

Abstract
To date, gene therapy with transiently derived lentivectors has been very successful to cure rare infant genetic diseases. However, transient manufacturing is unfeasible to treat adult malignancies because large vector lots are required. By contrast, stable manufacturing is the best option for high-incidence diseases since it reduces the production cost, which is the major current limitation to scale up the transient methods. We have previously developed the proprietary RD2-MolPack technology for the stable production of second-generation lentivectors, based on the RD114-TR envelope. Of note, opposite to vesicular stomatitis virus glycoprotein (VSV-G) envelope, RD114-TR does not need inducible expression thanks to lack of toxicity. Here, we present the construction of RD2- and RD3-MolPack cells for the production of self-inactivating lentivectors expressing green fluorescent protein (GFP) as a proof-of-concept of the feasibility and safety of this technology before its later therapeutic exploitation. We report that human T lymphocytes transduced with self-inactivating lentivectors derived from RD3-MolPack cells or with self-inactivating VSV-G pseudotyped lentivectors derived from transient transfection show identical T-cell memory differentiation phenotype and comparable transduction efficiency in all T-cell subsets. RD-MolPack technology represents, therefore, a straightforward tool to simplify and standardize lentivector manufacturing to engineer T-cells for frontline immunotherapy applications.
Introduction
More than a decade of research and development has drastically enriched the efficacy and safety of HIV-based lentiviral vector (LV) technology in gene therapy. This know-how has been proven to be clinically applicable, in a temporal order, first to AIDS gene therapy,1–6 then to the correction of rare genetic diseases,7,8 and more recently to cancer immunotherapy.9,10
Human hematopoietic stem/progenitor cells and peripheral blood (PB) T lymphocytes are highly relevant cells for gene therapy interventions of blood disorders. Up to now, these cells have been successfully transduced with vesicular stomatitis virus glycoprotein (VSV-G) pseudotyped LVs produced by transient transfection. However, this method is quite inadequate from a cost standpoint being the price of the good manufacturing practice-grade plasmids extremely high. Therefore, the implementation of stable LV packaging cells in the clinic represents a mandatory milestone to reduce the manufacturing cost and to further enhance the overall quality of the vectors. Thus far, several strategies have generated stable LV packaging cells differing, essentially, for the tools used to deliver the packaging genes, i.e., plasmids versus integrating vectors. Furthermore, another key element is the different nature of the pseudotyping envelope, i.e., VSV-G or other retrovirus-derived envelopes. Packaging cells can be divided into inducible and constitutive, depending upon the type of envelope loaded, i.e., VSV-G is generally loaded into inducible cells due to toxicity of its stable expression. The first category includes, for example, two packaging cells derived from the GPRG cells, the GPRG-EF1α-hγcOPT, that has been exploited for clinical production,11 and the 650MNDhWASp1.12 The latter contains also the Tat gene and it has been recently generated for clinical-grade production of WAS LV. In both systems, all viral genes are integrated by means of self-inactivating (SIN) γ-retroviral vectors γRVs) and the expression of the severely toxic VSV-G envelope is under a doxycycline-inducible system.11,12
The second category includes the STAR13 and the WinPac cells14 in which gag-pol genes are introduced by means of LTR-γRV and SIN-γRV, respectively. In both cell types, the nontoxic rd114 envelope and rev genes are integrated by plasmid.15
We have previously generated the RD2-MolPack-Chim3 cells for stable production of second-generation LTR-LVs expressing the anti-HIV chim3 therapeutic gene, a dominant-negative HIV Vif.16–18 The RD2-MolPack-Chim3 cells were obtained by serially loading traced HEK-293T cells with the vector coding genes. The HIV-1 gag, pol, rev, and hygro-resistance genes were delivered by a chimeric baculo-AAV vector, thereby obtaining the PK-7 clone. The tat and rd114-tr genes were later introduced in PK-7 cells by VSV-G pseudotyped SIN-LVs. We demonstrated that RD2-MolPack-Chim3 LVs outperformed VSV-G pseudotyped LVs in transducing human cord blood (CB)-derived hematopoietic stem cells,19 confirming previous findings.19–21
Here, we describe the RD2- and RD3-MolPack packaging systems fitting for the production of SIN-LVs to target human PB T lymphocytes, which are playing an ever stronger role in the rapidly expanding field of T-cell immunotherapy of cancer. We show comparable levels of T-cell transduction using LVs produced by either the RD3-MolPack technology or the standard VSV-G-based transiently-derived lentivectors. The data are very promising toward large-scale production for clinical indication involving a large cohort of patients.
Results
Construction of the SIN-RD114-TR LV
To construct Tat-independent third-generation RD3-MolPack packaging cells, the PK-7 clone must be loaded with the RD114-TR envelope. Similarly to others, 11–14 we decided to use integrating vector, i.e., a VSV-G-pseudotyped SIN-LV in order to introduce RD114-TR into PK-7 cells. We did not use plasmids to avoid, prior to each LV lot manufacturing, antibiotic reselection of the producer cells that is recommended when viral genes are delivered by plasmids. Towards this aim, we first designed the SIN-RD114-TR transfer vector (TV) (Figure 1a, part 1) in which the CMV-rd114-tr cDNA expression cassette is followed by an IRES-puro element. Unexpectedly, this vector was not functional, as demonstrated by western blot of the protein extracts derived from HEK-293T cells transfected with the SIN-RD114-TR TV plasmid (Figure 2a, lane 3). Likewise, northern blot showed that the 3.9-kb transcript of the internal expression cassette was barely visible compared to the 6.3-kb full-length RNA of the vector (Figure 2b, lane 3). This result suggests a functional defect of the expression cassette rather than a defect of the vector backbone itself. We reasoned that this problem might be caused by the absence of the β-globin intron (BGI) between the promoter and the RD114-TR cDNA, originally present in the phCMVRD114-TR plasmid.20 To test this hypothesis, we designed three new TVs, all carrying the BGI between the promoter and the ORF of the transgene: the SIN-RD114-TR-IN, SIN-RD114-TR-IN-antisense, and the SIN-RD114-TR-IN-RRE (Figure 1a, parts 2, 3, and 4). The CMV-BGI-rd114-tr cassette was inserted either in sense (Figure 1a, parts 2 and 4) or antisense orientation (Figure 1a, part 3) or with a second Rev Responsive Element (RRE) embedded in the BGI of the SIN-RD114-TR-IN-RRE (Figure 1a, part 4). The extra RRE was included to protect more efficiently the CMV cassette promoter from being spliced out. All TV plasmids were active after their transient transfection in HEK-293T cells (Figure 2c). However, when PK-7 cells were transduced with the VSV-G pseudotyped corresponding LVs, the cells transduced with the SIN-RD114-TR in sense orientation expressed the envelope (Figure 2d, lanes 1 and 3), but not those transduced with the cassette in antisense orientation (Figure 2d, lane 2). The SIN-RD114-TR-IN-antisense TV was therefore dropped.
Figure 1.

SIN LVs used in this study. (a) Schemes of the SIN-RD114-TR LVs. CMV, cytomegalovirus promoter; ΔU3, deleted U3; IN, intron; BGI, rabbit β-globin intron; RRE, Rev Responsive Element; A, polyA sequence; IRES, internal ribosome entry site; SD, splice donor; SA, splice acceptor; Ψ, packaging signal; WPRE, woodchuck hepatitis post-transcriptional regulatory element; cPPT, central polypurine tract. (b) Scheme of the SIN-GFP TV. AAV-ITR, adeno-associated virus-inverted terminal repeat; SV40-P, Simian Virus 40 promoter; zeoR, zeocin resistance gene.
