

In this review, Gascard and Tlsty focus on novel insights into the contributions of carcinoma-associated fibroblasts (CAFs) to disease progression. They also discuss the generation of CAFs and recent therapeutic approaches aimed at blunting or reverting detrimental protumorigenic phenotypes associated with CAFs.
Keywords: carcinoma-associated fibroblasts, CAF, microenvironment, field cancerization, stromal–epithelial interactions, CAF targeted therapies
Abstract
The tumor stroma is no longer seen solely as physical support for mutated epithelial cells but as an important modulator and even a driver of tumorigenicity. Within the tumor stromal milieu, heterogeneous populations of fibroblast-like cells, collectively termed carcinoma-associated fibroblasts (CAFs), are key players in the multicellular, stromal-dependent alterations that contribute to malignant initiation and progression. This review focuses on novel insights into the contributions of CAFs to disease progression, emergent events leading to the generation of CAFs, identification of CAF-specific biomarkers predictive of disease outcome, and recent therapeutic approaches aimed at blunting or reverting detrimental protumorigenic phenotypes associated with CAFs.
Stromal cells have typically been viewed as a heterogeneous group of connective tissue cells that form the supportive structure in which the functional cells of the tissue reside. In this context, the stroma plays an essential role in tissue architecture by providing physical support for the epithelium via a complex network of fibrillar proteins called the extracellular matrix (ECM), which contains primarily collagens, elastins, and fibronectin. However, the stroma also consists of a spectrum of cell types that fulfill very distinct but complementary functions in controlling tissue identity and homeostasis: fibroblasts that secrete ECM proteins as well as proteases that remodel it, adipocytes that act as an energy reserve, endothelial cells organized into blood vessels that transfer oxygen and nutrients to and from the tissue, and immune cells (T cells, B cells, dendritic cells, and macrophages) that mediate defense mechanisms against infections and other aggressions. Also resident within the stroma are mesenchymal stem cells (MSCs), a primordial, fibroblastoid cell type that is ill defined but is believed to give rise to mesodermal derivatives that include fibroblasts, endothelial cells, and adipocytes. Both fibroblasts and MSCs are isolated by identical methods and exhibit similar fibroblastic morphologies. These two cell types are so similar that some have maintained that they are indistinguishable by morphology, cell surface markers, differentiation potential, and immunologic properties (Hematti 2012). Adding to the complexity, MSCs, similar to fibroblasts, have been isolated from nearly every tissue type in the body and display heterogeneity (Sriram et al. 2015). In early studies, the deterministic potential of resident stromal fibroblasts was unmasked based on the use of elegant heterotopic recombinants. The recombination of wing epithelium with leg (fibroblastic) mesenchyme resulted in the generation of scales instead of feathers (Dhouailly et al. 1978). Thus, each of the components of the stroma described above is essential for proper tissue homeostasis, maintaining architecture, and, most importantly, physiologically appropriate functions (Kalluri and Zeisberg 2006; Tlsty and Coussens 2006; Bing and Trayhurn 2009).
As we now know, alterations of each of these stromal cell types can contribute to the tumorigenic process through remodeling of the stroma (Karagiannis et al. 2012) and widespread dysregulation of cell signaling and intercellular cross-talk (Kalluri and Zeisberg 2006; Tlsty and Coussens 2006; Bing and Trayhurn 2009; Ishii et al. 2016). Similar to reports for the malignant epithelium, the malignant stroma exhibits expression profiles that provide prognostic information independent of epithelial biomarkers (Finak et al. 2008; Ma et al. 2009; Ostman and Augsten 2009; Sharma et al. 2010). Novel mechanisms of action by which carcinoma-associated fibroblasts (CAFs) can promote tumorigenesis and impair drug sensitivity (through direct effects on malignant cells and through mobilization and recruitment of other tumor cell types), and the molecular mediators involved in these processes have been extensively characterized (Fig. 1). The knowledge gained through these studies has been paramount for the design of increasingly sophisticated therapies specifically targeting CAFs. These recent findings have led experts in the field to propose an emerging concept of fibroblasts as “architects of cancer pathogenesis” (Marsh et al. 2013).
Figure 1.
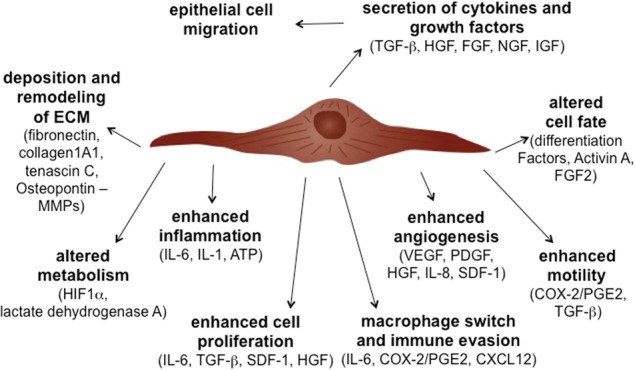
Multiple secreted factors and resultant phenotypes stimulated by CAFs. CAFs induce multiple phenotypes in neighboring tumor epithelial cells as well as other stromal cell types. Soluble factors secreted by CAFs have been involved in promoting each of these phenotypes.
This is in stark contrast to the historical view in which the scientific and patient communities have focused their attention on epithelial cancer cells, since the vast majority of human cancers exhibits epithelial abnormalities, and mutational changes in these cells are readily observable. However, the last 15 years have seen a shift in this momentum, driven, to a large extent, by a flourishing literature that has built a strong case for a major involvement of the stroma in restraining (Bissell and Hines 2011; Dumont et al. 2013) or facilitating cancer progression (Olumi et al. 1999; Kalluri and Zeisberg 2006; Tlsty and Coussens 2006; Finak et al. 2008; Dumont et al. 2013). Recent provocative reports have further extended this novel concept by unequivocally demonstrating that, in some cancers, the stroma goes beyond playing the role of a facilitator of the tumorigenic process. Alterations in stromal cell signaling and transcriptional program can precede (or act independently of) epithelial cell alterations and actually act as a driver of the tumorigenic process (DeFilippis et al. 2014; Scherz-Shouval et al. 2014; Hu et al. 2015; Procopio et al. 2015). As a result of this conceptual shift, we have witnessed a recent surge in therapeutic strategies aimed at reverting alterations associated with a protumorigenic stroma, in particular the activity of anti- and protumorigenic immune cells whose balance determines, to a large extent, disease outcome (Callahan and Wolchok 2015). Besides the widely recognized role of immune cells (i.e., immunotolerance) and endothelial cells (i.e., angiogenesis) in cancer progression, there is now strong evidence that CAFs are major players as well. Indeed, interactions of CAFs with cellular components of the immune system contribute, to a large extent, to the tumor-promoting role of CAFs through immunosuppression and sustained inflammation (Liao et al. 2009; Erez et al. 2010; Quante et al. 2011; Servais and Erez 2013). Moreover, the CAF status has an impact on the clinical behavior of a tumor (Chang et al. 2004, 2005; Kalluri and Zeisberg 2006; Tlsty and Coussens 2006; Finak et al. 2008; Berdiel-Acer et al. 2014), in particular early and targeted metastasis (Dumont et al. 2013; Zhang et al. 2013; Calon et al. 2014).
In this review, we focus specifically on key questions in the field that relate to the processes by which CAFs contribute to disease progression, processes that lead to the generation of CAFs, their identity, their cell of origin, the pivotal role of the dysregulation of signaling pathways that contributes to the acquisition of a CAF phenotype, and the promises and challenges associated with therapeutic regimens aimed at eliminating CAFs or at least reverting their detrimental CAF phenotype.
CAFs and disease progression
Early studies on stromal influences in malignancy involved the purification of fibroblast-like cells from human disease-free tissue and from invasive tumor tissue to compare their properties. A pioneering study using a prostate cancer murine xenograft model showed that while human prostate CAFs promote tumor growth, fibroblasts from disease-free tissue inhibit it instead (Olumi et al. 1999). Additionally, in this study, CAFs were found to secrete factors that enhance epithelial tumor cell proliferation and mutagenesis as well as angiogenesis. These secreted factors also inhibited epithelial cell apoptosis and functional cell–cell adhesion interactions. In this study, secreted factors could not only facilitate tumor progression of an immortalized but nontumorigenic cell but also initiate malignant phenotypes in morphologically and genotypically normal epithelial cells. Recently, using in vivo heterotypic cell recombinant studies, it was shown that human breast CAFs can promote early dissemination, tumorigenesis, and metastasis of breast cancer cells through deposition of a distinct ECM characterized by aligned collagen fibers (Dumont et al. 2013). CAFs promoted acquisition of a mesenchymal morphology associated with increased dissemination and metastasis in both premalignant and malignant mammary epithelial cells, whereas fibroblast-like cells from disease-free individuals favored maintenance of an epithelial morphology and constrained early dissemination, tumor growth, and metastasis. These findings support the idea that CAFs contribute to malignant disease progression and that normalizing the ECM organization within a tumor may be an effective therapeutic strategy. Similar coculture and heterotypic cell recombinant studies have been performed with cells from multiple tissue types, including breast and skin (Barcellos-Hoff and Ravani 2000; Maffini et al. 2004; Barcellos-Hoff and Medina 2005; Arendt et al. 2010).
To obtain clues as to the stromal pathways involved in CAF-dependent facilitation of malignant phenotypes, several studies have compared gene expression in disease-free fibroblasts and CAFs derived from various tissues (Sadlonova et al. 2009; Saadi et al. 2010; DeFilippis et al. 2012; Berdiel-Acer et al. 2014). In one of these studies conducted in breasts, the most dramatic change in expression between disease-free fibroblast and CAF populations was the repression of a cell surface protein, CD36. Strikingly, CD36 is typically expressed on all stromal cell components, adipocytes, fibroblasts, endothelial cells, and immune cells. When expressed, CD36-expressing stromal cells are in an anti-tumorigenic state. However, when reduced in expression, each stromal component acquires a distinct protumorigenic state. Thus, fibroblasts that have low expression of CD36 elaborate greater quantities of collagens and fibronectin than fibroblasts with high expression of CD36. Likewise, endothelial cells with low CD36 exhibit increased angiogenesis, preadipocytes with low CD36 fail to differentiate into adipocytes, and, finally, immune cells with low CD36 are in a M2 (protumorigenic) state rather than a M1 (anti-tumorigenic) state. This study also documents that CD36 controls a coordinated, multicellular program and that CD36 is necessary and sufficient to control a wide range of protumorigenic phenotypes (DeFilippis et al. 2012, 2014). These phenotypes, often observed in desmoplasia, include low adipocyte content, increased ECM proteins (collagen and fibronectin), an increased number of fibroblasts and vascular cells (DeFilippis et al. 2012), and potentially immune cells. CD36 is a widely expressed protein that modulates cell type-specific and ligand-specific functions, including adipocyte differentiation, angiogenesis, apoptosis, transforming growth factor-β (TGF-β) activation, cell–ECM interactions, and immune signaling (Silverstein and Febbraio 2009). Accompanying CD36 repression is the secretion of soluble factors, such as activin A, which has been implicated as a key effector in a DNA damage-inducible secretory program that acts in a cell-extrinsic fashion to generate many of the above phenotypes (Fig. 2; Fordyce et al. 2010, 2012). Extending these findings, recent evidence has been provided for CD36 repression as part of a dramatic switch in fibroblast identity. Fibroblasts characterized by a disappearance of caveloae exhibited a concomitant loss of CD36, cavin, and caveolin-1 expression and were shown to increase hypoxia-induced factor-1-α (HIF-1-α) and TGF-β signaling. These CD36-nonexpressing fibroblasts also exhibited a metabolic shift from mitochondrial oxidative phosphorylation to glycolysis, reflected by increased expression in monocarboxylate transporter 4 (MCT4). The consequences of this multifaceted signature, characteristic of the acquisition of a CAF phenotype, are far-reaching (Martinez-Outschoorn et al. 2014, 2015). Current studies are under way to investigate the mechanisms responsible for CD36 repression in CAFs. As discussed below, repression of CD36 may reflect changes in the epigenetic landscape.
