

Genomic analyses of myeloid malignancies have identified that mutations in genes encoding core spliceosomal proteins and accessory regulatory splicing factors are among the most common targets of somatic mutations. In this review, Inoue et al. describe our current understanding of the mechanistic and biological effects of spliceosomal gene mutations in myelodysplastic syndromes as well as the regulation of splicing throughout normal hematopoiesis.
Keywords: leukemia, SF3B1, SRSF2, U2AF1, mRNA splicing
Abstract
Genomic analyses of the myeloid malignancies and clonal disorders of hematopoiesis that may give rise to these disorders have identified that mutations in genes encoding core spliceosomal proteins and accessory regulatory splicing factors are among the most common targets of somatic mutations. These spliceosomal mutations often occur in a mutually exclusive manner with one another and, in aggregate, account for the most frequent class of mutations in patients with myelodysplastic syndromes (MDSs) in particular. Although substantial progress has been made in understanding the effects of several of these mutations on splicing and splice site recognition, functional connections linking the mechanistic changes in splicing induced by these mutations to the phenotypic consequences of clonal and aberrant hematopoiesis are not yet well defined. This review describes our current understanding of the mechanistic and biological effects of spliceosomal gene mutations in MDSs as well as the regulation of splicing throughout normal hematopoiesis.
Genomic analyses of the myeloid malignancies as well as the clonal disorders of hematopoiesis that may precede them have consistently identified that these conditions are enriched for mutations in genes encoding epigenetic regulatory proteins as well as proteins involved in mRNA splicing (Papaemmanuil et al. 2013; Haferlach et al. 2014; Jaiswal et al. 2014; Genovese et al. 2015). Since the initial discovery of spliceosomal gene mutations associated with hematological malignancies in 2011, it is now known that spliceosomal genes represent the most common targets of mutations in patients with myelodysplastic syndromes (MDSs), with these mutations occurring in ∼60%–70% of patients (Papaemmanuil et al. 2011; Yoshida et al. 2011; Graubert et al. 2012). Long known for being essential for producing mature mRNA molecules, splicing is also now widely understood to be a cotranscriptional and post-transcriptional mechanism essential for the regulation of gene expression in addition to modifying gene product function. Thus, alterations in RNA splicing caused by spliceosomal mutations add to the general transcriptional dysfunction that characterizes MDSs and related myeloid malignancies. Although much has been learned about the functional and therapeutic implications of epigenetic alterations in myeloid malignancy pathogenesis, understanding the functional roles of spliceosomal gene mutations is just beginning.
The discovery of spliceosomal mutations has raised an array of mechanistic, functional, and biological questions. The high frequencies of spliceosomal gene mutations as well as the evidence that many spliceosomal mutations confer an alteration of function rather than loss of function make understanding their functional effects critical. In this review, we describe what is currently known regarding mRNA splicing regulation in normal hematopoiesis as well as in the disordered clonal hematopoiesis characteristic of myeloid malignancies.
Splicing in normal hematopoiesis
Our current understanding of normal hematopoiesis and aberrancies in hematopoietic differentiation resulting in leukemias derives mostly from knowledge of the transcription factors that direct lineage commitment and post-translational modifications that alter protein function. It is estimated that >90% of multiexon human genes undergo alternative splicing (AS) to generate proteome diversity (Wang et al. 2008), yet relatively little is known about the regulation and functional roles of AS in hematopoiesis. Basic mechanisms of splicing regulation have been detailed in several recent reviews (Fu and Ares 2014; Lee and Rio 2015).
Recently, several studies used high-throughput mRNA sequencing (RNA-seq) to study splicing throughout normal hematopoiesis (Fig. 1A). These include RNA-seq of normal human hematopoietic stem cells (HSCs) plus seven other progenitor populations (Chen et al. 2014b), terminal human (Pimentel et al. 2016) and murine erythropoiesis (Edwards et al. 2016), terminal murine granulopoiesis (Wong et al. 2013), and terminal murine megakaryopoiesis (Edwards et al. 2016). One overlapping concept illustrated by these studies is that each stage of hematopoiesis is defined by cell type-specific differential splicing, which may not be associated with appreciable changes in parental gene expression. In this model, specific splicing events occur at defined stages of hematopoiesis, resulting in selective increases or decreases in inclusion of individual exons to alter the function and/or stability of the encoded proteins to define a cell's identity. Further studies of this model are needed, however, as only a small number of cell type-specific splicing events have been functionally defined in normal hematopoiesis (one of the best-characterized examples is shown in Fig. 1B).