Figure 2.
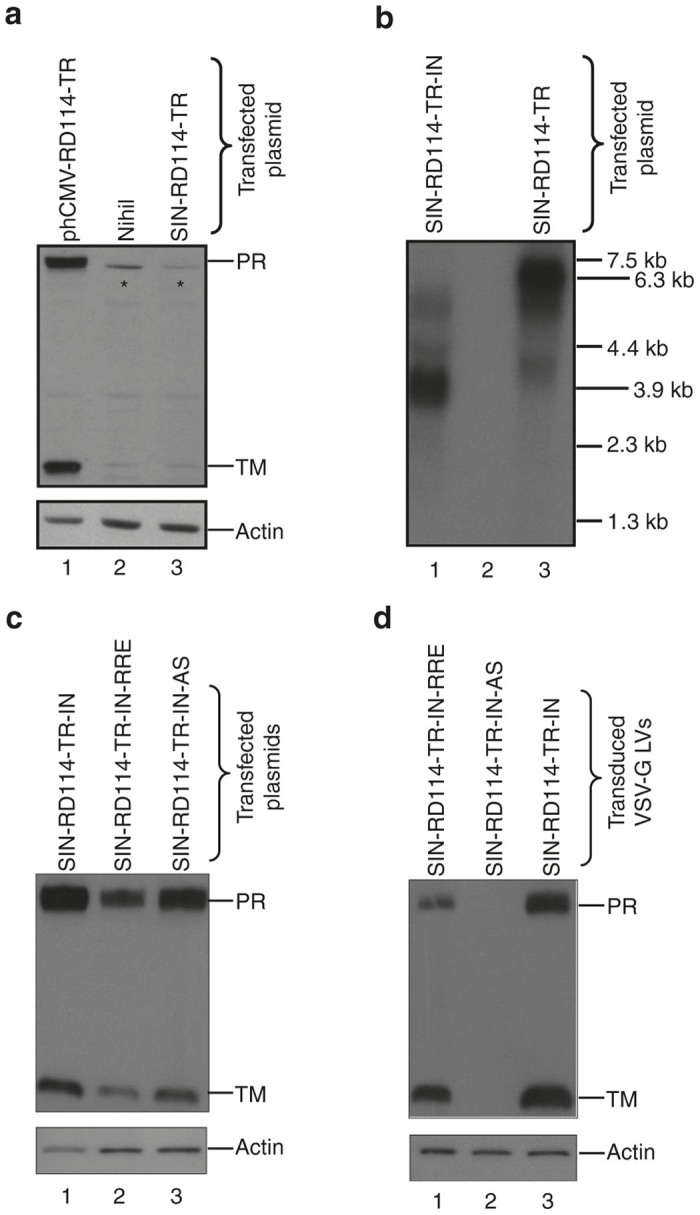
Characterization of the SIN-RD114-TR vectors. (a) Western blot analysis of cellular extracts (45 μg/sample) obtained from HEK-293T cells 72 hours after either mock-transfection (lane 2) or transfection with the indicated plasmids (plasmid DNA transfected, 2 μg/1 × 106 cells) (lanes 1 and 3). (b) Northern blot analysis of total RNA (7.5 μg/sample) extracted from HEK-293T 48 hours after transfection of the indicated plasmids (lanes 1 and 3). The size of the expected full length and internal cassette transcripts corresponds to 6.3- and 3.9-kb, respectively. The membrane was hybridized with a 550-bp RD114-TR-specific probe. (c) Western blot analysis of cellular extracts (35 μg/sample) obtained from HEK-293T cells 48 hours after transfection of the indicated plasmids (plasmid DNA transfected, 2 μg/1 × 106 cells). The membranes were hybridized with the anti-RD114-TR-specific Ab recognizing the precursor (PR, 75-kDa) and the transmembrane (TM, 18-kDa) subunit of the RD114-TR envelope and, after stripping, with the anti-actin Ab. (d) Western blot analysis of cellular extracts (35 μg/sample) obtained from PK-7 cells 72 hours after transduction with the indicated VSV-G-pseudotyped SIN-RD114-TR LVs. The membranes were hybridized as described for panel c. The asterisk (*) indicates nonspecific band.
Generation of different RD3-MolPack packaging cells
As both SIN-RD114-TR-IN and SIN-RD114-TR-IN-RRE TVs functioned properly when stably integrated (Figure 2d), we transduced PK-7 cells with both VSV-G pseudotyped transiently derived corresponding LVs to generate different RD3-MolPack packaging cells. After 14 days of puromycin selection, we measured the vector copy number (VCN) of the SIN-RD114-TR in several colonies. The PK-7-RD314 and PK-7-RD28 cells, containing 6 copies of the SIN-RD114-TR-IN and 12 copies of the SIN-RD114-TR-IN-RRE, respectively, were chosen as two independent RD3-MolPack packaging cells (Table 1).
Table 1. Vector copy numbera of integrated genes in RD-MolPack packaging and producer cells.
| RD-MolPack packaging | Gag/Pol/Rev | Tat | RD114-TR | RD-MolPack producer | SIN-GFP-zeo |
|---|---|---|---|---|---|
| PK-7-RD314 (RD114-TR-IN) | 2 | n.a. | 6.00 | RD3-MolPack1 | 15.7 |
| PK-7-RD28 (RD114-TR-IN-RRE) | 2 | n.a. | 12.0 | RD3-MolPack24 | 48.0 |
| PK-7-RD28 (RD114-TR-IN-RRE) | 2 | n.a. | 12.0 | RD3-MolPack28 | 119.0 |
| PK-7-Tat7-RD19 (RD114-TR-IN-RRE) | 2 | 6 | 13.0 | RD2-MolPack64 | 2.70 |
The vector copy number was calculated by quantitative polymerase chain reaction at least in three independent experiments using specific primers and probe sets, as reported in the Supplementary Material.
n.a., not applicable.
Derivation of several RD3-MolPack-SIN-GFP producer cells
The final step to obtain a prototypic third-generation RD3-MolPack producer cells for SIN-LVs, consisted in introducing the SIN-TV into either PK-7-RD314 or PK-7-RD28 packaging cells. Contrary to the LTR-driven TV, which can be integrated into packaging cells by transduction, SIN TV cannot be introduced by transduction because by doing so the TV is self-inactivated at the 5’LTR and the packaging signal is not incorporated into the genomic RNA. We decided therefore to integrate the SIN-TV by stable transfection. Toward this goal, we first modified a pCCL-based SIN-GFP (green fluorescent protein) TV by adding an antibiotic selection cassette downstream the 3’deleted (U3-LTR thereby obtaining the SIN-GFP-zeo plasmid (Figure 1b). This TV is also flanked by two adeno-associated virus (AAV)-inverted terminal repeats (ITRs) and contains an SV40P-zeocin expression cassette downstream the 3’U3-LTR (Figure 1b). This configuration allows one-plasmid rather than two-plasmid transfection, thus increasing the efficiency of transfection and antibiotic selection. We transfected the SIN-GFP-zeo plasmid in three independent RD-MolPack packaging cells: the third-generation PK-7-RD314 and PK-7-RD28 described above and the second-generation PK-7-Tat7-RD19 (i.e., RD2-MolPack) cells previously reported.19 We included RD2-MolPack cells in our analysis to verify whether Tat could somehow improve the performance of the SIN-LVs, as recently reported.12,22 After zeocin selection and screening of several colonies by physical and functional titer for each cell type (Table 2, picked/screened colonies), four independent producer cells were picked: (i) the RD3-MolPack1, derived from the PK-7-RD314 cells carrying the SIN-RD114-TR-IN and containing 16 copies of SIN-GFP-zeo; (ii–iii) the RD3-MolPack24 and RD3-MolPack28, both derived from the PK-7-RD28 cells, carrying the SIN-RD114-TR-IN-RRE and containing 48 and 119 copies of the TV, respectively; and (iv) the RD2-MolPack64, derived from the PK-7-Tat7-RD19 cells, carrying the SIN-RD114-TR-IN-RRE and containing 2.7 copies of the TV (Table 1).
