of the NO synthases (NOSs) system in the endothelium depending on blood vessel size; NOS mainly serves as a NO-generating system to elicit soluble guanylate cyclase (sGC)–cyclic guanosine monophosphate (cGMP)-mediated relaxations in large conduit vessels and a superoxide-generating system to cause EDH/H2O2-mediated responses in small resistance vessels (Fig. 2).21 In the clinical settings, it has been reported that chronic nitrate therapy could exert harmful effects in patients with myocardial infarction,22 and that antioxidant supplements are ineffective to prevent cardiovascular events.23 These lines of evidence suggest the potential importance of the physiological balance between NO and EDH/H2O2 through the diverse functions of endothelial NOSs system.
FIGURE 1.
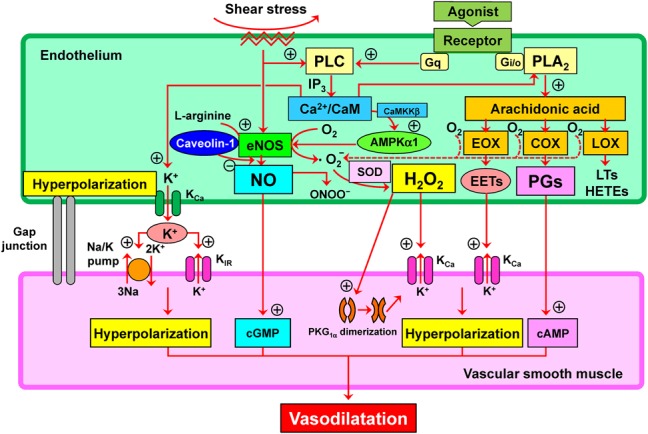
Mechanisms for synthesis and action of endothelium-derived relaxing factors in addition to vasodilator PGs and NO; several candidates could act as endothelium-dependent hyperpolarization (EDH) factor. PGs, NO, and EDH factor cause relaxations of underlying vascular smooth muscle through the mechanisms mediated by cyclic AMP (cAMP), cyclic GMP (cGMP), and hyperpolarization mediated by opening of Ca-activated K (KCa) channels. AMPKα1, α1-subunit of AMP-activated protein kinase; CaM, calmodulin; CaMKKβ, Ca2+/CaM-dependent protein kinase β; COX, cyclooxygenase; EETs, epoxyeicosatrienoic acids; eNOS, endothelial NO synthase; EOX, epoxygenase; HETEs, hydroxyeicosatetraenoic acids; H2O2, hydrogen peroxide; IP3, inositol trisphosphate; LOX, lipoxygenase; LTs, leukotrienes; ONOO−, peroxynitrite; PKG1α, 1α-subunit of protein kinase G; PLA2, phospholipase A2; PLC, phospholipase C.
FIGURE 2.
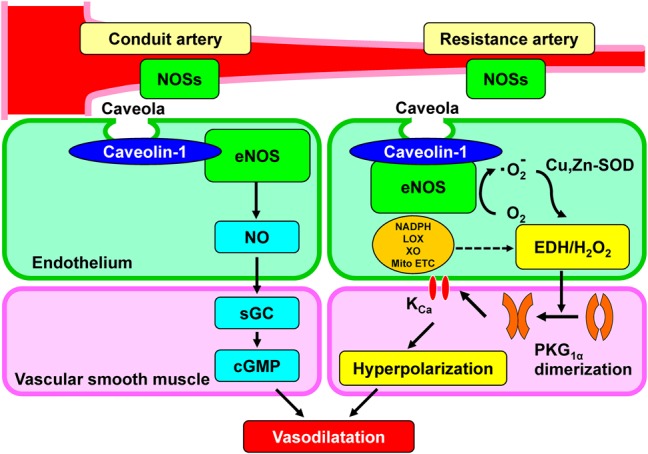
Diverse roles of endothelial nitric oxide synthases system in large conduit vessels, NO synthases (NOSs) mainly serve as a NO-generating system to cause vasodilatation through sGC–cGMP pathway, whereas in small resistance vessels, they act as a superoxide-generating system to evoke EDH-mediated responses through H2O2-induced PKG1α dimerization and subsequent activation of potassium channels, leading to hyperpolarization and vasodilatation. KCa, calcium-activated potassium channel; LOX, lipoxygenase; Mito ETC, mitochondrial electron transport chain; NADPH, reduced nicotinamide adenine dinucleotide phosphate oxidase; ONOO−, peroxynitrite; PKG1α, 1α-subunit of protein kinase G; XO, xanthine oxidase.