Figure 2.
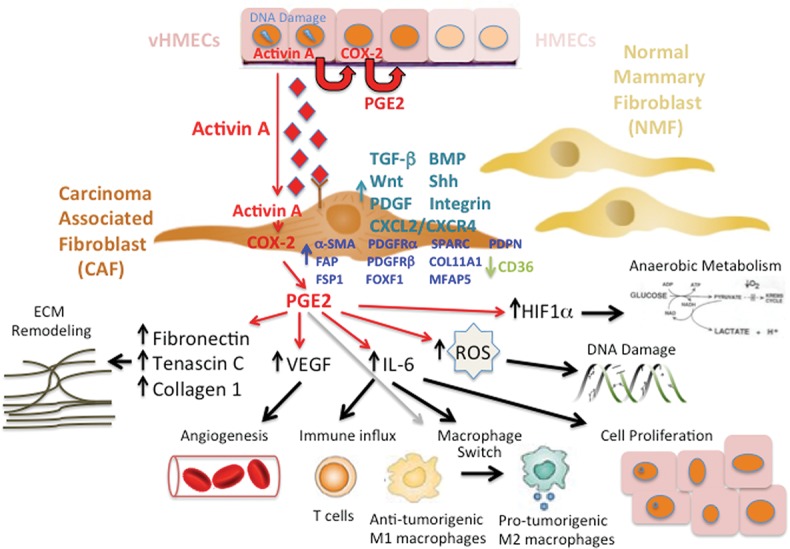
Birth of a CAF. Example of a p53/activin A/cyclooxygenase-2 (COX-2)-dependent DNA damage pathway originating in epithelial cells that elicits protumorigenic effects in neighboring fibroblasts through paracrine stimulation via activin A. Activation of fibroblasts by activin A triggers a spectrum of COX-2-driven protumorigenic phenotypes, including ECM remodeling, angiogenesis, immune influx, macrophage switch, cell proliferation, DNA damage, and acquisition of a hypoxic/glycolytic microenvironment. Also shown are the signaling pathways that are dysregulated and the CAF-specific proteins whose expression levels change upon acquisition of the CAF phenotype. Dysregulated signaling pathways include up-regulation of TGF-β, bone morphogenetic protein (BMP), Wnt, Sonic hedgehog (Shh), platelet-derived growth factor (PDGF), C-X-C motif ligand 12 (CXCL12)/CXCR4, and integrin-mediated signaling. Changes in protein expression include up-regulation of α-smooth muscle actin (α-SMA), fibroblast-activating protein (FAP), fibroblast-specific protein-1 (FSP1), PDGF receptor a (PDGFRa), PDGFRb, Forkhead box F1 (FOXF1), SPARC, Podoplanin (PDPN), and, more recently, collagen 11-α1 (COL11A1) and microfibrillar-associated protein 5 and down-regulation of CD36.
The above studies demonstrate that CAFs facilitate disease progression through cell-intrinsic dysregulated signaling and epigenetic events as well as through modulating cell-extrinsic events such as ECM remodeling (Cirri and Chiarugi 2011; Ungefroren et al. 2011), angiogenesis, immune status, and stromal metabolic state. Each group of investigators studying oncologic processes has their own measure of disease progression. Thus, clinicians use RECIST (response evaluation criteria in solid tumors) criteria, measuring tumor size and sensitivity or resistance to therapy (Eisenhauer et al. 2009), while pathologists evaluate grade (fraction of cells that proliferate and exhibit normal and abnormal tissue morphologies and specific cellular characteristics) and stage (spatial dispersal of mutated epithelial cells). On the molecular level, investigators measure endpoints that are tumor cell-intrinsic (changes in mutational burden, metabolic state, drug resistance index, and metastasis) and tumor cell-extrinsic (angiogenesis, stromal metabolic state, and immune status). Remarkably, recent literature has demonstrated that CAFs can modulate each of these multiple endpoints.
This rich, dynamic, and evolving perception is particularly well illustrated in the multiple mechanisms by which CAFs contribute to drug resistance (Castells et al. 2012). Typically, drug resistance is attributed to the accumulations of genetic and epigenetic changes in epithelial tumor cells influencing uptake, metabolism, and export of drug. However, emergent properties within the tumor stroma can actually contribute diverse mechanisms that facilitate drug resistance and survival of cells after therapy. CAFs can (1) alter cell–matrix interactions that control epithelial cell sensitivity to apoptosis, (2) secrete proteins that control epithelial cell survival and proliferation, (3) contribute to direct cell–cell interactions where cells can exchange membrane fragments (trogocytosis) and communicate, (4) create physical barriers that facilitate the acquisition of drug resistance, and (5) activate epigenetic plasticity in neighboring cells. Thus, CAFs alter the physical, dimensional, and chemical aspects of tissues in an ongoing dynamic and coevolutionary manner (Cuiffo and Karnoub 2012; Junttila and de Sauvage 2013).
Having documented CAF contributions to malignant phenotypes, one could ask whether fibroblast-like cells also contribute to premalignant phenotypes. The central role of CAFs in cancer progression is further illustrated by a recent study that demonstrates that CAFs promote ductal carcinoma in situ (DCIS) progression to invasive breast carcinoma via paracrine interleukin-6 (IL-6) signaling (Osuala et al. 2015). Multicellular three-dimensional (3D) structures composed of DCIS cells exhibit increased proliferation and migration when cultured with CAFs. Exposure of these multicellular structures to an IL-6-neutralizing antibody inhibits these phenotypes. Notably, selective knockdown of IL-6 in CAFs, but not in DCIS cells, abrogates these phenotypes. This report brings further mechanistic insights to a prior study reporting that DCIS lesions with a heightened DNA damage/activin A/cyclooxygenase-2 (COX-2) signature were associated with a more reactive stroma (Fordyce et al. 2010, 2012) and more frequent progression to invasive cancer (Kerlikowske et al. 2010).
Are CAFs detected only in association with malignant carcinomas? Do the functional characteristics of CAFs exist in fibroblast-like cells prior to formation of a carcinoma and, if so, how early in the process? Are CAFs created by interactions with carcinomas? Several studies have shed light on these questions and contributed to a stunning realization that stromal changes and the existence of CAF-like cells can precede the formation of malignant epithelial cells. While it is clear that coculturing fibroblasts and malignant epithelial cell lines can result in reciprocal gene expression changes (Rozenchan et al. 2009), characterization of tissues at high risk for carcinoma formation demonstrate that stromal changes associated with the CAF phenotype already exist and are therefore not dependent on signals from a carcinoma for their generation (Saadi et al. 2010; DeFilippis et al. 2014).
Evidence for the acquisition of a CAF phenotype prior to malignancy has been underscored by a recent study showing that fibroblasts derived from breast tissues obtained from healthy women with high mammographic density (and no breast cancer) exhibit desmoplastic/protumorigenic phenotypes compared with fibroblasts derived from breast tissues obtained from women with low mammographic density (DeFilippis et al. 2012, 2014). When fibroblasts from disease-free women of high and low mammographic density were compared, gene expression profiling identified CD36 as the gene most differentially expressed. Similar to the observations described above for the stromal components that repress CD36 and display multiple, coordinated, protumorigenic phenotypes, alterations in the expression of this master regulator, CD36, in the stromal components of healthy breast tissue prior to tumor formation are associated with increased risk for future tumor incidence and progression (DeFilippis et al. 2014).
In an additional example, microenvironment-induced loss of expression of a key repressor of canonical Notch signaling, CSL, in fibroblasts primes them for acquisition of a CAF phenotype. This loss occurs upon silencing of the tumor suppressor TP53 in the fibroblasts by fibroblast growth factor 2 (FGF2) secreted by neighboring epithelial cells (Hu et al. 2015; Procopio et al. 2015). These events in turn lead to subsequent spontaneous multifocal (epithelial) keratinocyte tumor formation in both mice and humans. Indeed, CAFs from hormone-independent breast tumors have been shown to express higher levels of FGF2 than their hormone-dependent counterparts. The secreted FGF2 promotes tumor cell proliferation in a hormone-independent fashion through activation of progesterone receptors (PRs) downstream from FGF receptor 2 (FGFR2) (Giulianelli et al. 2008).
Taken together, these findings led to the provocative conclusion that, although genetic changes in epithelial cells can drive carcinogenesis, nongenetic tissue changes in the stroma can also be the cause rather than just the consequence of the malignant process and thus contribute to altered tissue fields. These studies also hint at environmental influences that can create a protumorigenic stromal state prior to formation of a malignant lesion. These observations are consistent with the idea that stress-induced signals can trigger a coordinated multicellular program, creating a niche or field of tissue that is predisposed to malignant formation. This view of “field cancerization” or “tissue field effect” as an active evolving state in which cell–cell interactions and feedback loops integrate to produce a functional outcome is rapidly emerging as a powerful way of studying carcinogenesis and provides novel therapeutic targets. We define “tissue field effect” or “field cancerization” as a field of correlated (interdependent) cellular and/or molecular changes that predispose to the development and progression of multiple malignancies within that territory. The studies identifying CD36 and CSL modulations certainly qualify the consequent multicellular coordinated programs as “tissue field effect” or “field cancerization” events.
CAF: cell or state?