Table 2. Characterization of RD-MolPack-SIN-GFP producer cells.
| RD3-MolPack packaging cells | Picked/screened colonies | RD3-MolPack producer cells | Viability (%)a | Titer (TU/ml) | Titer (TU/cell/day) | p24Gag (ng/ml) | Infectivity (TU/ng) |
|---|---|---|---|---|---|---|---|
| PK-7-RD314 | 60b/45c | RD3-MolPack1e | 93 | 2.8 × 104 ± 9.2 × 103 | 0.0056 ± 0.0027 | 13.0 ± 6.20 | 3.8 × 103 ± 2.6 × 103 |
| PK-7-RD28 | 58/15d | RD3-MolPack24f | 90 | 3.7 × 105 ± 1.5 × 105 | 0.074 ± 0.039 | 38.3 ± 11.1 | 8.5 × 103 ± 1.5 × 103 |
| PK-7-RD28 | 11/7d | RD3-MolPack28g | 80 | 2.2 × 105 ± 5.4 × 104 | 0.043 ± 0.011 | 17.5 ± 3.40 | 1.2 × 104 ± 2.1 × 103 |
| RD2-MolPack packaging cells | Picked/screened colonies No. | RD2-MolPack producer cells | Viability (%)a | Titer (TU/ml) | Titer (TU/cell/day) | p24Gag (ng/ml) | Infectivity (TU/ng) |
| PK-7-Tat7-RD19 | 68/23d | RD2-MolPack64f | 91 | 7.0 × 104 ± 7.5 × 103 | 0.056 ± 0.009 | 40.4 ± 4.4 | 1.8 × 103 ± 1.4 × 102 |
Viability of the cells at the time of supernatant harvest (24 hours after cell seeding).
Number of picked-up colonies after zeocin selection.
Number of screened clones after limiting dilution cloning.
Number of picked/screened colonies after zeocin selection.
Mean ± standard error of the mean (SEM) of n = 3.
Mean ± SEM of n = 6.
Mean ± SEM of n = 3.
TU, transducing unit.
Characterization of the RD3-MolPack-SIN-GFP producers
To assess RD-MolPack LV potency, the clarified raw supernatant of each producer was titered in the reference T-cell line CEM A3.01 (Table 2). The titer of the RD3-MolPack1 LVs was the lowest, reflecting its low level of p24Gag (Table 2) and RD114-TR VCN (Table 1). For this reason, the RD3-MolPack1 was excluded from further analysis.
Since the titer of the other three producers, all carrying the SIN-RD114-TR-IN-RRE, was comparable in the order of 105 TU/ml (Table 2), we next assessed the integrity of the vector genes in each producer by Southern blot (Figure 3). The baculo-AAV chimeric vector expressing the gag-pol-rev genes did not undergo rearrangement after the later integration of the SIN-RD114-TR TV in all packaging and producer cells (Figure 3a). Similarly, the rd114-tr gene did not undergo rearrangement after the integration of the SIN-GFP-zeo TV in all cells (Figure 3b). However, the integrity of the SIN-GFP-zeo TV differed among the producer cells (Figure 3c). In fact, in contrast to RD3-MolPack24, showing only the expected 3.4-kb band, RD3-MolPack28 and RD2-MolPack64 cells contained also a strong shorter 3-kb band (correct 3.4-kb:shorter 3-kb band intensity ratio = 1:4) and a weak larger 7.5-kb band (correct 3.4-kb: larger 7.5-kb band intensity ratio = 1:1), respectively (Figure 3c, lanes 5 and 7 and Supplementary Table S1). Nevertheless, it is important to note that the rearranged integrants were not transferred to either CEM A3.01 or primary T target cells (Figure 3d, lanes 3, 5, 7, and 9), assuring the safety and integrity of the gene transfer process. Furthermore, in all samples, the intensity of the detected bands mirrored the VCN measured by qPCR. Because of the very high VCN = 119 (Table 1) and the rearranged profile of the SIN-GFP-zeo TV (Figure 3d), the RD3-MolPack28 cells were excluded from further analysis. We then concentrated our analysis on one RD2-MolPack derived producer (RD2-MolPack64) and one RD3-MolPack derived producer (RD3-MolPack24) in order to establish whether Tat contributed somehow in the production of the SIN-LVs. To this aim, we verified the expression of RD3-MolPack24 and RD2-MolPack64 vector proteins by western blot analysis of cellular and virion extracts (Supplementary Figure S1). All proteins were properly translated in packaging cells, producer cells and properly processed in the derived viral particles (Supplementary Figure S1a,b).
Figure 3.

Integrity of the vector genes in the packaging, producer and target cells. Southern blot analysis of genomic DNA (10 μg/sample) extracted from the indicated cells and hybridized with the specific probes: (a) 661-bp CMV probe; (b) 550-bp RD114-TR probe; (c and d) 339-bp GFP probe. In panel d, the CEM A3.01 and PB T lymphocytes were either mock-transduced (lane 7) or transduced at MOI = 25.The genomic DNA was extracted 10 days after transduction.
Transduction of activated T lymphocytes with the RD3-MolPack24
We previously demonstrated that RD114-TR-pseudotyped LVs derived from RD-MolPack technology performed better than VSV-G-pseudotyped LVs produced transiently in transducing CB-derived human CD34+ cells.18 Now, we focused our attention on PB T lymphocytes as key cells for the treatment of hematological malignancies. We found that CD3/CD28-activated PB CD3+ T lymphocytes were 92% transduced by the RD3-MolPack24 LVs at multiplicity of infection (MOI) = 25 (n = 3) and >75% at MOI = 1.5 (n = 3) both at 6 and 14 days after transduction (Figure 4a). Accordingly, the VCN remained stable up to 14 days after transduction, ranging from 7.4 to 4, at MOI = 25 (n = 3) and MOI = 1.5 (n = 3), respectively (Figure 4b).
Figure 4.