In this review, we will briefly summarize the current knowledge on the diverse functions of endothelial NOSs system: NO and EDH/H2O2. As the former is extensively reviewed recently3 and the space allowed for this article is limited, we will put more focus on the latter with particular reference to clinical implication.
Identification of EDH/H2O2
After the original reports on EDH factors in 1988,5,6 3 sets of early notions and observations suggesting the similarities between NO and EDH led us to hypothesize that a putative EDH factor could be a non-NO vasodilator substance [possibly reactive oxygen species (ROS)] derived from endothelial NOSs system. First, not only NO-mediated but also EDH-mediated responses are susceptible to vascular injuries caused by various atherosclerotic factors, such as aging, hypertension, diabetes mellitus, and dyslipidemia, with a resultant microvascular dysfunction, and conversely, the treatment of those risk factors restores both NO-mediated and EDH-mediated relaxations.15,24 Second, calcium/calmodulin is required for the generation of both NO by eNOS and EDH-mediated responses.25 Third, it is reasonable to consider that endothelial cells use a simple molecule (like NO) rather than complex substances in controlling and adjusting vascular tone in a moment-to-moment manner in response to various physiological demands. In 2000, using eNOS-knockout (eNOS-KO) mice, we identified that endothelium-derived H2O2 is an EDH factor in mouse mesenteric arteries13; EDH-mediated relaxation and hyperpolarization of underlying VSMC were inhibited by catalase, a specific H2O2 inhibitor, in small mesenteric arteries from wild-type mice and were significantly reduced in eNOS-KO mice.13 This is also the case in human mesenteric26 and coronary27 arteries, porcine coronary arteries,28 canine coronary arteries,19,20 and piglet pial arterioles,29 although EDH-independent vasodilating mechanisms by H2O2 have also been reported in other vascular beds.30,31 Notably, the estimated concentration of endothelium-derived H2O2 as an EDH factor is in micromolar order,28,32 which is a much lower concentration than that observed in various pathological conditions.33,34 Among the possible sources and mechanisms involved in the generation of H2O2 in the endothelium,34,35 Cu–Zn superoxide dismutase (SOD) plays a key role in the synthesis of EDH/H2O2; eNOS produces superoxide anions when synthesizing NO from l-arginine and oxygen under physiological conditions, whereas Cu–Zn-SOD dismutates those superoxide anions into H2O2, and Cu–Zn-SOD-KO mice show markedly impaired EDH-mediated relaxation and hyperpolarization in mesenteric arteries and coronary circulation without alterations in vasodilator properties of VSMC.36 Based on the observations that the EDH/H2O2-mediated responses are resistant to NOS inhibitors and upregulation of eNOS cofactor tetrahydrobiopterin has no effects on the responses, superoxide anions relevant to EDH/H2O2 are not derived from pathologically uncoupled eNOS.37 This is the case at least in normal mouse mesenteric arteries.37 Other sources of superoxide anions have been proposed in H2O2-mediated vasodilatation; in human coronary arterioles, mitochondrial respiratory chain-derived and nicotinamide adenine dinucleotide phosphate oxidase-derived H2O2 is associated with flow-mediated dilation and bradykinin-induced relaxation, respectively.38,39 To date, several mechanisms have been proposed for H2O2-induced vasodilatation.40 Among them, Burgoyne et al41 demonstrated a precise mechanism by which EDH/H2O2 relaxes underlying VSMC. Briefly, H2O2 induces an interprotein disulfide formation between two 1α isoforms of cGMP-dependent protein kinases (PKG1α) to enhance the kinase activity through their phosphorylation. The activated PKG1α subsequently stimulates potassium channels, leading to hyperpolarization and vasodilatation in mouse mesenteric arteries17 and also in human coronary arterioles (Fig. 2).42,43 This oxidant-mediated signaling is essential for blood pressure control because the “redox-dead” knock-in mice of Cys42Ser PKG1α, whose mutant PKG1α is unable to be activated by H2O2-induced dimerization because of the absence of its redox-sensitive sulfur, show markedly impaired EDH-mediated relaxation in resistance arteries ex vivo and systemic hypertension in vivo.17 Taken together, endothelium-derived H2O2 functions as an important endogenous second messenger to elicit EDH-meditated relaxations, modulate vascular tone, and maintain vascular homeostasis.