As noted above, a precise molecular definition of CAF does not exist. Instead, CAFs are defined operationally. CAFs can represent the major cellular component of tumor stroma where they are the main source for connective tissue components of the ECM and various classes of proteolytic enzymes. Particularly in breast and pancreatic carcinomas, CAFs can compose up to 80% of the tumor mass (Olive et al. 2009) as a result of an extensive desmoplastic process; i.e., a dramatic deposition of ECM proteins (Kalluri and Zeisberg 2006; Pietras and Ostman 2010). Attempts to define CAFs are most often aimed at identifying morphological characteristics and expression of specific markers (Table 1). Whereas mature fibroblasts exhibit a thin, wavy, and small spindle morphology, CAFs are often described as immature fibroblasts and appear as large, plump spindle-shaped cells with prominent nucleoli. Tissues that contain a high proportion (>50%) of immature fibroblasts correlate with increased microvessel density, tumor-associated macrophages, epithelial–mesenchymal transition (EMT), and poor prognosis (Ha et al. 2014). The most widely used (and earliest) reference for acquisition of a CAF phenotype is the myocyte marker α-smooth muscle actin (α-SMA) (Ronnov-Jessen et al. 1995; Kalluri and Zeisberg 2006; Ozdemir et al. 2014). Additional markers expressed in CAFs include fibroblast-specific protein-1 (FSP1; also called S100A4), fibroblast-activating protein (FAP), vimentin, platelet-derived growth factor receptor-α (PDGFR-α), PDGFR-β, the chondroitin sulfate proteoglycan neuron-glial antigen-2 (NG2), and prolyl-4-hydroxylase (Augsten 2014; Ohlund et al. 2014). These markers, along with osteonectin, podoplanin, metalloproteinases (MMPs), and the transcription factor Forkhead box F1 (FOXF1), have been proposed to be predictive of disease outcome based on independent association of protein expression with patient survival (Ha et al. 2014). However, most of these proteins are not CAF-specific (Augsten 2014; Ohlund et al. 2014). The recent identification of proteins whose expression is exquisitely restricted to CAFs and exhibits remarkable prognostic value, asporin (Maris et al. 2015), collagen 11-α1 (COL11A1) (Vazquez-Villa et al. 2015), and microfibrillar-associated protein 5 (MFAP5) (Yeung et al. 2014) may improve the reliable identification of CAFs, not to mention their potential value as candidate biomarkers and therapeutic targets.
Table 1.
List of CAF-specific and nonspecific markers
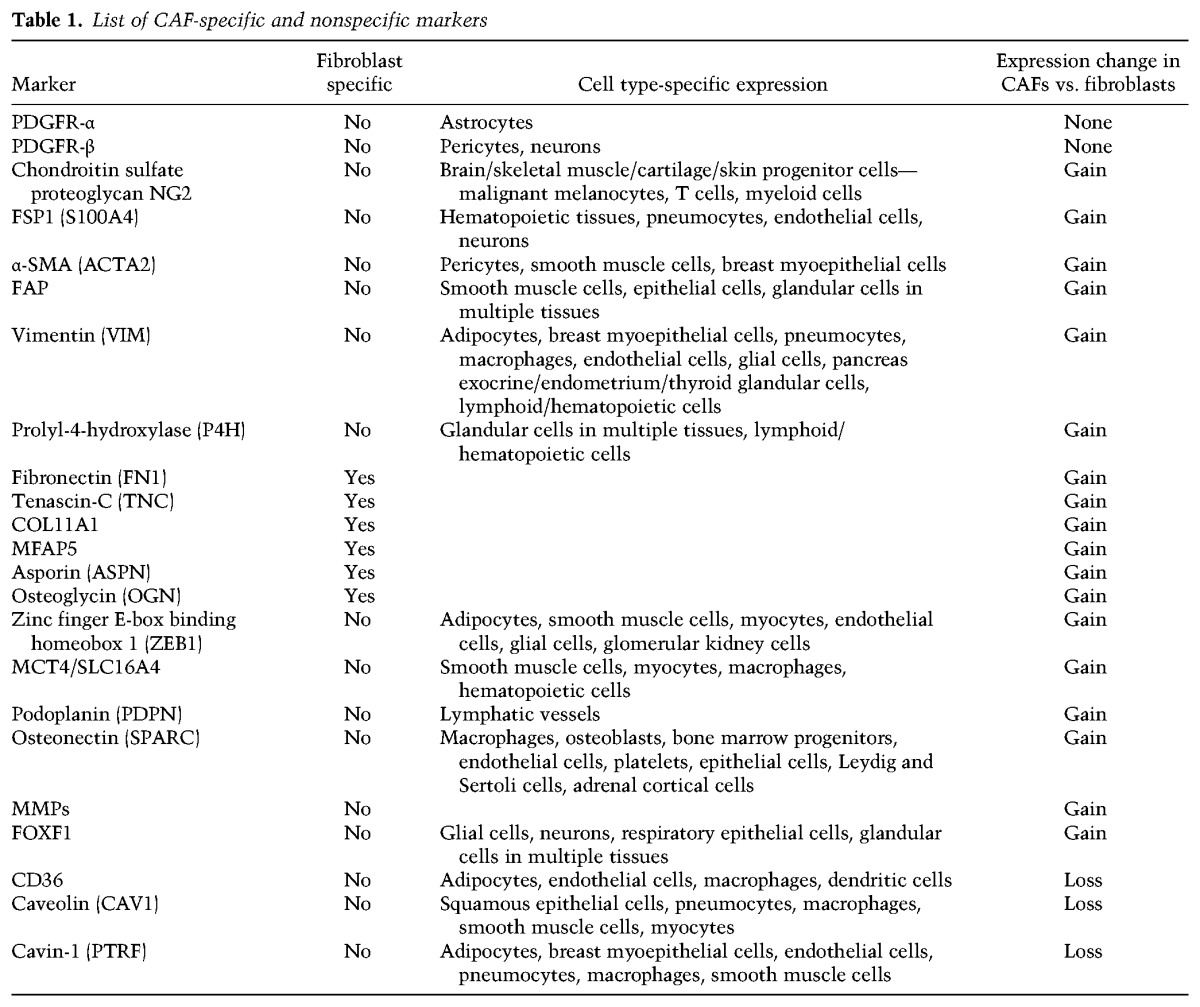
To add to the challenge, CAFs coexist as heterogeneous populations defined by partly overlapping marker expression patterns, making it difficult to conclusively define the role of CAFs based on these markers and identify the individual factors involved in clinically relevant tumor–stroma interactions (Augsten 2014; Ohlund et al. 2014; Paulsson and Micke 2014). Thus, two subsets of CAFs have been identified by immunohistochemistry in mouse models of pancreatic and breast cancer: a FSP1-negative subtype coexpressing α-SMA, PDGFR-β, and NG2 and a FSP1-positive subtype lacking expression of α-SMA, PDGFR-β, and NG2 (Sugimoto et al. 2006). The phenotypic heterogeneity of CAFs extends to their multifaceted functionality (Augsten 2014). CAF subtypes have been defined based on their anti-tumorigenic (F1 subtype) or protumorigenic (F2 subtype) activity (Bhowmick et al. 2004; Ozdemir et al. 2014) or distinct tumor type-specific gene and protein expression profiles (Orimo and Weinberg 2007). For example, CAFs derived from human epidermal growth factor receptor 2-positive (Her2+) breast tumors exhibit significantly higher expression levels of cytoskeleton and integrin signaling genes/proteins compared with CAFs derived from triple-negative (estrogen receptor negative [ER−]/PR−/Her2−) or ER+ breast tumors (Tchou et al. 2012). This finding is in accordance with the observed enhanced migration of breast cancer cells cocultured with CAFs derived from Her2+ tumors and may account in part for the poorer prognosis of Her2+ breast tumors. In a separate study, a significantly higher number of CAFs and higher microvessel density, vasohibin-1/CD31 ratio, and insulin growth factor-1 (IGF-1) transcript levels but lower intratumoral microvessel nestin expression have been observed in breast intralobular carcinomas (ILCs) compared with intraductal carcinomas (IDCs), suggesting higher proliferation of CAFs and endothelial cells but less mature newly formed microvessels in ILC than in IDC (Nakagawa et al. 2016). Thus, CAFs display functional differences depending on the tumor environment in which they evolve (Orimo and Weinberg 2007). This observation has important ramifications for prevention, risk stratification, diagnostics, and treatment, as discussed later.
CAF heterogeneity may result from the multiple origins of CAFs (α-SMA+ cells). While a majority of CAFs are thought to arise from activation of resident fibroblasts (see below), studies in genetically modified mouse models have demonstrated that about one-third of CAFs derive from other sources: bone marrow (BM)-derived cells (Quante et al. 2011; Lecomte et al. 2012; McDonald et al. 2015), adipocytes (Bochet et al. 2013), endothelial cells (Zeisberg et al. 2007), or even, perhaps, epithelial cells (Radisky et al. 2007) that undergo mesenchymal transition when instructed by systemic factors secreted by the tumor (Chen et al. 2015a). This complexity has led some investigators to propose that a CAF is a “cell state” rather than a “cell type” (Madar et al. 2013). In turn, the resulting CAF subsets fuel the tumorigenic process through secretion of specialized factors that mediate various aspects of stromal alterations. The concept that fibroblast specialization depends on the cell of origin is illustrated by a study using a murine model of skin carcinoma that had been irradiated and engrafted with BM from green fluorescent protein (GFP) transgenic mice. The investigators showed that one-third of BM-derived GFP+ cells infiltrating the tumor were CAFs expressing NG2 or α-SMA, whereas ∼90% of Thy1+ fibroblasts originated from resident GFP− cells (Lecomte et al. 2012). Notably, only α-SMA+ cells derived from GFP+ BM cells produced proinvasive stromal MMP-13.
The birth of a CAF: When does a fibroblast become a CAF?
In light of these morphological, molecular, and functional diversities, one pressing question that comes to mind is: When does a fibroblast become a CAF, or how does a fibroblast-like cell acquire tumor-promoting properties? The nomenclature suggests that CAFs are found only in the context of tumors. However, conceptually, the term CAF could also refer to any fibroblast with protumorigenic activity. In addition to the CD36 and CSL studies in non-tumor-containing tissue described above, a study has reported that fibroblasts isolated from patients with rheumatoid arthritis (nonmalignant samples) are as capable as CAFs in promoting formation of invasive mammary tumors when coinjected with DCIS cells in mice (Hu et al. 2008). Acquisition of a tumor-promoting activity phenotype by a fibroblast (through intrinsic and extrinsic mechanisms) therefore seems to be an earlier event than originally thought. This leads to the next question: Which processes contribute to this acquisition of a CAF phenotype?