Transduction efficiency of activated peripheral blood (PB) T lymphocytes with either RD3-MolPack24 or VSV-G-pseudotyped SIN-GFP-zeo LVs. (a) Peripheral blood mononuclear cells (PBMC) were preactivated with CD3/CD28 Dynabeads for 48 hours and cultured in the presence of IL-7 and IL-15. T lymphocytes were then transduced with the RD3-MolPack24 LVs at the indicated MOI and 6 and 14 days after transduction, GFP expression was evaluated by FACS analysis to calculate transduction efficiency (MOI 25 day 6 p.t., range 89.2–94.5%, n = 3, MOI 25 day 14 p.t., range 83.5–96.3%, n = 4; MOI 3 day 6 p.t., range 82.4–96.9%, n = 3, MOI 3 day 14 p.t., range 84.8–97.8%, n = 4; MOI 1.5 day 6 p.t., range 63.4–90.9%, n = 3, MOI 1.5 day 14 p.t., range 65–92.5%, n = 4). (b) Quantification of the SIN-GFP-zeo vector copy number (VCN) in T lymphocytes of panel a by quantitative polymerase chain reaction (q-PCR) using specific primer-probe sets recognizing the packaging (Ψ) signal of the integrated vector and the genomic telomerase gene as control (MOI 25 day 6 p.t., range 6.2–8.5, n = 3, MOI 25 day 14 p.t., range 3.9–7.9, n = 4; MOI 3 day 6 p.t., range 4.6–11.9, n = 3, MOI 3 day 14 p.t., range 3.7–12.7, n = 3; MOI 1.5 day 6 p.t., range 2.7–7.3, n = 3, MOI 1.5 day 14 p.t., range 2.1–6.5, n = 3). (c) PBMC were preactivated as in (a) and then transduced with 36 ng of p24Gag equivalents of either RD3-MolPack24 LVs or VSV-G-pseudotyped LVs carrying the SIN-GFP-zeo TV. Six and 14 days after transduction, the GFP expression was evaluated by FACS analysis (RD114-TR day 6 p.t., range 66.7–90.9%, n = 3, RD114-TR day 14 p.t. range 84.8–89.7%, n = 2; VSV-G day 6 p.t. range 92.4–98.8%, n = 3; VSV-G day 14 p.t., range 91.9–96.4%, n = 2). (d) Quantification of the SIN-GFP-zeo VCN in T lymphocytes of panel (c) as described in (b) (RD114-TR day 6 p.t., range 2.6–5.2%, n = 3, RD114-TR day 14 p.t. range 3.7–5.3%, n = 2; VSV-G day 6 p.t. range 15.3–31.8%, n = 3; VSV-G day 14 p.t., range 7.7–15.8%, n = 2). Results are mean ± standard error of the mean of n = 3 independent experiments, unless otherwise indicated. *P ≤ 0.05; **P ≤ 0.01.
We then compared the transduction efficiency of 36-ng p24Gag equivalents of either RD3-MolPack24 LVs (infectivity = 9 × 103 TU/ng p24Gag) or transiently derived VSV-G-pseudotyped SIN-GFP-zeo LVs (infectivity = 3 × 105 TU/ng p24Gag) on CD3/CD28-activated T-cells. Six and 14 days after transduction, the percentage of GFP+ cells was statistically significant higher in the total CD3+ T-cell population transduced with the VSV-G LVs than that transduced with the RD114-TR LVs (Figure 4c). Similarly, 6 days post-transduction the SIN-GFP VCN in VSV-G-LV-transduced cells was higher than that in RD114-TR LV-transduced cells (Figure 4d). In contrast, 14 days after transduction, the VCN of the SIN-GFP TV T-cells transduced with both type of LVs was not statistically significant different (Figure 4d, n = 2). This result indicates that the VCN reduction observed in VSV-G-LV-transduced cells derives from the loss of the nonintegrated TVs over time and that after 14 days the VCN is comparable if we take into consideration the high SD of VSV-G samples. We then matched the two LVs by equal MOI that by definition should return same values. At MOI = 25 that is at saturation for both LVs in terms of percentage of transduction, we observed results similar to those obtained by matching for physical particles (Supplementary Figure S2a,b). At MOI = 1.5 that is not at saturation for both LVs, we did not detect statistically significant difference of either the percentage of transduction or VCN 14 days after transduction (Supplementary Figure S2c,d).
Next, we investigated the possible difference in the level of transduction of CD3+/CD8− and CD3+/CD8+ subsets. We observed that RD114-TR LVs transduced slightly better the CD3+/CD8+ subset than the CD3+/CD8− one, whereas the opposite results applied for the VSV-G LVs (Figure 5a, n = 4). Furthermore, for both LVs, the relative frequency of total CD3+/CD8+ and CD3+/CD8− was identical to mock-transduced cells (Figure 5b, n = 4). Most importantly, T-cell memory subpopulations of the CD3+/CD8+ and CD3+/CD8− subsets were transduced with identical efficiency by the two differently pseudotyped LVs (Figure 5c,d, n = 4). Yet, transduction with both LVs did not affect the relative frequency of the T-cell differentiation phenotype, which was similar to mock-transduced cells (Figure 5e,f, n = 4). As expected, when T-cells were transduced at the same MOI 25 with the two types of LVs, no major difference was observed (Supplementary Figure S3, n = 3).
Figure 5.

Transduction efficiency of activated PB T lymphocyte subsets with either RD3-MolPack24 or VSV-G-pseudotyped SIN-GFP-zeo LVs. (a) PBMC were preactivated with CD3/CD28 Dynabeads for 48 hours and cultured in the presence of IL-7 and IL-15. T lymphocytes were then transduced with 36-ng p24Gag equivalent of RD3-MolPack24 LVs and 6 days after transduction, GFP expression was evaluated in CD3+/CD8+ and CD3+/CD8− T-cell subsets (a), and in T-cell memory subpopulations of CD3+/CD8+ and CD3+/CD8− T-cell subsets (c) and (d) by FACS analysis. (b) Relative frequency of CD3+/CD8+ and CD3+/CD8− T cell subsets in LV- and mock-transduced cells. (e and f) Relative frequency of T central memory (TCM), T-stem-cell memory (TSCM), T effector memory (TEM) and T effector memory RA+ (TEMRA) in either CD3+/CD8+ (e) or CD3+/CD8− Tcell subsets (f) of LV- and mock-transduced cells. Results are mean ± standard error of the mean of n = 4 independent experiments (day 6 p.t.). *P ≤ 0.05; **P ≤ 0.01.
Cell metabolism of RD3-MolPack24 and RD2-MolPack64 cells
To investigate the future potential manufacturing scalability of RD2- and RD3-MolPack technology for the LV production in bioreactor, we seeded both RD2-MolPack64 and RD3-MolPack24 cells at 1.5 × 104/cm2 in standard flask and monitored several parameters for 7 days of continuous culture (Supplementary Figure S4). Interestingly, while the two systems were comparable in terms of LV production when evaluated either as TU/cell or TU/ml (Supplementary Figure S4a,b), they differed in terms of metabolism and growth. The RD3-MolPack64 cells grew faster (T = 25.7 ± 1.4 hours versus T = 32.7 ± 2.9 SEM hours) (Supplementary Figure S4c,e) and after 4–7 days of culture their viability decreased, increasing at the same time, the number of viable cells in suspension (Supplementary Figure S4d,f).
Medium scale growth of RD3-MolPack24 cells and LV concentration
Once established that RD3-MolPack24 cells were the best candidate for adherent growth, we optimized their culture conditions and LV concentration toward medium–large-scale production. We observed almost fourfold increment of the titer (from 4.0 × 105 to 1.5 × 106 TU/ml) after optimization of cell culture and harvest conditions (see Materials and Methods) and a slightly higher titer when the clarified supernatants were centrifuged at low rather than high speed (Table 3). We then cultured the cells in the multilayer HYPERFlask. When the cells reached high density (i.e., 2.2–2.3 × 105 cells/cm2 equal to 4 × 108 cells/HYPERFlask), we collected in six independent harvests, almost 3 l of supernatant containing 1.39 × 109 total TU with a mean infectivity of 3.52 ± 0.58 × 103 TU/ng p24Gag (Supplementary Table S2).14 To confirm these results, we carried out four independent runs obtaining similar results (Supplementary Table S3). Taken together, these results are encouraging for future setting up of large-scale manufacturing either in multi-stack vessels or in bioreactor.