Diverse Roles of Endothelial NOS System
There are 3 NOS isoforms, including neural NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3).3,44 Although 3 NOS isoforms are expressed in cardiovascular system, eNOS is the dominant NOS isoform in blood vessels.45 NOSs have been known to generate superoxide anions from reductase domain under physiological conditions,46 where superoxide anions are converted into H2O2 to cause EDH-mediated responses. Because heme reduction rate in eNOS is much slower than that in the other NOS isoforms, reductase domain-mediated superoxide generation is a significant alternative in eNOS.46 Based on these observations, it is conceivable that eNOS is the most important isoform in generating H2O2/EDH in the endothelium. Indeed, as mentioned above, genetic deletion of eNOS gene in mice causes impaired EDH-mediated relaxations associated with systemic hypertension.47 Although singly eNOS-KO mice exhibit abolished NO-mediated relaxations in the aorta and markedly reduced (but not abolished) EDH-mediated relaxations in the mesenteric artery, the remaining relaxations are still sensitive to catalase.13 We speculated that the 3 NOSs compensate each other to maintain endothelium-dependent responses. To clarify the roles of endothelial NOSs in EDH/H2O2-mediated responses, we generated doubly n/eNOS-KO and triply n/i/eNOS-KO mice.48 Interestingly, the EDH-mediated relaxations are progressively reduced in accordance with the number of NOS genes ablated; as compared with wild-type mice, the EDH/H2O2-mediated relaxations of small mesenteric arteries are reduced almost by half in singly eNOS-KO mice, further diminished in doubly n/eNOS-KO mice, and are finally completely abolished in the triply n/i/eNOS-KO mice without functional alterations of underlying VSMC.21 In contrast, NO-mediated relaxations are totally absent in all 3 genotypes of NOS-KO mice.21 These findings provide a novel concept on the diverse roles of endothelial NOSs system; in large conduit vessels, they mainly serve as a NO-generating system to cause vasodilatation through sGC–cGMP pathway, whereas in resistance vessels, they act as a superoxide-generating system to evoke EDH-mediated responses through H2O2-induced PKG1α dimerization and subsequent activation of KCa channels, leading to hyperpolarization and vasodilatation (Fig. 2). Furthermore, at least in mice under physiological condition, the extent of eNOS bound to cavelion-1 (a negative regulator of eNOS) is greater in mesenteric arteries than in the aorta, and thus, eNOS is functionally suppressed in resistance vessels through a cavelin-1–dependent mechanism, switching its function from NO synthase to EDH/H2O2-generating enzyme (Fig. 2).49
Mechanistic Insight into Vessel Size-dependent Contribution of NO and EDH/H2O2
As mentioned above, NO and EDH/H2O2 share the diverse roles in modulating vascular tone in a distinct vessel size-dependent manner; NO plays a dominant role in conduit arteries and EDH/H2O2 in resistance vessels.14 Mechanistic insight into vessel size-dependent contribution of NO and EDH/H2O2 has been recently demonstrated. As compared with large conduit vessels, eNOS is functionally inhibited by caveolin-1, and relaxation responses of VSMC to H2O2 are enhanced through PKG1α-mediated mechanism in resistance vessels in mice.49,50 Indeed, mouse resistance vessels have less NO production and less antioxidant capacity, allowing PKG1α to be more sensitive to H2O2-induced activation and subsequent hyperpolarization and relaxation of VSMC.50 Furthermore, endothelial AMP-activated protein kinase (AMPK) modulates EDH-mediated responses in resistance arteries, but not in conduit arteries, to regulate blood pressure and coronary flow responses in mice in vivo.51
Interactions Between NO and EDH