Recent genetic studies conducted in CAFs derived from human ovarian and breast tumors have demonstrated that unlike epithelial cancer cells, copy number alterations, loss of heterozygosity, and/or the number of TP53 mutations are extremely rare in CAFs (4%–5% of samples). Such rare alterations therefore cannot constitute the basis for the widespread tumor-promoting phenotypes exemplified by CAFs (Qiu et al. 2008; Hosein et al. 2010; Corver et al. 2011).
In contrast, epigenetic alterations (i.e., DNA methylation, histone modifications, and nucleosome structure) and changes in expression of noncoding RNAs (i.e., microRNAs and long noncoding RNAs) account for many of the (early) gene expression changes observed upon acquisition of a CAF state (Kang et al. 2015). For example, TGF-β type II receptor (TGFBR2) is epigenetically silenced in 70% of prostate cancer patients and in host mouse prostate fibroblasts in a murine xenograft model as a result of secretion of IL-6 (Banerjee et al. 2014). Notably, this epigenetic event coincides with elevated DNA methyltransferase 1 (DNMT1) activity and histone H3 Lys9 trimethylation (H3K9me3), supporting the occurrence of more frequent epigenetic events in CAFs. Another study demonstrated a pivotal role for the proinflammatory cytokine leukemia inhibitory factor (LIF) in epigenetic-driven activation of fibroblasts into CAFs via constitutive activation of the Janus kinase 1 (JAK1)/signal transducer and activator of transcription 3 (STAT3) pathway following STAT3 acetylation and up-regulation of DNMT1 and DNMT3b activities (Albrengues et al. 2015). Combined inhibition of DNMT activities and JAK signaling results in long-term reversion of the CAF phenotype into a nonprotumorigenic fibroblast phenotype. Histone methylation (H3K4me3) and acetylation (H3K9ac) are essential for up-regulation of protumorigenic/desmoplastic genes such as tissue inhibitor of metalloprotease-1 (TIMP1), ACT2(α-SMA), TGFB1, and collagen 1 (COL1A1) during hepatic stellate cell transdifferentiation (Kim and Shukla 2005; Perugorria et al. 2012). Epithelial cells cocultured with CAFs exhibit hypermethylation and repressive chromatin histone marks (H3K27me3 and H3K9me2) at the promoter of the tumor suppressor cystatin M (CST6) and other loci as well as aberrant protein kinase AKT1 activation and repression of the 4-phosphatase INNP4B (Lin et al. 2008). Notably, acquisition of these latter phenotypes requires direct cell–cell contact, underlining the complexity of the crosstalk between cancer cells and CAFs. One may also speculate that the widespread repression of CD36 described in the breast stroma may result from epigenetic reprogramming. In favor of this hypothesis, macroH2A1, a histone variant that is relocated across the genome in response to oncogene-induced senescence and DNA damage (Chen et al. 2015b), has been shown to regulate CD36 expression in mouse livers (Boulard et al. 2010).
Acquisition of a CAF phenotype also involves binding of specific microRNAs to the 3′ untranslated region of selected mRNAs and induction of their degradation. Thus, down-regulation of miR-31 and miR-214 and up-regulation of miR-155 in normal ovarian fibroblasts induce functional conversion into CAFs; the reverse experiment has been shown to reverse this conversion (Mitra et al. 2012). Genes up-regulated in both microRNA-reprogrammed normal fibroblasts and CAFs are highly enriched in chemokines, in particular C-C motif ligand 5 (CCL5), a direct target of miR-214. The regulation of microRNA expression controls key pathways involved in acquisition of a CAF phenotype. For example, up-regulation of miR-21 in fibroblasts results in deregulation of TGF-β1 signaling and TGF-β1-induced conversion to CAFs through inhibition of translation of the TGF-β inhibitor Smad 7 (Li et al. 2013). Up-regulation of the hypoxia-induced miR-210 in young fibroblasts triggers senescence and conversion into CAFs able to promote EMT in cancer cells, support angiogenesis, and recruit monocytes/macrophages (Taddei et al. 2014). Phenotypes resulting from changes in expression of specific microRNAs in CAFs can also contribute to drug resistance. Esophageal cancer cells exposed to conditioned medium (CM) from normal fibroblasts transfected with miR-27a/b are more cisplatin-resistant than cells exposed to naïve CM (Tanaka et al. 2015). Overexpression of miR-27a/b in normal fibroblasts results in up-regulation of α-SMA and TGF-β (i.e., acquisition of a CAF phenotype), a phenotype that is CAF-specific, since transfection of miR-27a/b in esophageal cancer cells has no significant impact on chemosensitivity. As predicted, cancer cell chemosensitivity is restored after CAF exposure to a TGF-β-neutralizing antibody.
MicroRNA biology illustrates the extensive involvement and functional consequences of cross-talk between tumor cells and CAFs—the effects are reciprocal. One study found that CAFs mediate the epigenetic silencing of miR-200b (a microRNA family that plays a central role in the regulation of EMT during metastasis) in gastric cancer cells, stimulating invasion and peritoneal dissemination in a murine model (Kurashige et al. 2015). Similarly, CAFs induce down-regulation of miR-205 in prostate cancer cells, thus feeding an oxidative stress/proinflammatory axis leading to EMT. In this system, the re-expression of miR-205 in cancer cells prevents and reverts CAF-induced EMT and fibroblast activation by cancer cells (Gandellini et al. 2014; Pennati et al. 2014; Taddei et al. 2014). Down-regulation of miR-26b in CAFs derived from ER+ breast tumors results in enhanced cancer cell migration and invasion (Verghese et al. 2013). This mechanism may also be at the root of the intriguing progressive loss of ER expression seen in some breast tumors. Indeed, two microRNAs secreted by CAFs, miR-221/222, have been directly linked to ER repression, a finding that may explain mechanistically how the mitogen-associated protein kinase (MAPK) pathway represses ER in breast cancer cells (Shah et al. 2015). Notably, these microRNAs are part of a microRNA signature indicative of hyperactive MAPK signaling and are significantly associated with reduced recurrence-free and overall survival. As an example of acquisition of a CAF phenotype potentiated by neighboring cancer cells, fibroblasts convert into CAFs after uptake of miR-155-containing microvesicles secreted by neighboring pancreas cancer cells (Pang et al. 2015). The down-regulation of the anti-proliferative/proapoptotic tumor protein 53-inducible nuclear protein 1 (TP53INP1) by miR-155 in fibroblasts is the likely event contributing to this switch. Uptake of miR-155 by CAFs may also account for the dramatic repression of CD36 reported in breast stromal cells (DeFilippis et al. 2012), since miR-155 has been shown to regulate CD36 in the liver (Lin et al. 2015). The dependency of such protumorigenic phenotypes on miR-155 would substantiate a previous report highlighting the involvement of mir-155 in breast cancer progression (Chou and Werb 2012; Pang et al. 2015). Last, muscle-specific miR-133b has been recently identified as a soluble factor secreted by activated fibroblasts to support paracrine activation of nonactivated fibroblasts and promote tumor progression (Doldi et al. 2015). The above studies provide a strong body of evidence that the modulation of epigenetic control and targeted alterations of microRNAs represent novel therapeutic targets in the tumor microenvironment.
Acquisition of a CAF state: therapeutic opportunities
We discussed the synergistic cross-talk between cancer cells, CAFs, and other stromal cells that together drive neoplastic progression. The realization of these biological links justifies the design of clinical studies based on the targeting of CAF (and, more generally, reversal of the attributes of a protumorigenic microenvironment) in addition to targeting cancer cells in order to create a tumor-resistant environment that inhibits malignant phenotypes in diseased epithelial cells (Takebe et al. 2013; Togo et al. 2013; Zhou et al. 2015). Such clinical studies can now be envisioned based on the findings from experimental studies with agents directed against fibroblast-specific proteins (FAP, COL11A1, and MFAP5) or signaling pathways exemplified in CAFs (TGF-β, hedgehog, Notch, FGF, PDGF, or C-X-C motif ligand 2 [CXCL2]/CXCR4 signaling). As shown in Figure 3 and Table 2, fibroblast-directed therapy can be envisioned as either “ablating” CAFs by interfering with their survival or “normalizing” them by interfering with secreted protumorigenic signals (Kalluri and Zeisberg 2006; Ishii et al. 2016).
Figure 3.
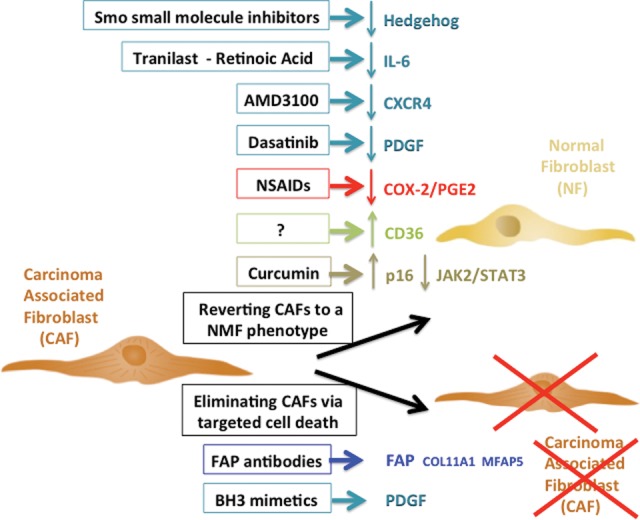
CAF-driven therapeutic opportunities. Drugs specifically targeting CAFs fall into two categories: drugs aimed at (1) reverting the CAF phenotype to a “normal” fibroblast phenotype by targeting pathways responsible for this phenotype (hedgehog, PDGF, or IL-6 CXCR4 signaling), by using anti-fibrotic agents (nonsteroidal anti-inflammatory drugs [NSAIDs] or tranilast) that will modulate ECM remodeling, or by using drugs with wider spectrum of activity such as curcumin; or (2) eliminating CAFs by delivering proapoptotic molecules using “carriers” recognizing CAF-specific proteins (FAP-specific antibodies or PDGF BH3 mimetics). Drugs listed here are those with the most promising outcome, as they specifically target CAFs, for the most part spare healthy epithelial and stromal cells, and have limited/no secondary proinflammatory effects that could spark secondary tumors. Drugs targeting the TGF-β, BMP, activin A, or Wnt pathways are also in development but are more challenging in the clinical setting, as they fulfill key physiological functions in a large variety of cell types.
Table 2.