Table 3. RD3-MolPack24 LV potency after cell culture optimization and concentration.
| Condition | Titer (TU/ml) | Titer (TU/cell/day) | p24Gag (ng/ml) | Infectivity (TU/ng) |
|---|---|---|---|---|
| Raw supernatanta | 3.7 × 105 ± 1.5 × 105 | 0.074 ± 0.039 | 42.2 ± 12.7 | 8.1 × 103 ± 1.8 × 103 |
| Optimized culture conditionsb | 1.8 × 106 ± 3.5 × 105 | 0.38 ± 0.075 | 256 ± 23.2 | 6.2 × 103 ± 1.2 × 103 |
| Low-speed concentrationc | 2.4 × 108 ± 1.9 × 108 | n.a. | 2.7 × 104 ± 2.1 × 104 | 7.8 × 103 ± 3.0 × 103 |
| High-speed concentrationd | 8.6 × 107 ± 3.8 × 107 | n.a. | 1.8 × 104 ± 0.5 × 104 | 4.8 × 103 ± 2.2 × 103 |
As in Table 2.
After optimization of the culture conditions as described in Materials and Methods. Mean ± standard error of the mean (SEM) of n = 6.
After 100 × concentration at low-speed centrifugation. Mean ± SEM of n = 3. Yield of concentration step: total TU = 79.3 ± 24.7%; total p24Gag = 59.4 ± 7.6%.
After 100 × concentration at high-speed centrifugation. Mean ± SEM of n = 3. Yield of concentration step: total TU = 53.0 ± 17.3%; total p24Gag = 62.7 ± 4.8%.
n.a., not applicable.
Possible mobilization of SIN-RD114-TR TV from RD3-MolPack24 cells
Finally, we verified the safety of RD3-MolPack24 cells in terms of potential mobilization of the envelope. The mobilization of the envelope could lead, in fact, to the formation of possible recombinant vectors. In order to do so, we first calculated the theoretical mobilization frequency of the SIN-RD114-TR-IN-RRE TV into RD3-MolPack24 LVs. We used the estimated mobilization frequency of a generic SIN-LV reported by Hanawa et al.23 corresponding to 1:1,000–3,000 (0.1–0.03%) of the truly integrated SIN-LVs. As the truly integrated VCN of the SIN-RD114-TR-IN-RRE is 12 in RD3-MolPack24 cells, the anticipated frequency is 1.2–0.4% (Supplementary Table S4). We then calculated the real mobilization frequency by a validated reverse transcription-quantitative polymerase chain reaction (RT-qPCR) method, which grounds on the calculation of the percentage of SIN-RD114-TR-IN-RRE genome contamination respect to the sample genome, i.e., SIN-GFP-zeo. We estimated that the real mobilization frequency of SIN-RD114-TR in three independent LV lots corresponded to 0.17 ± 0.015% that is well below the theoretical calculated value (Supplementary Table S4). Finally, we calculated also the expected mobilization frequency of RD114-TR in transduced cells. Based on the real frequency of mobilization into the LVs and on the assumption that the possible mobilized RD114-TR genome transduces target cells similarly to SIN-GFP-zeo genome, the expected mobilized VCN on target cells corresponded to 247.9–82.6 range when cells were transduced at MOI = 10. However, 15 days after transduction, the true VCN in T-cells was undetectable (Supplementary Table S4). These data are consistent with our previous observations indicating that integration of the envelope gene by a SIN-LV is not a safety concern in the RD-MolPack system.18
Discussion
This study is the continuation and extension of a long-term project on the construction of stable packaging cells for the manufacturing of RD114-TR-pseudotyped lentiviral vectors. We earlier reported on the RD2-MolPack-Chim3 cells producing LTR-driven second-generation LVs for anti-HIV gene therapy.18 Here, we describe the RD-MolPack technology for the constitutive production of third-generation SIN-LV.
In both RD2- and RD3-MolPack systems, we stably integrated the RD114-TR envelope via viral vectors, which are naturally more stable than plasmids. Plasmids require in fact cyclic antibiotic selection to maintain high level of expression over time. The use of integrating vectors is an approach similar to that developed for the GPRG cells, in which SIN γ-retroviral vectors were used.11,12,24 We rather used VSV-G-pseudo-typed SIN-LVs to increase the efficiency of the delivery. The initial design of the SIN-RD114-TR TV in which the internal cassette lacks the BGI permitted us to discover that the expression of RD114-TR necessarily needs an intron downstream its driving promoter. We speculate that the presence of the BGI is necessary to attenuate the inhibitory effect of some “instability sequences” which, similarly to other genes,25 are embedded within the RD114-TR ORF. In this contest, it is worth mentioning that codon-optimized rd114-tr gene is transcribed and translated abundantly even in the absence of the BGI. GeneOptimizer Assisted Sequence analysis performed by GENEART AG (Regensburg, Germany) determined, in fact, that a lot of codons with a bad codon usage were spread along the rd114-tr gene, giving reason of our assumption (Zucchelli et al., manuscript in preparation).
Another interesting observation is the lack of expression of the SIN-RD114-TR-IN-AS in transduced cells. We explain this finding postulating the formation of nonfunctional double-strand RNA molecules between the full-length genomic RNA and the RNA of the internal cassette in antisense orientation.
Another key step during the construction of third-generation producer cells is the integration of the SIN TV that is generally achieved by stable transfection of the plasmid DNA. In fact, a SIN TV cannot be integrated by transduction because the 5’LTR becomes inactive once integrated. A clever strategy for stable transfection has been previously described, which is based on the formation of concatamers24 or monomers12 deriving by the ligation of the TV, devoid of plasmid backbone, to a zeo-resistance cassette.12,24 We have addressed this issue in a similar manner by incorporating the zeo-cassette into the TV plasmid itself. However, the zeo-cassette cannot be transferred to target cells because it is downstream the 3’U3-LTR. This TV configuration is therefore suitable for clinical application for which antibiotic resistance genes are not supposed to be delivered in target cells.
Regarding the integrity of the TV, we have demonstrated that even in the unfortunate case that ca. 80% of the TV is rearranged, as for the RD3-MolPack28 cells, the integrity of the gene transferred is not affected. In fact, in CEM A3.01 target cells only the expected band is visible even after long exposure of the Southern blot membrane. To explain this result, we reasoned that, depending on the nature of the rearrangement, the strong shorter proviral DNA that is present in the producer, but not in the target cells, can be either (i) not transcribed, or (ii) transcribed, but not encapsidated, or (iii) encapsidated, but not transferred/integrated to/in target cells. Whatever is its fate, the integrity and identity of gene transfer is not affected. Furthermore, in our previous report, we did not observed formation of RCL when the therapeutic gene Chim3 was produced in RD2-MolPack.18 In this regard, based on the considerable efforts both in term of cost and time, we decided to carry out RCL assay only with RD-MolPack packaging cells expressing therapeutic genes.
Concerning the possible mobilization of the SIN-RD114-TR-IN-RRE vector, we have confirmed our previous data showing an extremely low mobilization level of the SIN-RD114-TR-IN-RRE vector into viral particles. More importantly, we have also demonstrated the absence of mobilization in target cells even when they were transduced with MOI higher than that usually applied in clinical protocols. It is important to note that RT-qPCR analysis does not distinguish between true mobilized gRNA and mRNA passively incorporated into the virions. Being the length of the amplicons very short (93-bp for RD114-TR), this method estimates also the contribution of molecules inert in term of mobilization potential thereby adding false-positive signal.
Regarding the comparison between VSV-G-pseudo-typed transiently derived and RD3-MolPack24 lentiviral vectors in T lymphocytes, our results are very encouraging. Although the infectivity of the RD-MolPack LVs was two logs inferior to that of the LVs transiently produced, the transduction efficiency was almost comparable. Notably, the T memory differentiation phenotype is not affected when the cells are transduced by either type of LVs.