It has been previously reported that NO donors attenuate EDH-mediated responses in porcine coronary arteries in vitro52 and canine coronary microcirculation in vivo.53 Furthermore, NO exerts a negative-feedback effect on endothelium-dependent relaxations through cGMP-mediated desensitization in canine coronary arteries ex vivo.54 Multiple mechanisms may be involved in the negative interactions between NO and EDH. Among them, desensitization of VSMC to H2O2 is likely to be involved because H2O2-induced PKG1α dimerization, a central mechanism of H2O2-induced vasodilatation, is inhibited by cGMP-dependent activation of PKG.50 Moreover, pharmacological inhibition of sGC sensitizes conduit vessels to H2O2-induced vasodilatation in mice.50 These observations support the notion that excessive endothelium-derived NO desensitizes blood vessels to EDH/H2O2-mediated relaxations. In addition, the actions of other EDH factors may also be inhibited through interaction with NO. For instance, Bauersachs et al52 showed that exogenous NO donors attenuate EDH-mediated relaxations in vitro. A possible mechanism involved in this phenomenon is the inhibitory effect of NO on EET production through inhibition of cytochrome P450 epoxygenase activity.52 More recently, Mustafa et al55 have reported that NO exerts a direct inhibitory effect on cystathionine γ-lyase activity in vitro. Considering that cystathionine γ-lyase is a biosynthetic enzyme of H2S that has been suggested to be one of EDH factors in mouse mesenteric arteries,12,55 and it is conceivable that this mechanism is also involved in the negative feedback of NO on EDH-mediated relaxations. These results are consistent with the widely accepted notion that EDH functions as a compensatory vasodilator system when NO-mediated relaxations are hampered. Thus, EDH dominance in microcirculation is a vital mechanism to maintain sufficient tissue perfusion in the setting of pathological conditions where NO-mediated responses are jeopardized.14
Dual Roles of ROS
Endothelium-derived ROSs, including superoxide anions, NO, peroxynitrite, hydroxyl radicals, and H2O2, modulate vascular tone through multiple mechanisms in health and disease.14,56,57 Although these ROSs have been regarded to be primarily harmful, the vasoprotective roles of H2O2 have attracted much attention as endothelium-derived H2O2 causes endothelium-dependent vasodilatation and contributes to microvascular homeostasis at its physiological concentrations.26,32,33,56 As predicted previously in the commentary article58 on our original EDH/H2O2 report,13 H2O2 is a physiological signaling molecule serving as an EDH factor especially in microcirculations to modulate blood pressure,17 metabolic coronary vasodilatation,20 and metabolic functions.18 These findings shed new light on the significance of maintaining EDH/H2O2-mediated relaxations in microcirculations and may provide a clue for better understanding of the harmful effects of antioxidant supplements.23,59
Endothelium-derived H2O2 is mainly generated by the dismutation of superoxide anions, which come from various sources in the endothelium, including nicotinamide adenine dinucleotide phosphate oxidase oxidase, mitochondrial electron transport chain, xanthine oxidase, lipoxygenase, and NOS.33,60 Although the precise mechanisms underlying the physiological regulation of ROS production in the endothelium have not yet fully understood, recent studies have provided novel potential mechanisms relevant to modulation of endothelium-dependent responses. For instance, microRNAs, which are small noncoding RNAs regulating gene expressions through degradation or translational repression of mRNA, have emerged as important regulators in cardiovascular system.61 Among the key players in EDH/H2O2-mediated responses, miR-103/107 have been shown to target caveolin-1 to downregulate its expression,62 and miR155 is substantially involved in the negative regulation of NO production and endothelium-dependent vasodilatation by directly targeting eNOS.63 Moreover, a class of transient receptor potential channels plays important role in regulating intracellular Ca2+ concentration and membrane potentials to control vascular tone and thereby blood flow through EDH-mediated mechanisms.2 Notably, several transient receptor potential subfamilies serve as redox sensor to be controlled by endogenous ROS including H2O2 and NO.64,65 Further studies are needed to understand how endothelium-derived ROS are finely regulated to participate in endothelium-dependent responses under physiological conditions.