Clinical trials testing drugs targeting CAFs

Potential therapies aimed at eliminating CAFs
Agents targeting FAP
FAP is a membrane-bound serine protease selectively expressed in fibroblasts. Its up-regulation in CAFs makes it a prime target. FAP silencing inhibits stromagenesis, tumor growth, and angiogenesis in lung and colon cancer murine models (Santos et al. 2009) and suppresses cell proliferation, migration, and invasion (metastasis) of ovarian squamous cell carcinoma cells in vitro and in vivo as a result of inactivation of phosphatase and tensin homolog (PTEN)/phosphoinositide 3-kinase (PI3K)/AKT and Ras extracellular signal-regulated kinase (ERK) signaling (Wang et al. 2014). Although antibodies targeting FAP are promising candidate drugs in animal models (Loeffler et al. 2006; Ostermann et al. 2008), clinical trials assessing two antibodies (Table 2) have not shown the expected efficacy (Hofheinz et al. 2003). The depletion of FAP+ stromal cells (CAFs) has uncovered an unexpected benefit in the latest pancreatic ductal adenocarcinoma (PDAC) therapeutic regimens based on the use of two immunological checkpoint antagonists that promote the function of T cells: anti-cytotoxic T-lymphocyte-associated protein 4 (α-CTLA-4) and anti-programmed cell death ligand 1 (α-PD-L1). Indeed, CAFs exert immunosuppressive activity through secretion of the chemokine CXCL12, which is known to coat cancer cells and enable tumor immune evasion (Fig. 1). As predicted, a CXCL12 receptor inhibitor synergized with α-CTLA-4 and α-PD-L1 to clear cancer cells (Feig et al. 2013). The initial development of small-molecule inhibitors of FAP enzymatic activity has been a suboptimal strategy, since FAP tumorigenic activity depends greatly on tumor microenvironment and cancer type (i.e., FAP can act as a tumor promoter or tumor suppressor). Alternative strategies have sought to develop drugs that would be activated only in high-FAP-expressing cells where FAP acts as tumor promoter, sparing cells in which it fulfills a beneficial tumor suppressor activity (Brennen et al. 2012). Ongoing clinical trials are assessing the therapeutic value of human CD8+ T cells engineered to target FAP-expressing cells (Schuberth et al. 2013) and two FAP-antibody/cytokine fusion proteins, RO6874281 and RO6874813 (Table 2). The diagnostic and therapeutic value of radio-immunoconjugates that bind to FAP with high affinity and are internalized rapidly in cancer cells expressing high levels of FAP (such as CAFs) has been validated in vitro and in a murine preclinical model in vivo (Fischer et al. 2012).
FGF5
FGF5, a FGF that exhibits oncogenic properties, is up-regulated in cancer cells and neighboring stromal cells, including fibroblasts and macrophages (Kornmann et al. 1997). The therapeutic efficacy of vaccines against FGF5 is currently being tested (Table 2).
MCT4
Studies have described early phenotypes that accompany acquisition of the CAF phenotype, including a metabolic switch characterized by an up-regulation of MCT4 (Martinez-Outschoorn et al. 2014, 2015). A recent clinical trial will evaluate the effect of metformin on reverting this switch using MCT4 repression as a readout (Table 2).
BH3 mimetics
CAFs are primed for cell death as a result of heightened PDGF signaling (Rizvi et al. 2014). Cell death can be triggered by BH3 mimetics, an approach that can be exploited for therapeutic purposes. Given the addiction of CAFs for PDGF signaling, Rizvi et al. (2014) reasoned that PDGF isomers (that are abundantly expressed in desmoplastic stroma of cholangiocarcinoma) could specifically prime CAFs for targeted BH3 mimetic-driven cell death. As predicted, PDGF-primed myofibroblasts exhibited an increase in the proapoptotic protein Puma and underwent full-blown apoptosis via Puma-mediated Bak activation after exposure to the BH3 mimetic navitoclax or the B-cell lymphoma 2 (Bcl-2)-specific BH3 mimetic ABT-199. This treatment reduced tumor formation and tumor burden in a murine model of cholangiocarcinoma (a highly desmoplastic carcinoma).
Potential therapies aimed at reverting the CAF state to a non-tumor-promoting fibroblastic state
The benefit of a therapeutic regimen including a stromal (CAF) target may be best evaluated in highly desmoplastic, aggressive, and metastatic cancers that do not benefit significantly from standard systemic therapies, such as PDAC.
Epigenetic modifiers
Treatment of an aggressive mouse model of PDAC with the DNA demethylating agent 5-aza-2′-deoxycytidine (5-AZA) slowed PDAC progression and markedly extended survival when applied early. Transient administration inhibited tumor growth when initiated later, without adverse side effects. Escaping tumors contained areas of sarcomatoid transformation but with disappearance of CAFs (Shakya et al. 2013). Studies have also investigated the potential predictive, diagnostic, and therapeutic value of specific microRNAs within PDAC, taking advantage of microRNA stability even in a tissue with high proteolytic activity. Potential candidates, including miR-21, miR-196a, and miR-217, may serve as long-awaited tools for screening and optimized treatment of PDAC (Steele et al. 2011). Targeting miR-21 in CAFs also seems a promising strategy in breast cancer (Ren et al. 2016). These studies provide a rationale for the design of future combination therapies including hypomethylating agents.
A large number of clinical trials have been built on the concept that targeting prodesmoplastic signaling pathways will not only eliminate protumorigenic stromal components but also improve drug delivery to cancer cells. Indeed, it is often underappreciated that most of the drugs described below act simultaneously on cancer cells and stromal cells (in particular CAFs). Moreover, they are most often evaluated as part of multidrug therapy clinical trials.
TGF-β inhibitors
TGF-β (and TGF-β family members) plays a key role in acquisition of a CAF phenotype. Multiple clinical trials based on the use of either TGF-β antibodies, TGF-β kinase inhibitors, or TGF-β antisense oligonucleotides have been initiated (Table 2). Although a few drugs are now assessed as part of phase III trials, the main challenge is that both pro- and anti-tumoral effects have been assigned to TGF-β receptors on fibroblasts (Berking et al. 2001; Tuxhorn et al. 2002; Bhowmick et al. 2004; Cheng et al. 2007). Targeting the bone morphogenetic protein (BMP) pathway raises similar challenges.
Hedgehog inhibitors
Hedgehog signaling antagonists target primarily stromal cells in vitro and in vivo, since cancer cells are mostly hedgehog-independent (Yauch et al. 2008). However, this promising approach has also proven challenging despite the approval of vismodegib for treatment of basal cell carcinoma (Table 2).
Notch inhibitors
Two γ-secretase inhibitors, R04929097 and MK0752, have been evaluated for the treatment of breast and pancreatic cancer (Table 2). Research and development on RO4929097 has been terminated despite promising phase I/II results. A pilot study aimed at evaluating MK0752 in the treatment of early-stage breast cancer is ongoing. Six recently characterized Gli inhibitors (GANT58 and GANT61 HPI-1, HPI-2, HPI-3, and HPI-4) have not been tested yet in the clinical setting.
PDGFR inhibitors
Targeting the PDGFR pathway, which is specifically heightened in CAFs, has proven to be very promising. Crenolanib is a selective and potent inhibitor of PDGFR currently being evaluated extensively in phase II trials for the treatment of gastrointestinal stromal tumors, gliomas, and hematopoietic malignancies (Table 2).
FGFR inhibitors
Earlier, we discussed the importance of FGF signaling in the synergistic roles played by cancer cells and stromal cells in tumorigenesis. Most inhibitory drugs bind to FGFRs and blunt their activity, leading to inhibition of proliferation and cell death in FGFR-overexpressing tumor cells. A second type of drugs consists of antibody–drug conjugates. Several drugs with more or less specificity for each FGFR are currently being tested in clinical trials (Table 2).
Multitarget (receptor) tyrosine kinase inhibitors
This family of drugs that target multiple tyrosine kinase receptors (expressed in cancer cells and/or stromal cells) is the most promising, with several drugs currently being assessed as part of phase III or phase IV clinical trials (Table 2). Screening of a panel of small-molecule kinase inhibitors on CAF growth has identified five potent compounds, including three PDGFR inhibitors active at nanomolar concentrations. Testing of four FDA-approved PDGFR inhibitors (dasatinib, nilotinib, sorafenib, and imatinib) on 37 CAF strains identified dasatinib as the most effective compound (Haubeiss et al. 2010). Tumor cells incubated with CM from CAFs preincubated with dasatinib exhibited reduced proliferation, confirming that dasatinib could blunt CAF tumor-promoting activity (Haubeiss et al. 2010). Promising results have also been obtained in a mouse model of cervical cancer (Jain et al. 2008). In light of the importance of the PDGF/PDGFR pathway in CAF biology, blockage of PDGFR activation represents a major opportunity for the development of novel pharmaceutical compounds and the design of combined therapies (Gialeli et al. 2014).
CXCL12/CXCR4 inhibitors
CAFs promote cancer cell invasion through CXCL12/CXCR4 signaling-induced FAK/β-1 integrin clustering in cancer cells. The superior efficacy of a CXCR4 antagonist (that targets CAFs) over a FAK inhibitor (that targets cancer cells) on inhibiting the invasiveness of gastric cancer cells supports the idea that targeting CAFs is a promising therapeutic strategy (Izumi et al. 2016). The clinical efficacy of CXCR4 competitive ligands and antibodies is currently being tested (Table 2).
Anti-fibrotic agents
Recent evidence has documented the long-term, protumorigenic, adverse effects of a fibrotic/inflammatory stroma. Using a mouse model of postpartum breast cancer, Lyons et al. (2011) have deciphered the mechanisms by which mammary gland involution, which is characterized by a dramatically reactive stroma with abundant fibrillar collagen and high COX-2 expression, drives tumor progression. COX-2 inhibition with NSAIDs during mammary gland involution reduced stromal reactivity as well as primary tumor growth and lung metastasis. Similarly, immune-driven “regression” of high-grade DCIS of the breast, also characterized by reactive stromal fibrosis, has been paradoxically associated with increased risk of subsequent invasive carcinoma (Wasserman and Parra-Herran 2015). The use of anti-fibrotic agents to curb tumor stromal alterations (CAF phenotypes) in combined therapies therefore seems a logical strategy (Table 2). COX-2 inhibitors, such as celecoxib, are tested in multiple combined therapy-based clinical trials. Promising results have been obtained in lymphoma, lung cancer, and melanoma mouse models treated with the anti-fibrotic agent tranilast (Ohshio et al. 2015). This treatment resulted in reduced tumor infiltration with immunosuppressive cells (regulatory T cells and myeloid-derived suppressor cells) and activation of tumor-associated antigen-specific CD8+ T cells. Reactivation of the host systemic anti-tumor immune response resulted from a decrease in secretion of stromal-derived factor-1 (SDF-1), prostaglandin E2, and TGF-β by CAFs. The synergistic effect of CAF targeted therapy with cell-based cancer immunotherapies (cell-based vaccines) provides a rationale for the development of novel therapeutic regimens (for review, see Slany et al. 2015).