Finally, we have established that the two RD2- and RD3-MolPack systems hold different growing potentials that render them suitable for either suspension or adherent large-scale cultures, respectively. Very recently, second-generation producer cells expressing Tat, analogous to RD2-MolPack64, was transferred to good manufacturing practice facility for SIN-LV production for WAS gene therapy.12 Furthermore, in alike contest, the incorporation of Tat in the GPRTG-EF1α-hγcOPT producer cells generating SIN-LV for X-SCID gene therapy, was found to improve the performance of the cells and increase the transduction of hCD34+ cells by SIN-LVs.22 In the RD-MolPack contest, we have observed that the expression of Tat correlates with the tendency of the cells to grow in suspension (this study and ref. 18). On the one hand, this feature is an advantage for large-scale production in bioreactor. On the other hand, it could be linked to the oncogenic properties of Tat, as postulated by several groups.26–29
In conclusion, we believe that the RD3-MolPack packaging technology is ready to satisfy the increasing demand of clinical grade large batch vectors toward the routine treatment of rare and acquired diseases affecting children and adults.
Materials and Methods
Plasmids
The SIN-RD114-TR TV (Figure 1a, part 1) was constructed by the BamHI excision of the RD114-TR open reading frame from the phCMV-RD114-TR vector.20 The excised fragment was then inserted in the AflII site of the pGEM-CMV-IRES-puro plasmid obtaining thereby the pGEM-CMV-RD114-TR-IRES-puro intermediate plasmid.
The CMV-RD114-TR-IRES-puro cassette was MluI excised from the intermediate plasmid and finally cloned in the MluI site of the pCCL-SIN-poly-MluI vector previously described in Stornaiuolo et al.18 The SIN-RD114-TR-IN TV (Figure 1a, part 2) was constructed by the EcoRI excision of the RD114-TR-IRES-puro-WPRE cassette from the plasmid SIN-RD114-TR-IN-RRE previously described in Stornaiuolo et al.18 The fragment was then cloned in the EcoRI site of the pCCL-SIN-CMV-BGI-RD114-TR replacing, thereby, the RD114-TR-WPRE cassette with the RD114-TR-IRES-puro-WPRE cassette. The SIN-RD114-TR-IN-antisense (Figure 1a, part 3) was constructed by cloning into the MluI site of the SIN-poly-MluI vector, the expression cassette CMV-BGI-RD114-TR-IRES-puro-WPRE, previously built-up in the pIRESpuro3 plasmid backbone, in antisense orientation. Finally, the SIN-RD114-TR-IN-RRE (Figure 1a, part 4) was previously described in Stornaiuolo et al.18
The pMD.G plasmid encodes the vesicular stomatitis envelope glycoprotein (VSV-G). The SIN-GFP-zeo TV (Figure 1b) was generated by the initial excision of the SIN-GFP cassette from the pCCLsin.PPT.hPGK.eGFP.WPRE.Amp construct, kindly provided by L. Naldini (TIGET, OSR, Milan, Italy), by PvuII partial digestion. The excised fragment was then cloned into the PvuII site of the PBSAAVzeo plasmid, generating an intermediate SIN-GFP-zeo construct. After PstI cut, the SIN-GFP-zeo cassette was cloned into the pGEM3.1MluI plasmid thereby generating the SIN-GFP-zeo TV. The third-generation packaging pCMV-R8.9 construct encoding the HIV gag, pol, rev genes, were kindly provided by L. Naldini (TIGET, OSR, Milan).
Cells
Human embryo kidney-293T (HEK-293T) cells and its derivatives were propagated in Iscove’s modified Dulbecco’s modified Eagle medium (IMDM) supplemented with 10% fetal calf serum (FCS) and Penicillin-Streptomycin-Glutamine (PSG) (Lonza, Basel, Switzerland). CEM A3.01 T cells were grown in RPMI 1640 supplemented with 10% FCS and PSG.
Human peripheral blood mononuclear cells were isolated from healthy donors after centrifugation on a Ficoll-Hypaque gradient (Lymphoprep; Stemcell Technologies, Vancouver, Canada) and then frozen in aliquots. Peripheral blood mononuclear cells were subsequently thawed and cultured in RPMI 1640 supplemented with 10% FCS, PSG, and IL-7 and IL-15 (5 ng/ml, each) (Miltenyi Biotec, Bergisch Gladbach, Germany) after preactivation with CD3/CD28 Dynabeads (Life Technologies Italia, Monza, Italy) for 48 hours as described in Kaneko et al.30
Transient and continuous LV production, concentration, cell transduction, and titer calculation
LVs were obtained by transient cotransfection of HEK-293T cells with the following plasmids: the packaging constructs pCMV-R8.9 (third-generation), the pMD.G plasmid encoding the VSV-G envelope or the SIN-RD114-TR plasmids encoding the RD114-TR envelope, and the third-generation pCCLsin.PPT.hPGK.eGFP.WPRE.Amp (SIN-GFP) TV. Supernatants were harvested 48 hours after transfection.
Continuous LV production was routinely obtained by cultivating RD-MolPack cells in different cell culture devices: standard flasks, a small disposable bioreactor, and HYPERFlask. To evaluate the productivity of the RD-MolPack cells, 2.5 × 106 cells/cm2 in 0.5 ml/cm2 IMDM supplemented with 10% FBS and PSG were seeded in 24-well plates and after 24 hours supernatant was harvested. RD-MolPack LVs were also produced by inoculating 2.5 × 107 RD-MolPack cells in the CELLine AD 1 000 bioreactor (INTEGRA Biosciences AG, Zizers, Switzerland). This disposable bioreactor contains a cell compartment, containing a PET matrix in which the cells grow, and a medium compartment separated by a 10-kDa semipermeable membrane. The cell compartment medium (15-ml of complete medium) was replaced every 48 hours, corresponding to a single harvest, whereas the medium compartment (1,000-ml IMDM supplemented with 1% FCS) was replaced weekly for up to 3 months.
In selected experiments, RD-MolPack cells were cultured in Corning HYPERFlask (Corning Life Science, Lowell, MA) starting the harvests when the total number of cells reached approximately 4 × 108. In all production systems, the harvested supernatant was clarified by 0.45-μm filtration and stored at −80 °C until use. When indicated, the supernatants were concentrated 100-fold by either low speed (3,761 × g at +4 °C for 16 hours in a Multifuge 32-R centrifuge) or high-speed centrifugation (50,000 × g at +4 °C for 2 hours in a Beckman L-80 ultracentrifuge). The viral pellets were resuspended in IMDM medium supplemented with 10% FBS and frozen at −80 °C for deferred use.31
The transduction of CEM A3.01 cell line and activated T lymphocytes was carried out by one cycle of spinoculation at 1,024 × g for 2 hours at 37 °C in the presence of polybrene (8 μg/ml) (Sigma-Aldrich, St Louis, MO). The transduction efficiency was monitored by flow cytometry analysis (FACS Canto II Instrument, BD Bioscience, San Jose, CA) of GFP (SIN-GFP) by the DIVA software (BD Bioscience). The LV titer was calculated on CEM A3.01 cells using the 5–25% range of transduction efficiency as previously described.17
The physical particles (pp) production was estimated by measuring the p24Gag released in the supernatants by the Alliance HIV-1 p24 Antigen ELISA kit (Perkin Elmer, Waltham, MA) following manufacturer’s instructions. It is assumed that 1 ng of p24Gag corresponds to 1 × 107 pp.