Clinical Implications
It is difficult to accurately assess the in vivo importance of EDH in humans because the contribution of EDH is determined only after the blockade of both vasodilator PGs and NO.1–3 However, evaluation of endothelial function in humans has attracted much attention in the clinical settings. Endothelial dysfunction is often noted in patients with atherosclerotic risk factors and cardiovascular diseases; antecedent exposure to various risk factors disables endothelial cells to produce sufficient NO, leading to the initial step toward inflammatory responses and atherosclerosis.1 Although NO-mediated relaxations are easily impaired in the early stage of atherosclerotic conditions, EDH-mediated responses are fairly preserved or even temporarily enhanced to maintain vascular homeostasis.2,40 This is well exemplified in the studies showing that enhanced EDH-mediated vasodilation compensates for reduced NO-mediated responses in hypercholesterolemic subjects.15,66 In other clinical studies, endothelial dysfunction, as evaluated by impaired digital reactive hyperemia in peripheral arterial tonometry or flow-mediated dilation of the brachial artery, are associated with future cardiovascular events in patients with coronary artery diseases.67,68 These observations suggest that endothelial functions in peripheral vascular beds could predict future cardiovascular events. In addition, H2O2 also has potent vasodilator properties in coronary resistance vessels, such that impaired EDH/H2O2-mediated vasodilatation could lead to coronary microvascular dysfunction.69 Coronary vascular resistance is mostly determined by the prearterioles (approximately 100–500 μm in diameter) and arterioles (less than 100 μm in diameter) where the contribution of EDH-mediated responses is greater than NO-mediated ones.69 Thus, it is important to maintain the vessel size-dependent contribution of NO and EDH for the treatment of cardiovascular disease. Indeed, a number of clinical studies have failed to show that chronic nitrate therapy, as a NO donor, has beneficial prognostic effects for patients with ischemic heart disease.22,70 Similarly, long-term antioxidant therapy for patients with hypertension failed to lower systemic blood pressure or improve mortality rate.23,59 These results in the clinical studies indicate the importance of the physiological balance between NO and EDH/H2O2 in humans. Notably, standard therapeutic agents used for the treatment of cardiovascular diseases in the current era share the pleiotropic effects on endothelial function by enhancing NO-mediated vasodilatations, including angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and statins.44 Further studies are warranted for improving EDH-mediated responses in microcirculations to develop a novel therapeutic strategy beyond NO.
CONCLUSIONS
Experimental and clinical studies in our and other laboratories have demonstrated that endothelial NOSs have diverse functions to maintain cardiovascular homeostasis, where the physiological balance between NO and EDH is important. The new research avenue of EDH that was opened a quarter century ago by Vanhoutte et al has greatly contributed to our better understanding of endothelial functions and to develop novel diagnostic and therapeutic strategies in cardiovascular medicine.
Footnotes
Supported in part by the Grant-in-Aid for Tohoku University Global COE for Conquest of Signal Transduction Diseases with Network Medicine, the Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Tokyo, Japan, and the Grants-in-Aid for Scientific Research from the Ministry of Health, Labour, and Welfare, Tokyo, Japan.
The authors report no conflicts of interest.