Agents blocking secretion of cytokines by CAFs
Retinoic acid inhibits EMT in cancer cells (i.e., their migration) as a result of inhibition of IL-6 secretion by CAFs (Guan and Chen 2014).
Agents inducing selective senescence in CAFs
Curcumin efficiently reverts the CAF phenotype and blunts paracrine procarcinogenic potential in primary breast CAFs, even at low doses. The observed decreases in α-SMA expression, secretion of protumorigenic factors (SDF-1, IL-6, MMP-2, MMP-9, and TGF-β), and cell motility result from up-regulation of p16INK4A and other tumor suppressor proteins and concomitant inactivation of the JAK2/STAT3 pathway, leading to DNA damage-independent senescence. This line of treatment is attractive in many aspects: Curcumin is a pharmacologically safe natural product; it is active at low doses; its effects are maintained even after drug withdrawal and cell propagation; it triggers DNA damage-independent senescence only in proliferating cells (including CAFs), thus sparing healthy cells; and, last, the p16INK4A-dependent senescence that it triggers is not accompanied by an inflammatory secretory phenotype (Hendrayani et al. 2013).
Challenges to targeting CAFs as a therapeutic approach
One of the main messages of this review is that detailed and customized characterization of CAF/stroma profiles is paramount for the identification of novel targets that will be the basis for prognostic and diagnostic tools as well as novel therapies (Micke and Ostman 2004). An exciting field of research will consist of customizing the evaluation of the stroma's tumor-promoting ability for cancer patients to optimize therapeutic strategies to target both cancer cells and their permissive stroma. Therapeutic strategies targeting tumor cell–stroma interactions and sparing, whenever possible, healthy cells should be favored to prevent the emergence of acquired resistance (Meads et al. 2009; Li et al. 2015).
A second major message of this review is that the stromal and epithelial components of a tissue, malignant or healthy, act in an integrated, reciprocal fashion. Therefore, simultaneous targeting of tumor stroma and epithelial cancer cells with combination therapies may be a successful therapeutic approach for treating various cancers and have the added benefit of preventing drug resistance. Indeed, drug resistance often arises from a coerced epithelial–stromal metabolic coupling, in which cancer cells induce aerobic glycolysis and proliferation in fibroblasts via oxidative stress. In turn, proliferating CAFs protect cancer cells against apoptosis by providing nutrients to cancer cells (Martinez-Outschoorn et al. 2011b). Furthermore, standard chemotherapies transform phenotypically and metabolically characterized breast fibroblasts into CAFs, creating a hypoxic, glycolytic, autophagic, and proinflammatory microenvironment that in turn activates stemness through Sonic hedgehog (Shh)/GLI signaling, antioxidant response, and interferon-mediated signaling in breast cancer cells (Peiris-Pages et al. 2015). Hypoxic senescent fibroblasts can promote prostate cancer aggressiveness by inducing EMT, secreting energy-rich compounds to support cancer cell growth, promoting M2 macrophage polarization, and promoting angiogenesis through recruitment of BM-derived endothelial precursor cells and proliferation and invasion of mature endothelial cells (Taddei et al. 2014). Such unforeseen effects could trigger secondary tumorigenic processes facilitated by recruitment of immunosuppressive cells. Altered metabolic interactions are the basis for tamoxifen resistance in ER+ MCF7 breast cancer cells (Martinez-Outschoorn et al. 2011a). A combination of tamoxifen and dasatinib (a drug that targets PDGF signaling, which CAFs greatly depend on) overcomes fibroblast-induced tamoxifen resistance. This drug combination has the added benefit of imposing an anti-oxidant effect on both fibroblasts and cancer cells, thus reducing the risk of tumor–stroma coevolution. Fibronectin secretion by CAFs has been reported to synergize with a heightened β1-integrin signaling pathway in tamoxifen-resistant breast cancer cells to mediate acquisition of an EMT phenotype (Yuan et al. 2015). CAFs have been also reported to mediate resistance to anti-angiogenic therapy (Pietras et al. 2008) or MMP-mediated cetuximab resistance in head and neck squamous cell carcinoma cells (Johansson et al. 2012). They can adopt decoy strategies such as expression of CD44, a cell surface protein normally expressed in epithelial cells, to support stemness and drug resistance of malignant cancer cells (Kinugasa et al. 2014). The simultaneous targeting of tumor stroma and epithelial cancer cells with combination therapies may be a successful therapeutic approach for treating various cancers while overcoming drug resistance. In that context, it is greatly underappreciated that the most promising drugs not only inhibit the growth of epithelial tumor cells but often also target signaling pathways required for CAF function. Consequently, endpoints that specifically read out the status of the stroma during therapy are lacking.
Future drug discovery will be complicated by the fact that a CAF targeted strategy can be a double-edged sword. Indeed, recent studies have shown that targeting CAFs may result in protumorigenic effects depending on the target and the tissue context (Ozdemir et al. 2014; Koliaraki et al. 2015; Wagner 2016).
Future perspectives
For decades, investigators have studied cancer at its most basic level, isolating cells and processes. More recently, some have suggested that studying malignancy and cellular contributions to disease processes would benefit from considering malignancies as emergent structures and processes. Emergent structures are more than the sum of their parts. Indeed, they exhibit unique properties that arise from the collective behavior of these parts. Tissues are emergent structures that arise from cellular components that themselves are emergent structures. In an emergent entity, architectural, temporal, and dynamic aspects of tissues are integrated via feedback loops. Conceptualizing CAFs as emergent structures that constitute a major component of a more encompassing emergent structure (a malignancy) may aid in our understanding of how CAFs come to be and contribute to the biology of malignancy. This alternative perspective can most easily be seen in competing theories of carcinogenesis. For decades, the majority of the cancer research field has embraced the somatic mutation theory as the template for cancer biology and potential therapeutic approaches. According to this somatic mutation theory, malignancies have monoclonal origins, the accumulation of mutations providing selective advantage to a clone of cells. This view has been challenged through the years by an alternative interpretation of the data that hypothesizes that malignancy can also result from a dysregulated organ/tissue homeostasis, which generates an abnormal but organized field supporting multifocal emergence of malignancy. Understanding the relative contribution of these two ideas is important, since targeting a dominant clone with driver mutations dictates a different therapeutic approach than attacking “field cancerization” by targeting an entire field of interacting, constantly dynamically evolving cells. Studying CAFs in the context of “field cancerization” or “tissue field effects” may hold the answer to some of the most persistent problems in clinical research today—recurring metastases and drug resistance.
Acknowledgments
We thank Dr. Joseph Caruso (member of the Tlsty laboratory, University of California at San Francisco) for critical reading of the manuscript. This work was supported by National Institutes of Health/National Cancer Institute grants PO1 CA107584, R01 CA097214, U54 CA143803, R01 CA187800, R01 CA197977, and R35 CA197674; Pfizer Research Foundation grant WS2067777; Susan G. Komen for the Cure Foundation Scientific Advisory Council grant SAC 100011; and California Breast Cancer Research Program grants 14OB-0165 and 18OB-0060 to T.D.T.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.279737.116.
References
- Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, Philippe C, Herraiz Serrano C, Benamar S, Croce O, et al. 2015. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun 6: 10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Rudnick JA, Keller PJ, Kuperwasser C. 2010. Stroma in breast development and disease. Semin Cell Dev Biol 21: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augsten M. 2014. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol 4: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee J, Mishra R, Li X, Jackson RS II, Sharma A, Bhowmick NA. 2014. A reciprocal role of prostate cancer on stromal DNA damage. Oncogene 33: 4924–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Medina D. 2005. New highlights on stroma–epithelial interactions in breast cancer. Breast Cancer Res 7: 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Ravani SA. 2000. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res 60: 1254–1260. [PubMed] [Google Scholar]
- Berdiel-Acer M, Sanz-Pamplona R, Calon A, Cuadras D, Berenguer A, Sanjuan X, Paules MJ, Salazar R, Moreno V, Batlle E, et al. 2014. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol Oncol 8: 1290–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P, Herlyn M. 2001. Transforming growth factor-β1 increases survival of human melanoma through stroma remodeling. Cancer Res 61: 8306–8316. [PubMed] [Google Scholar]
- Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. 2004. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303: 848–851. [DOI] [PubMed] [Google Scholar]
- Bing C, Trayhurn P. 2009. New insights into adipose tissue atrophy in cancer cachexia. Proc Nutr Soc 68: 385–392. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Hines WC. 2011. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 17: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, Dray C, Guiet R, Maridonneau-Parini I, Le Gonidec S, et al. 2013. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res 73: 5657–5668. [DOI] [PubMed] [Google Scholar]
- Boulard M, Storck S, Cong R, Pinto R, Delage H, Bouvet P. 2010. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenetics Chromatin 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. 2012. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J Natl Cancer Inst 104: 1320–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Wolchok JD. 2015. Clinical activity, toxicity, biomarkers, and future development of CTLA-4 checkpoint antagonists. Semin Oncol 42: 573–586. [DOI] [PubMed] [Google Scholar]
- Calon A, Tauriello DV, Batlle E. 2014. TGF-β in CAF-mediated tumor growth and metastasis. Semin Cancer Biol 25: 15–22. [DOI] [PubMed] [Google Scholar]
- Castells M, Thibault B, Delord JP, Couderc B. 2012. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci 13: 9545–9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. 2004. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol 2: E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van't Veer LJ, Bartelink H, et al. 2005. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci 102: 3738–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, Sun Y. 2015a. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med 13: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. 2015b. MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol Cell 59: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng N, Chytil A, Shyr Y, Joly A, Moses HL. 2007. Enhanced hepatocyte growth factor signaling by type II transforming growth factor-β receptor knockout fibroblasts promotes mammary tumorigenesis. Cancer Res 67: 4869–4877. [DOI] [PubMed] [Google Scholar]
- Chou J, Werb Z. 2012. MicroRNAs play a big role in regulating ovarian cancer-associated fibroblasts and the tumor microenvironment. Cancer Discov 2: 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirri P, Chiarugi P. 2011. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res 1: 482–497. [PMC free article] [PubMed] [Google Scholar]