Determination of VCN by quantitative (q)-PCR
The VCN of integrated vectors was established by quantitative polymerase chain reaction (q-PCR) using an ABI Prism 7900HT FAST Real-Time PCR system (Applied Biosystems, Foster City, CA) and was analyzed by SDS 2.3 software (Applied Biosystems). The genomic DNA was extracted with the QIAamp Gentra Puregene Cell Kit (QIAGEN GmbH, Hilden, Germany) according to manufacturer’s instructions and 100 ng were used for each reaction (each sample was run in triplicates). Standard amplification curves were obtained by serial dilutions (from 106 to 101 copies, R2 > 0.99, slope range: from −3.7 to −3.2.) of either the specific plasmid DNA or gDNA extracted from a cell line with defined VCN of the analyzed transgene. The sequences of the primer and probe sets used in this study are reported in Supplementary Table S5. The PCR conditions were the following: 2 minutes at 50 °C and 15 minutes at 95 °C, followed by 40 cycles of 15 seconds at 95 °C, and 1 minute at 60 °C.
Southern and northern blot assays
The genomic DNA (gDNA) was isolated by the QIAamp Gentra Puregene Cell Kit (QIAGEN) according to the manufacturer’s instructions. After overnight digestion with the restriction enzymes indicated in Supplementary Figure S5, 10 μg gDNA/sample was run on 0.8% agarose gel an then blotted by Southern capillary transfer onto nylon membranes (GE Healthcare Europe GmbH, Milan, Italy). The membranes were then hybridized to 1.5 × 106 dpm/ml of 32P-random primed labeled of either 661-bp CMV-, or 550-bp RD114-TR- or 339-bp GFP-specific probe, in PerfectHyb PLUS hybridization buffer.
The total cellular RNA was isolated by the TRIzol reagent (Life Technologies) according to manufacturer’s instructions. Five micrograms/sample were run on denaturing formaldehyde/MOPS 1.0% agarose gel, blotted by capillary transfer onto nylon membranes, and then hybridized to 2 × 106 dpm/ml of 32P-random primed labeled of either 550-bp RD114-TR, or 270-bp Ψ specific probe, in PerfectHyb PLUS hybridization buffer.
After extensive washing, the membranes were exposed to X-ray films at −80 °C or to Typhoon Instrument (GE Healthcare Europe GmbH). The intensity of the bands were quantified by the Quantity One 4.6.2 1D Analysis Software (Bio-Rad, Hercules, CA).
Western blot assay
Cellular and viral proteins, the latter derived from isolated cell-free VLPs or LVs, were prepared as previously described17 and then separated by SDS-PAGE on 4–15% Mini-PROTEAN TGX precast gels (Bio-Rad). The primary antibodies were the following: the anti-HIV serum, obtained from an AIDS patient, and kindly donated by G. Poli (OSR, Milan, Italy) was diluted 1:1,000; the anti-TM RD114-TR rabbit serum, kindly provided by F.L. Cosset (INSERM, Paris, France),32 was diluted 1:1,000; the anti-Rev mouse antibody, from Santa Cruz Biotechnology (Dallas, TX, sc-69730), was diluted 1:500; the anti-Tat mouse antibody, obtained from the NIH AIDS Research and Reference Reagent Program (Germantown, MD), was diluted 1:500; the anti-actin rabbit antibody, (A 2066) (Sigma-Aldrich), was diluted 1:2,500. The secondary horseradish peroxidase-linked antibodies were the following: anti-human (NA933V) and anti-rabbit (NA934V) (GE Healthcare, Europe GmbH) were diluted 1:5,000; the anti-mouse (A2066) (Sigma-Aldrich) was diluted 1:10,000. For the chemiluminescence reaction, we used the ECL-Western Blotting Detection Reagents (RPMN2106) (GE Healthcare).
FACS analysis
Immune phenotypic analysis of ex-vivo activated T-cells were carried out by staining with VioGreen-conjugated anti-CD3 monoclonal antibody, APC-Vio770-conjugated anti-CD8 monoclonal antibody, VioBlue-conjugated anti-CD62L monoclonal antibody, PE-Vio770-conjugated anti-CD45RA monoclonal antibody, and APC-conjugated anti-CD95 monoclonal antibody (Miltenyi Biotec). Dead cells were excluded from analysis by 7-AAD (BD Bioscience) staining. Fluorescence-activated cell sorting (FACS) analysis was performed by the FACS Canto II Instrument and the DIVA software (BD Bioscience).
RD3-MolPack24 and RD2-MolPack64 cell metabolism
RD3-MolPack24 and RD2-MolPack64 cells were seeded at 1.5 × 104 cells/cm2 density in T25 flasks. Cells were counted daily for a week and at each time point, supernatants were harvested to measure glucose consumption and lactate production by the YSI 2700 SELECT Biochemistry Analyzer (YSI, Yellow Springs, OH) and the functional and physical titers as described above.
Analysis of possible SIN-RD114-TR mobilization
The possible mobilization frequency of the SIN-RD114-TR TV from the RD3-MolPack24 cells was measured by calculating the percentage of the SIN-RD114-TR gene compared to the GFP gene (100%) in the derived RD3-MolPack24 viral particles. gRNA was extracted by the QIAmp Viral RNA purification kit (Qiagen) and then retro-transcribed to cDNA by the First-Strand Synthesis System (Life Technologies). Quantitative PCR was performed on the cDNAs by using either the RD114-TR or GFP-specific primers and probes (Supplementary Table S5). The percentage of possible SIN-RD114-TR mobilized DNA compared to the total SIN-GFP DNA (100%) was quantified according to the following formula: 2-(Ct SIN-RD114-TR - Ct SIN-GFP). Three different lots of supernatant were analyzed, each containing 2.5 × 1010 pp (2.5 × 103 ng p24Gag). Each supernatant lot was divided in three samples and retrotranscribed into cDNA. Each cDNA was then analyzed in triplicate.
The expected mobilization of SIN-RD114-TR-IN-RRE in target T-cells was expressed as VCN of SIN-RD114-TR-IN-RRE/15,000 cells and calculated with the following formula: MOI × (% of mobilization of SIN-RD114-TR-IN-RRE in RD3-MolPack24 LVs/100) × (% of transduction/100) × 15,000 cells. The detected mobilization of SIN-RD114-TR-IN-RRE into T-cells was measured by calculating the VCN of SIN-RD114-TR-IN-RRE/15,000 cells. Q-PCR was carried out on cellular lysates of T-cells at 15 days post-transduction, using telomerase and RD114-TR specific primer and probe sets (Supplementary Table S5). Two different lots of RD3-MolPack24 LV supernatants were used to transduce T-cells in two independent experiments at MOI 25 and 10 to obtain >90% transduced cells.
Statistical analysis
Mean values ± standard error of the mean are indicated unless otherwise stated. The results were compared by using the paired Student t-test. A P ≤ 0.05 is considered significant.
Acknowledgments
The authors wish to thank Mary Collins and Khaled Sanber (University College London, Cancer Institute, London, UK) for sharing information on the optimization of packaging cell culture conditions.
All authors are employees of MolMed S.p.A.
The first two authors contributed equally to this work.