REFERENCES
- 1.Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. [DOI] [PubMed] [Google Scholar]
- 2.Feletou M, Vanhoutte PM. EDHF: an update. Clin Sci (Lond). 2009;117:139–155. [DOI] [PubMed] [Google Scholar]
- 3.Feletou M, Kohler R, Vanhoutte PM. Nitric oxide: orchestrator of endothelium-dependent responses. Ann Med. 2012;44:694–716. [DOI] [PubMed] [Google Scholar]
- 4.Vanhoutte PM, Shimokawa H, Tang EH, et al. Endothelial dysfunction and vascular disease. Acta Physiol. 2009;196:193–222. [DOI] [PubMed] [Google Scholar]
- 5.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization of canine coronary smooth muscle. Br J Pharmacol. 1988;93:515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br J Pharmacol. 1988;95:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarization: no longer an F-word! J Cardiovasc Pharmacol. 2013;61:91–92. [DOI] [PubMed] [Google Scholar]
- 8.Fisslthaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–497. [DOI] [PubMed] [Google Scholar]
- 9.Campbell WB, Gebremedhin D, Pratt PF, et al. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. [DOI] [PubMed] [Google Scholar]
- 10.Griffith TM, Chaytor AT, Edwards DH. The obligatory link: role of gap junctional communication in endothelium-dependent smooth muscle hyperpolarization. Pharmacol Res. 2004;49:551–564. [DOI] [PubMed] [Google Scholar]
- 11.Edwards G, Dora KA, Gardener MJ, et al. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. [DOI] [PubMed] [Google Scholar]
- 12.Tang G, Yang G, Jiang B, et al. H2S is an endothelium-derived hyperpolarizing factor. Antioxid Redox Signal. 2013;19:1634–1646. [DOI] [PubMed] [Google Scholar]
- 13.Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimokawa H. 2014 Williams Harvey Lecture: importance of coronary vasomotion abnormalities-from bench to bedside. Eur Heart J. 2014;35:3180–3193. [DOI] [PubMed] [Google Scholar]
- 15.Urakami-Harasawa L, Shimokawa H, Nakashima M, et al. Importance of endothelium-derived hyperpolarizing factor in human arteries. J Clin Invest. 1997;100:2793–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimokawa H, Yasutake H, Fujii K, et al. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol. 1996;28:703–711. [DOI] [PubMed] [Google Scholar]
- 17.Prysyazhna O, Rudyk O, Eaton P. Single atom substitution in mouse protein kinase G eliminates oxidant sensing to cause hypertension. Nat Med. 2012;18:286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakajima S, Ohashi J, Sawada A, et al. Essential role of bone marrow for microvascular endothelial and metabolic functions in mice. Circ Res. 2012;111:87–96. [DOI] [PubMed] [Google Scholar]
- 19.Yada T, Shimokawa H, Hiramatsu O, et al. Hydrogen peroxide, an endogenous endothelium-derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107:1040–1045. [DOI] [PubMed] [Google Scholar]
- 20.Yada T, Shimokawa H, Hiramatsu O, et al. Important role of endogenous hydrogen peroxide in pacing-induced metabolic coronary vasodilation in dogs in vivo. J Am Coll Cardiol. 2007;50:1272–1278. [DOI] [PubMed] [Google Scholar]
- 21.Takaki A, Morikawa K, Tsutsui M, et al. Crucial role of nitric oxide synthases system in endothelium-dependent hyperpolarization in mice. J Exp Med. 2008;205:2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kojima S, Matsui K, Sakamoto T, et al. Long-term nitrate therapy after acute myocardial infarction does not improve or aggravate prognosis. Circ J. 2007;71:301–307. [DOI] [PubMed] [Google Scholar]
- 23.Bjelakovic G, Nikolova D, Gluud LL, et al. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–857. [DOI] [PubMed] [Google Scholar]
- 24.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39:725–732. [DOI] [PubMed] [Google Scholar]