- Corver WE, Ter Haar NT, Fleuren GJ, Oosting J. 2011. Cervical carcinoma-associated fibroblasts are DNA diploid and do not show evidence for somatic genetic alterations. Cell Oncol (Dordr) 34: 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuiffo BG, Karnoub AE. 2012. Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell Adh Migr 6: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, Berman HK, Gauthier ML, Zhao J, Hu D, et al. 2012. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov 2: 826–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilippis RA, Fordyce C, Patten K, Chang H, Zhao J, Fontenay GV, Kerlikowske K, Parvin B, Tlsty TD. 2014. Stress signaling from human mammary epithelial cells contributes to phenotypes of mammographic density. Cancer Res 74: 5032–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhouailly D, Rogers GE, Sengel P. 1978. The specification of feather and scale protein synthesis in epidermal-dermal recombinations. Dev Biol 65: 58–68. [DOI] [PubMed] [Google Scholar]
- Doldi V, Callari M, Giannoni E, D'Aiuto F, Maffezzini M, Valdagni R, Chiarugi P, Gandellini P, Zaffaroni N. 2015. Integrated gene and miRNA expression analysis of prostate cancer associated fibroblasts supports a prominent role for interleukin-6 in fibroblast activation. Oncotarget 6: 31441–31460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont N, Liu B, Defilippis RA, Chang H, Rabban JT, Karnezis AN, Tjoe JA, Marx J, Parvin B, Tlsty TD. 2013. Breast fibroblasts modulate early dissemination, tumorigenesis, and metastasis through alteration of extracellular matrix characteristics. Neoplasia 15: 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. 2009. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45: 228–247. [DOI] [PubMed] [Google Scholar]
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. 2010. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 17: 135–147. [DOI] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. 2013. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci 110: 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, et al. 2008. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med 14: 518–527. [DOI] [PubMed] [Google Scholar]
- Fischer E, Chaitanya K, Wuest T, Wadle A, Scott AM, van den Broek M, Schibli R, Bauer S, Renner C. 2012. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin Cancer Res 18: 6208–6218. [DOI] [PubMed] [Google Scholar]
- Fordyce C, Fessenden T, Pickering C, Jung J, Singla V, Berman H, Tlsty T. 2010. DNA damage drives an activin a–dependent induction of cyclooxygenase-2 in premalignant cells and lesions. Cancer Prev Res (Phila) 3: 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordyce CA, Patten KT, Fessenden TB, Defilippis R, Hwang ES, Zhao J, Tlsty TD. 2012. Cell-extrinsic consequences of epithelial stress: activation of protumorigenic tissue phenotypes. Breast Cancer Res 14: R155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandellini P, Giannoni E, Casamichele A, Taddei ML, Callari M, Piovan C, Valdagni R, Pierotti MA, Zaffaroni N, Chiarugi P. 2014. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid Redox Signal 20: 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gialeli C, Nikitovic D, Kletsas D, Theocharis AD, Tzanakakis GN, Karamanos NK. 2014. PDGF/PDGFR signaling and targeting in cancer growth and progression: focus on tumor microenvironment and cancer-associated fibroblasts. Curr Pharm Des 20: 2843–2848. [DOI] [PubMed] [Google Scholar]
- Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, Novaro V, Gongora A, Baldi A, Molinolo A, et al. 2008. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: a role for the FGF-2/FGFR-2 axis. Int J Cancer 123: 2518–2531. [DOI] [PubMed] [Google Scholar]
- Guan J, Chen J. 2014. Tumor microenvironment: the promising target for tumor therapy. Cancer Cell Microenviron 1: e81. [Google Scholar]
- Ha SY, Yeo SY, Xuan YH, Kim SH. 2014. The prognostic significance of cancer-associated fibroblasts in esophageal squamous cell carcinoma. PLoS One 9: e99955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubeiss S, Schmid JO, Murdter TE, Sonnenberg M, Friedel G, van der Kuip H, Aulitzky WE. 2010. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol Cancer 9: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hematti P. 2012. Mesenchymal stromal cells and fibroblasts: a case of mistaken identity? Cytotherapy 14: 516–521. [DOI] [PubMed] [Google Scholar]
- Hendrayani SF, Al-Khalaf HH, Aboussekhra A. 2013. Curcumin triggers p16-dependent senescence in active breast cancer-associated fibroblasts and suppresses their paracrine procarcinogenic effects. Neoplasia 15: 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, et al. 2003. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26: 44–48. [DOI] [PubMed] [Google Scholar]
- Hosein AN, Wu M, Arcand SL, Lavallee S, Hebert J, Tonin PN, Basik M. 2010. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res 70: 5770–5777. [DOI] [PubMed] [Google Scholar]
- Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A, Violette S, Nikolskaya T, Nikolsky Y, et al. 2008. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 13: 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, Wu Y, Qin J. 2015. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS One 10: e0125625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii G, Ochiai A, Neri S. 2016. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev 99: 186–196. [DOI] [PubMed] [Google Scholar]
- Izumi D, Ishimoto T, Miyake K, Sugihara H, Eto K, Sawayama H, Yasuda T, Kiyozumi Y, Kaida T, Kurashige J, et al. 2016. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin β1 clustering and invasiveness in gastric cancer. Int J Cancer 138: 1207–1219. [DOI] [PubMed] [Google Scholar]
- Jain RK, Lahdenranta J, Fukumura D. 2008. Targeting PDGF signaling in carcinoma-associated fibroblasts controls cervical cancer in mouse model. PLoS Med 5: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson AC, Ansell A, Jerhammar F, Lindh MB, Grenman R, Munck-Wikland E, Ostman A, Roberg K. 2012. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol Cancer Res 10: 1158–1168. [DOI] [PubMed] [Google Scholar]
- Junttila MR, de Sauvage FJ. 2013. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501: 346–354. [DOI] [PubMed] [Google Scholar]
- Kalluri R, Zeisberg M. 2006. Fibroblasts in cancer. Nat Rev Cancer 6: 392–401. [DOI] [PubMed] [Google Scholar]
- Kang N, Shah VH, Urrutia R. 2015. Membrane-to-nucleus signals and epigenetic mechanisms for myofibroblastic activation and desmoplastic stroma: potential therapeutic targets for liver metastasis? Mol Cancer Res 13: 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagiannis GS, Poutahidis T, Erdman SE, Kirsch R, Riddell RH, Diamandis EP. 2012. Cancer-associated fibroblasts drive the progression of metastasis through both paracrine and mechanical pressure on cancer tissue. Mol Cancer Res 10: 1403–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerlikowske K, Molinaro AM, Gauthier ML, Berman HK, Waldman F, Bennington J, Sanchez H, Jimenez C, Stewart K, Chew K, et al. 2010. Biomarker expression and risk of subsequent tumors after initial ductal carcinoma in situ diagnosis. J Natl Cancer Inst 102: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Shukla SD. 2005. Histone h3 modifications in rat hepatic stellate cells by ethanol. Alcohol Alcohol 40: 367–372. [DOI] [PubMed] [Google Scholar]
- Kinugasa Y, Matsui T, Takakura N. 2014. CD44 expressed on cancer-associated fibroblasts is a functional molecule supporting the stemness and drug resistance of malignant cancer cells in the tumor microenvironment. Stem Cells 32: 145–156. [DOI] [PubMed] [Google Scholar]
- Koliaraki V, Pasparakis M, Kollias G. 2015. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J Exp Med 212: 2235–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann M, Ishiwata T, Beger HG, Korc M. 1997. Fibroblast growth factor-5 stimulates mitogenic signaling and is overexpressed in human pancreatic cancer: evidence for autocrine and paracrine actions. Oncogene 15: 1417–1424. [DOI] [PubMed] [Google Scholar]
- Kurashige J, Mima K, Sawada G, Takahashi Y, Eguchi H, Sugimachi K, Mori M, Yanagihara K, Yashiro M, Hirakawa K, et al. 2015. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 36: 133–141. [DOI] [PubMed] [Google Scholar]
- Lecomte J, Masset A, Blacher S, Maertens L, Gothot A, Delgaudine M, Bruyere F, Carnet O, Paupert J, Illemann M, et al. 2012. Bone marrow-derived myofibroblasts are the providers of pro-invasive matrix metalloproteinase 13 in primary tumor. Neoplasia 14: 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhang D, Wang Y, Sun P, Hou X, Larner J, Xiong W, Mi J. 2013. MiR-21/Smad 7 signaling determines TGF-β1-induced CAF formation. Sci Rep 3: 2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Hu SQ, Xiao L. 2015. The cancer-associated fibroblasts and drug resistance. Eur Rev Med Pharmacol Sci 19: 2112–2119. [PubMed] [Google Scholar]
- Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. 2009. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One 4: e7965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HJ, Zuo T, Lin CH, Kuo CT, Liyanarachchi S, Sun S, Shen R, Deatherage DE, Potter D, Asamoto L, et al. 2008. Breast cancer–associated fibroblasts confer AKT1-mediated epigenetic silencing of Cystatin M in epithelial cells. Cancer Res 68: 10257–10266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Jia J, Du T, Li W, Wang X, Wei J, Zeng H, Yao L, Chen X, Zhuang J, et al. 2015. Overexpression of miR-155 in the liver of transgenic mice alters the expression profiling of hepatic genes associated with lipid metabolism. PLoS One 10: e0118417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. 2006. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest 116: 1955–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons TR, O'Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. 2011. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med 17: 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Dahiya S, Richardson E, Erlander M, Sgroi DC. 2009. Gene expression profiling of the tumor microenvironment during breast cancer progression. Breast Cancer Res 11: R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar S, Goldstein I, Rotter V. 2013. ‘Cancer associated fibroblasts’—more than meets the eye. Trends Mol Med 19: 447–453. [DOI] [PubMed] [Google Scholar]
- Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. 2004. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci 117: 1495–1502. [DOI] [PubMed] [Google Scholar]
- Maris P, Blomme A, Palacios AP, Costanza B, Bellahcene A, Bianchi E, Gofflot S, Drion P, Trombino GE, Di Valentin E, et al. 2015. Asporin is a fibroblast-derived TGF-β1 inhibitor and a tumor suppressor associated with good prognosis in breast cancer. PLoS Med 12: e1001871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh T, Pietras K, McAllister SS. 2013. Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta 1832: 1070–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, et al. 2011a. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 10: 2521–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, Lisanti MP. 2011b. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 43: 1045–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Outschoorn U, Sotgia F, Lisanti MP. 2014. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol 41: 195–216. [DOI] [PubMed] [Google Scholar]
- Martinez-Outschoorn UE, Sotgia F, Lisanti MP. 2015. Caveolae and signalling in cancer. Nat Rev Cancer 15: 225–237. [DOI] [PubMed] [Google Scholar]