References
- MacGregor, RR (2001). Clinical protocol. A phase 1 open-label clinical trial of the safety and tolerability of single escalating doses of autologous CD4 T cells transduced with VRX496 in HIV-positive subjects. Hum Gene Ther 12: 2028–2029. [PubMed] [Google Scholar]
- Levine, BL, Humeau, LM, Boyer, J, MacGregor, RR, Rebello, T, Lu, X et al. (2006). Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA 103: 17372–17377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebas, P, Stein, D, Binder-Scholl, G, Mukherjee, R, Brady, T, Rebello, T et al. (2013). Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 121: 1524–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto, DL, Krishnan, A, Li, L, Li, H, Li, S, Rao, A et al. (2010). RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med 2: 36ra43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto, DL (2015). Stem cell gene therapy for HIV: strategies to inhibit viral entry and replication. Curr HIV/AIDS Rep 12: 79–87. [DOI] [PubMed] [Google Scholar]
- Stan, R and Zaia, JA (2014). Practical considerations in gene therapy for HIV cure. Curr HIV/AIDS Rep 11: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti, A, Biasco, L, Scaramuzza, S, Ferrua, F, Cicalese, MP, Baricordi, C et al. (2013). Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341: 1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi, A, Montini, E, Lorioli, L, Cesani, M, Fumagalli, F, Plati, T et al. (2013). Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158. [DOI] [PubMed] [Google Scholar]
- Maus, MV, Grupp, SA, Porter, DL and June, CH (2014). Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123: 2625–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain, M, Brentjens, R, Rivière, I and Park, J (2015). CD19 CAR Therapy for Acute Lymphoblastic Leukemia. Am Soc Clin Oncol Educ Book 35: e360–e363. [DOI] [PubMed] [Google Scholar]
- Greene, MR, Lockey, T, Mehta, PK, Kim, YS, Eldridge, PW, Gray, JT et al. (2012). Transduction of human CD34+ repopulating cells with a self-inactivating lentiviral vector for SCID-X1 produced at clinical scale by a stable cell line. Hum Gene Ther Methods 23: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielgosz, MM, Kim, YS, Carney, GG, Zhan, J, Reddivari, M, Coop, T et al. (2015). Generation of a lentiviral vector producer cell clone for human Wiskott-Aldrich syndrome gene therapy. Mol Ther Methods Clin Dev 2: 14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, Y, Takeuchi, Y, Martin, F, Cosset, FL, Mitrophanous, K and Collins, M (2003). Continuous high-titer HIV-1 vector production. Nat Biotechnol 21: 569–572. [DOI] [PubMed] [Google Scholar]
- Sanber, KS, Knight, SB, Stephen, SL, Bailey, R, Escors, D, Minshull, J et al. (2015). Construction of stable packaging cell lines for clinical lentiviral vector production. Sci Rep 5: 9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, AJ Jr, Fegen, D, Ward, M and Bank, A (2010). RD114 envelope proteins provide an effective and versatile approach to pseudotype lentiviral vectors. Exp Biol Med (Maywood) 235: 1269–1276. [DOI] [PubMed] [Google Scholar]
- Porcellini, S, Alberici, L, Gubinelli, F, Lupo, R, Olgiati, C, Rizzardi, GP et al. (2009). The F12-Vif derivative Chim3 inhibits HIV-1 replication in CD4+ T lymphocytes and CD34+-derived macrophages by blocking HIV-1 DNA integration. Blood 113: 3443–3452. [DOI] [PubMed] [Google Scholar]
- Porcellini, S, Gubinelli, F, Alberici, L, Piovani, BM, Rizzardi, GP and Bovolenta, C (2010). Chim3 confers survival advantage to CD4+ T cells upon HIV-1 infection by preventing HIV-1 DNA integration and HIV-1-induced G2 cell-cycle delay. Blood 115: 4021–4029. [DOI] [PubMed] [Google Scholar]
- Stornaiuolo, A, Piovani, BM, Bossi, S, Zucchelli, E, Corna, S, Salvatori, F et al. (2013). RD2-MolPack-Chim3, a packaging cell line for stable production of lentiviral vectors for anti-HIV gene therapy. Hum Gene Ther Methods 24: 228–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nunzio, F, Piovani, B, Cosset, FL, Mavilio, F and Stornaiuolo, A (2007). Transduction of human hematopoietic stem cells by lentiviral vectors pseudotyped with the RD114-TR chimeric envelope glycoprotein. Hum Gene Ther 18: 811–820. [DOI] [PubMed] [Google Scholar]
- Sandrin, V, Boson, B, Salmon, P, Gay, W, Nègre, D, Le Grand, R et al. (2002). Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 100: 823–832. [DOI] [PubMed] [Google Scholar]
- Neff, T, Peterson, LJ, Morris, JC, Thompson, J, Zhang, X, Horn, PA et al. (2004). Efficient gene transfer to hematopoietic repopulating cells using concentrated RD114-pseudotype vectors produced by human packaging cells. Mol Ther 9: 157–159. [DOI] [PubMed] [Google Scholar]
- Bonner, M, Ma, Z, Zhou, S, Ren, A, Chandrasekaran, A, Gray, JT, et al. (2015). Development of a second generation stable lentiviral packaging cell line in support of clinical gene therapy protocols. 18th ASGCT Annual Meeting, New Orleans, Abstract 81 Abstracts2View™ site. http://www.abstracts2viewcom/asgct/indexphp.
- Hanawa, H, Persons, DA and Nienhuis, AW (2005). Mobilization and mechanism of transcription of integrated self-inactivating lentiviral vectors. J Virol 79: 8410–8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Throm, RE, Ouma, AA, Zhou, S, Chandrasekaran, A, Lockey, T, Greene, M et al. (2009). Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 113: 5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoko, N, Baralle, M, Tisminetzky, S and Buratti, E (2011). InTRONs in biotech. Mol Biotechnol 48: 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombrino, E, Rossi, E, Ballon, G, Terrin, L, Indraccolo, S, Chieco-Bianchi, L et al. (2004). Human immunodeficiency virus type 1 Tat protein modulates cell cycle and apoptosis in Epstein-Barr virus-immortalized B cells. Exp Cell Res 295: 539–548. [DOI] [PubMed] [Google Scholar]
- Kim, RH, Yochim, JM, Kang, MK, Shin, KH, Christensen, R and Park, NH (2008). HIV-1 Tat enhances replicative potential of human oral keratinocytes harboring HPV-16 genome. Int J Oncol 33: 777–782. [PubMed] [Google Scholar]
- Huynh, D, Vincan, E, Mantamadiotis, T, Purcell, D, Chan, CK and Ramsay, R (2007). Oncogenic properties of HIV-Tat in colorectal cancer cells. Curr HIV Res 5: 403–409. [DOI] [PubMed] [Google Scholar]
- Kim, YS, Wielgosz, MM, Hargrove, P, Kepes, S, Gray, J, Persons, DA et al. (2010). Transduction of human primitive repopulating hematopoietic cells with lentiviral vectors pseudotyped with various envelope proteins. Mol Ther 18: 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko, S, Mastaglio, S, Bondanza, A, Ponzoni, M, Sanvito, F, Aldrighetti, L et al. (2009). IL-7 and IL-15 allow the generation of suicide gene-modified alloreactive self-renewing central memory human T lymphocytes. Blood 113: 1006–1015. [DOI] [PubMed] [Google Scholar]
- Strang, BL, Ikeda, Y, Cosset, FL, Collins, MK and Takeuchi, Y (2004). Characterization of HIV-1 vectors with gammaretrovirus envelope glycoproteins produced from stable packaging cells. Gene Ther 11: 591–598. [DOI] [PubMed] [Google Scholar]
- Sandrin, V, Muriaux, D, Darlix, JL and Cosset, FL (2004). Intracellular trafficking of Gag and Env proteins and their interactions modulate pseudotyping of retroviruses. J Virol 78: 7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.