- 25.Nagao T, Illiano S, Vanhoutte PM. Calmodulin antagonists inhibit endothelium-dependent hyperpolarization in the canine coronary artery. Br J Pharmacol. 1992;107:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matoba T, Shimokawa H, Kubota H, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun. 2002;290:909–913. [DOI] [PubMed] [Google Scholar]
- 27.Miura H, Bosnjak JJ, Ning G, et al. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–40. [DOI] [PubMed] [Google Scholar]
- 28.Matoba T, Shimokawa H, Morikawa K, et al. Electron spin resonance detection of hydrogen peroxide as an endothelium-derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol. 2003;23:1224–1230. [DOI] [PubMed] [Google Scholar]
- 29.Lacza Z, Puskar M, Kis B, et al. Hydrogen peroxide acts as an EDHF in the piglet pial vasculature in response to bradykinin. Am J Physiol. 2002;283:H406–H411. [DOI] [PubMed] [Google Scholar]
- 30.Hatoum OA, Binion DG, Miura H, et al. Role of hydrogen peroxide in ACh-induced dilation of human submucosal intestinal microvessels. Am J Physiol. 2005;288:H48–H54. [DOI] [PubMed] [Google Scholar]
- 31.Chaytor AT, Edwards DH, Bakker LM, et al. Distinct hyperpolarizing and relaxant roles for gap junctions and endothelium-derived H2O2 in NO-independent relaxations of rabbit arteries. Proc Natl Acad Sci U S A. 2003;100:15212–15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yada T, Shimokawa H, Hiramatsu O, et al. Cardioprotective role of endogenous hydrogen peroxide during ischemia-reperfusion injury in canine coronary microcirculation in vivo. Am J Physiol. 2006;291:H1138–H1146. [DOI] [PubMed] [Google Scholar]
- 33.Burgoyne JR, Oka S, Ale-Agha N, et al. Hydrogen peroxide sensing and signaling by protein kinases in the cardiovascular system. Antioxid Redox Signal. 2013;18:1042–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroder E, Eaton P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: issues and considerations. Curr Opin Pharmacol. 2008;8:153–159. [DOI] [PubMed] [Google Scholar]
- 35.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morikawa K, Shimokawa H, Matoba T, et al. Pivotal role of Cu,Zn-superoxide dismutase in endothelium-dependent hyperpolarization. J Clin Invest. 2003;112:1871–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takaki A, Morikawa K, Murayama Y, et al. Roles of endothelial oxidases in endothelium-derived hyperpolarizing factor responses in mice. J Cardiovasc Pharmacol. 2008;52:510–517. [DOI] [PubMed] [Google Scholar]
- 38.Larsen BT, Bubolz AH, Mendoza SA, et al. Bradykinin-induced dilation of human coronary arterioles requires NADPH oxidase-derived reactive oxygen species. Arterioscler Thromb Vasc Biol. 2009;29:739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Zhao H, Li H, et al. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. [DOI] [PubMed] [Google Scholar]
- 40.Shimokawa H. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pflugers Arch. 2010;459:915–922. [DOI] [PubMed] [Google Scholar]
- 41.Burgoyne JR, Madhani M, Cuello F, et al. Cysteine redox sensor in PKGIα enables oxidant-induced activation. Science. 2007;317:1393–1397. [DOI] [PubMed] [Google Scholar]
- 42.Zhang DX, Borbouse L, Gebremedhin D, et al. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110:471–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Bubolz AH, Mendoza S, et al. H2O2 is the transferrable factor mediating flow-induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forstermann U, Li H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol. 2011;164:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. J Biol Chem. 2001;276:14533–14536. [DOI] [PubMed] [Google Scholar]
- 47.Huang PL, Huang Z, Mashimo H, et al. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377:239–242. [DOI] [PubMed] [Google Scholar]