- McDonald LT, Russell DL, Kelly RR, Xiong Y, Motamarry A, Patel RK, Jones JA, Watson PM, Turner DP, Watson DK, et al. 2015. Hematopoietic stem cell-derived cancer-associated fibroblasts are novel contributors to the pro-tumorigenic microenvironment. Neoplasia 17: 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meads MB, Gatenby RA, Dalton WS. 2009. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer 9: 665–674. [DOI] [PubMed] [Google Scholar]
- Micke P, Ostman A. 2004. Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 45Suppl 2: S163–S175. [DOI] [PubMed] [Google Scholar]
- Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, Lengyel E. 2012. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov 2: 1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Miki Y, Miyashita M, Hata S, Takahashi Y, Rai Y, Sagara Y, Ohi Y, Hirakawa H, Tamaki K, et al. 2016. Tumor microenvironment in invasive lobular carcinoma: possible therapeutic targets. Breast Cancer Res Treat 155: 65–75. [DOI] [PubMed] [Google Scholar]
- Ohlund D, Elyada E, Tuveson D. 2014. Fibroblast heterogeneity in the cancer wound. J Exp Med 211: 1503–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, Daigo Y, Ogasawara K. 2015. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci 106: 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, et al. 2009. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. 1999. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 59: 5002–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, Weinberg RA. 2007. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol Ther 6: 618–619. [DOI] [PubMed] [Google Scholar]
- Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, Kerjaschki D, Rettig WJ, Adolf GR. 2008. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res 14: 4584–4592. [DOI] [PubMed] [Google Scholar]
- Ostman A, Augsten M. 2009. Cancer-associated fibroblasts and tumor growth—bystanders turning into key players. Curr Opin Genet Dev 19: 67–73. [DOI] [PubMed] [Google Scholar]
- Osuala KO, Sameni M, Shah S, Aggarwal N, Simonait ML, Franco OE, Hong Y, Hayward SW, Behbod F, Mattingly RR, et al. 2015. Il-6 signaling between ductal carcinoma in situ cells and carcinoma-associated fibroblasts mediates tumor cell growth and migration. BMC Cancer 15: 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25: 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang W, Su J, Wang Y, Feng H, Dai X, Yuan Y, Chen X, Yao W. 2015. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci 106: 1362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson J, Micke P. 2014. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol 25: 61–68. [DOI] [PubMed] [Google Scholar]
- Peiris-Pages M, Sotgia F, Lisanti MP. 2015. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog–GLI signalling in breast cancer cells. Oncotarget 6: 10728–10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennati M, Lopergolo A, Profumo V, De Cesare M, Sbarra S, Valdagni R, Zaffaroni N, Gandellini P, Folini M. 2014. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochem Pharmacol 87: 579–597. [DOI] [PubMed] [Google Scholar]
- Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, White SA, Burt AD, Oakley F, Tsukamoto H, et al. 2012. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 56: 1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras K, Ostman A. 2010. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 316: 1324–1331. [DOI] [PubMed] [Google Scholar]
- Pietras K, Pahler J, Bergers G, Hanahan D. 2008. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med 5: e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, Goruppi S, Menietti E, Ostano P, Ala U, et al. 2015. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol 17: 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, Trivett M, Thompson ER, Ramakrishna M, Gorringe KL, et al. 2008. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet 40: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, et al. 2011. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19: 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Kenny PA, Bissell MJ. 2007. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 101: 830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Zhou X, Liu X, Jia HH, Zhao XH, Wang QX, Han L, Song X, Zhu ZY, Sun T, et al. 2016. Reprogramming carcinoma associated fibroblasts by AC1MMYR2 impedes tumor metastasis and improves chemotherapy efficacy. Cancer Lett 374: 96–106. [DOI] [PubMed] [Google Scholar]
- Rizvi S, Mertens JC, Bronk SF, Hirsova P, Dai H, Roberts LR, Kaufmann SH, Gores GJ. 2014. Platelet-derived growth factor primes cancer-associated fibroblasts for apoptosis. J Biol Chem 289: 22835–22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. 1995. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest 95: 859–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenchan PB, Carraro DM, Brentani H, de Carvalho Mota LD, Bastos EP, e Ferreira EN, Torres CH, Katayama ML, Roela RA, Lyra EC, et al. 2009. Reciprocal changes in gene expression profiles of cocultured breast epithelial cells and primary fibroblasts. Int J Cancer 125: 2767–2777. [DOI] [PubMed] [Google Scholar]
- Saadi A, Shannon NB, Lao-Sirieix P, O'Donovan M, Walker E, Clemons NJ, Hardwick JS, Zhang C, Das M, Save V, et al. 2010. Stromal genes discriminate preinvasive from invasive disease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proc Natl Acad Sci 107: 2177–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadlonova A, Bowe DB, Novak Z, Mukherjee S, Duncan VE, Page GP, Frost AR. 2009. Identification of molecular distinctions between normal breast-associated fibroblasts and breast cancer-associated fibroblasts. Cancer Microenviron 2: 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AM, Jung J, Aziz N, Kissil JL, Pure E. 2009. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest 119: 3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherz-Shouval R, Santagata S, Mendillo ML, Sholl LM, Ben-Aharon I, Beck AH, Dias-Santagata D, Koeva M, Stemmer SM, Whitesell L, et al. 2014. The reprogramming of tumor stroma by HSF1 is a potent enabler of malignancy. Cell 158: 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, Soltermann A, Jungel A, Marroquin Belaunzaran O, Stahel R, et al. 2013. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med 11: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais C, Erez N. 2013. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol 229: 198–207. [DOI] [PubMed] [Google Scholar]
- Shah SH, Miller P, Garcia-Contreras M, Ao Z, Machlin L, Issa E, El-Ashry D. 2015. Hierarchical paracrine interaction of breast cancer associated fibroblasts with cancer cells via hMAPK-microRNAs to drive ER-negative breast cancer phenotype. Cancer Biol Ther 16: 1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya R, Gonda T, Quante M, Salas M, Kim S, Brooks J, Hirsch S, Davies J, Cullo A, Olive K, et al. 2013. Hypomethylating therapy in an aggressive stroma-rich model of pancreatic carcinoma. Cancer Res 73: 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Beck AH, Webster JA, Espinosa I, Montgomery K, Varma S, van de Rijn M, Jensen KC, West RB. 2010. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res Treat 123: 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstein RL, Febbraio M. 2009. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2: re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany A, Bileck A, Muqaku B, Gerner C. 2015. Targeting breast cancer-associated fibroblasts to improve anti-cancer therapy. Breast 24: 532–538. [DOI] [PubMed] [Google Scholar]
- Sriram G, Bigliardi PL, Bigliardi-Qi M. 2015. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur J Cell Biol 94: 483–512. [DOI] [PubMed] [Google Scholar]
- Steele CW, Oien KA, McKay CJ, Jamieson NB. 2011. Clinical potential of microRNAs in pancreatic ductal adenocarcinoma. Pancreas 40: 1165–1171. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Mundel TM, Kieran MW, Kalluri R. 2006. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 5: 1640–1646. [DOI] [PubMed] [Google Scholar]
- Taddei ML, Cavallini L, Comito G, Giannoni E, Folini M, Marini A, Gandellini P, Morandi A, Pintus G, Raspollini MR, et al. 2014. Senescent stroma promotes prostate cancer progression: the role of miR-210. Mol Oncol 8: 1729–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe N, Ivy P, Timmer W, Khan N, Schulz T, Harris PJ. 2013. Review of cancer-associated fibroblasts and therapies that interfere with their activity. Tumor Microenviron Ther 1: 19–36. [Google Scholar]
- Tanaka K, Miyata H, Sugimura K, Fukuda S, Kanemura T, Yamashita K, Miyazaki Y, Takahashi T, Kurokawa Y, Yamasaki M, et al. 2015. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 36: 894–903. [DOI] [PubMed] [Google Scholar]
- Tchou J, Kossenkov AV, Chang L, Satija C, Herlyn M, Showe LC, Pure E. 2012. Human breast cancer associated fibroblasts exhibit subtype specific gene expression profiles. BMC Med Genomics 5: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlsty TD, Coussens LM. 2006. Tumor stroma and regulation of cancer development. Annu Rev Pathol 1: 119–150. [DOI] [PubMed] [Google Scholar]
- Togo S, Polanska UM, Horimoto Y, Orimo A. 2013. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers (Basel) 5: 149–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuxhorn JA, McAlhany SJ, Yang F, Dang TD, Rowley DR. 2002. Inhibition of transforming growth factor-β activity decreases angiogenesis in a human prostate cancer-reactive stroma xenograft model. Cancer Res 62: 6021–6025. [PubMed] [Google Scholar]
- Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R. 2011. Interaction of tumor cells with the microenvironment. Cell Commun Signal 9: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Villa F, Garcia-Ocana M, Galvan JA, Garcia-Martinez J, Garcia-Pravia C, Menendez-Rodriguez P, Gonzalez-del Rey C, Barneo-Serra L, de Los Toyos JR. 2015. COL11A1/(pro)collagen 11A1 expression is a remarkable biomarker of human invasive carcinoma-associated stromal cells and carcinoma progression. Tumour Biol 36: 2213–2222. [DOI] [PubMed] [Google Scholar]
- Verghese ET, Drury R, Green CA, Holliday DL, Lu X, Nash C, Speirs V, Thorne JL, Thygesen HH, Zougman A, et al. 2013. MiR-26b is down-regulated in carcinoma-associated fibroblasts from ER-positive breast cancers leading to enhanced cell migration and invasion. J Pathol 231: 388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner EF. 2016. Cancer: fibroblasts for all seasons. Nature 530: 42–43. [DOI] [PubMed] [Google Scholar]
- Wang H, Wu Q, Liu Z, Luo X, Fan Y, Liu Y, Zhang Y, Hua S, Fu Q, Zhao M, et al. 2014. Downregulation of FAP suppresses cell proliferation and metastasis through PTEN/PI3K/AKT and Ras–ERK signaling in oral squamous cell carcinoma. Cell Death Dis 5: e1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman JK, Parra-Herran C. 2015. Regressive change in high-grade ductal carcinoma in situ of the breast: histopathologic spectrum and biologic importance. Am J Clin Pathol 144: 503–510. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, et al. 2008. A paracrine requirement for hedgehog signalling in cancer. Nature 455: 406–410. [DOI] [PubMed] [Google Scholar]
- Yeung TL, Leung CS, Mok SC. 2014. CAF reprogramming inhibits ovarian cancer progression. Cell Cycle 13: 3783–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, Cheng H, Luo H, Fu W, Li Z, et al. 2015. Acquisition of epithelial–mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and β1-integrin signaling pathway in tumor cells. Breast Cancer Res 17: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. 2007. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67: 10123–10128. [DOI] [PubMed] [Google Scholar]
- Zhang XH, Jin X, Malladi S, Zou Y, Wen YH, Brogi E, Smid M, Foekens JA, Massague J. 2013. Selection of bone metastasis seeds by mesenchymal signals in the primary tumor stroma. Cell 154: 1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Yang K, Andl T, Wickett RR, Zhang Y. 2015. Perspective of targeting cancer-associated fibroblasts in melanoma. J Cancer 6: 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]