- 48.Morishita T, Tsutsui M, Shimokawa H, et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 2005;102:10616–10621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ohashi J, Sawada A, Nakajima S, et al. Mechanisms for enhanced endothelium-derived hyperpolarizing factor-mediated responses in microvessels in mice. Circ J. 2012;76:1768–1779. [DOI] [PubMed] [Google Scholar]
- 50.Burgoyne JR, Prysyazhna O, Rudyk O, et al. cGMP-dependent activation of protein kinase G precludes disulfide activation: implications for blood pressure control. Hypertension. 2012;60:1301–1308. [DOI] [PubMed] [Google Scholar]
- 51.Enkhjargal B, Godo S, Sawada A, et al. Endothelial AMP-activated protein kinase regulates blood pressure and coronary flow responses through hyperpolarization mechanism in mice. Arterioscler Thromb Vasc Biol. 2014;34:1505–1513. [DOI] [PubMed] [Google Scholar]
- 52.Bauersachs J, Popp R, Hecker M, et al. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–3347. [DOI] [PubMed] [Google Scholar]
- 53.Nishikawa Y, Stepp DW, Chilian WM. Nitric oxide exerts feedback inhibition on EDHF-induced coronary arteriolar dilation in vivo. Am J Physiol. 2000;279:H459–H465. [DOI] [PubMed] [Google Scholar]
- 54.Olmos L, Mombouli JV, Illiano S, et al. cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am J Physiol. 1995;268:H865–H870. [DOI] [PubMed] [Google Scholar]
- 55.Mustafa AK, Sikka G, Gazi SK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Satoh K, Godo S, Saito H, et al. Dual roles of vascular-derived reactive oxygen species–with a special reference to hydrogen peroxide and cyclophilin A. J Mol Cell Cardiol. 2014;73:50–56. [DOI] [PubMed] [Google Scholar]
- 57.Wong WT, Tian XY, Huang Y. Endothelial dysfunction in diabetes and hypertension: cross talk in ras, BMP4, and ROS-dependent COX-2-derived prostanoids. J Cardiovasc Pharmacol. 2013;61:204–214. [DOI] [PubMed] [Google Scholar]
- 58.Vanhoutte PM. Endothelium-derived free radicals: for worse and for better. J Clin Invest. 2001;107:23–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjelakovic G, Nikolova D, Gluud C. Antioxidant supplements to prevent mortality. JAMA. 2013;310:1178–1179. [DOI] [PubMed] [Google Scholar]
- 60.Shao D, Oka S, Brady CD, et al. Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol. 2012;52:550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trajkovski M, Hausser J, Soutschek J, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649–653. [DOI] [PubMed] [Google Scholar]
- 63.Sun HX, Zeng DY, Li RT, et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012;60:1407–1414. [DOI] [PubMed] [Google Scholar]
- 64.Kozai D, Ogawa N, Mori Y. Redox regulation of transient receptor potential channels. Antioxid Redox Signal. 2014;21:971–986. [DOI] [PubMed] [Google Scholar]
- 65.Nishida M, Hara Y, Yoshida T, et al. TRP channels: molecular diversity and physiological function. Microcirculation. 2006;13:535–550. [DOI] [PubMed] [Google Scholar]
- 66.Ozkor MA, Murrow JR, Rahman AM, et al. Endothelium-derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation. 2011;123:2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matsuzawa Y, Sugiyama S, Sugamura K, et al. Digital assessment of endothelial function and ischemic heart disease in women. J Am Coll Cardiol. 2010;55:1688–1696. [DOI] [PubMed] [Google Scholar]
- 68.Kitta Y, Obata JE, Nakamura T, et al. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J Am Coll Cardiol. 2009;53:323–330. [DOI] [PubMed] [Google Scholar]
- 69.Crea F, Lanza G, Camici P. Mechanisms of coronary microvascular dysfunction. In: Coronary Microvascular Dysfunction. Milan, Italy: Springer; 2014:31–47. [Google Scholar]
- 70.Ambrosio G, Del Pinto M, Tritto I, et al. Chronic nitrate therapy is associated with different presentation and evolution of acute coronary syndromes: insights from 52,693 patients in the global registry of acute coronary events. Eur Heart J. 2010;31:430–438. [DOI] [PubMed] [Google Scholar]