Figure 1.

RNA-seq studies of hematopoiesis reveal stage-specific regulation of splicing during normal hematopoiesis. (A) Summary of RNA-seq studies of normal hematopoiesis performed to date with examples of models proposed regulating terminal erythropoiesis and late-stage granulopoiesis. Cell populations from mice or humans that have yet to be subjected to splicing analysis by RNA-seq analysis are opaquely shaded. (B) Examples of differentiation stage-specific splicing of EPB41, which encodes protein 4.1R, a vital component of the red blood cell membrane cytoskeleton. Exon 16, which encodes a portion of the spectrin/actin-binding domain, is skipped in early erythroid progenitors but included in mature erythroblasts (exons 14–15 are not thought to be expressed in hematopoietic cells and are largely restricted to expression in the brain [Conboy et al. 1991; Huang et al. 1993]). A series of studies has elucidated the cis elements and trans-acting splicing factors that orchestrate the stage-specific splicing of exon 16, as shown. Normally occurring alterations in expression of the RNA-binding proteins hnRNPA/B, SRSF1, and FOX1/2 throughout erythropoiesis are responsible for the exon 16 splicing switch. This occurs via inhibition of splicing by hnRNPA/B binding to an exon splicing silencer (ESS) (Hou et al. 2002) as well as promotion of exon inclusion by SRSF1 (Yang et al. 2005) and FOX1/2 (Deguillien et al. 2001; Ponthier et al. 2006) when they bind to exonic splicing enhancers (ESEs) and intronic splicing enhancers (ISEs), respectively, as shown.
Stage-specific regulation of splicing has been well characterized in the development of other tissue types, such as neuronal differentiation (for recent reviews, see Licatalosi and Darnell 2010; Zheng and Black 2013) and heart development (van den Hoogenhof et al. 2016). As just one example, the development of post-mitotic neurons from neuronal precursor cells (NPCs) is regulated in part by a change in the expression levels of two splicing regulatory proteins: the polypyrimidine (Py) tract-binding proteins 1 and 2 (PTBP1 and PTBP2, also known as PTB and nPTB). In NPCs, PTBP1 levels are elevated and repress PTBP2 levels by promoting expression of an isoform of PTBP2 that undergoes nonsense-mediated decay (NMD). However, as NPCs differentiate, PTBP1 is silenced, resulting in up-regulation of PTBP2 and subsequent promotion of a neural AS program (Boutz et al. 2007; Keppetipola et al. 2012). Whether analogous splicing regulatory mechanisms similarly regulate hematopoietic differentiation remains an open question.
The concept of differentiation stage-specific switches in RNA processing with corresponding effects on protein structure has been most clearly defined within hematopoiesis from studies of the terminal stages of granulopoiesis (Wong et al. 2013) and erythropoiesis (Edwards et al. 2016; Pimentel et al. 2016), stages of blood development associated with distinct morphological changes and changes in the proteome (including cell surface protein expression). RNA-seq analysis of promyelocytes, myelocytes, and mature granulocytes isolated from murine bone marrow (BM) revealed a striking pattern of intron retention (IR) that increased during granulopoiesis (Wong et al. 2013). Many intron-containing mRNAs were apparently efficiently exported from the nucleus and subject to degradation by NMD due to premature termination codons (PTCs) contained within the retained introns. For example, accumulation of IR transcripts was observed in Lmnb1, which encodes a key nuclear lamin protein, resulting in down-regulation of Lmnb1. Enforced expression of intronless Lmnb1 resulted in aberrant granulocytes with altered nuclear structure and volume. Increased IR in the final stages of granulopoiesis was coincident with decreased levels of mRNAs encoding several core spliceosomal proteins, including members of the U1 and U2 small nuclear ribonucleoprotein (snRNP) complexes, potentially contributing to inefficient intron excision (whether snRNA levels are similarly lower remains to be determined).
Analogous to the above studies of granulopoiesis, RNA-seq analysis of five populations of human erythroid precursors representing the last four cell divisions before enucleation similarly revealed striking increases in levels of intron-containing transcripts in the final two stages of erythropoiesis (Pimentel et al. 2016). In contrast to the report by Wong et al. (2013) that many intron-containing transcripts were apparently exported to the cytoplasm during granulopoiesis, Pimentel et al. (2016) found that many intron-containing transcripts were retained in the nucleus during erythropoiesis. Because these transcripts were retained in the nucleus, they were not degraded by NMD, which is translation-dependent and occurs in the cytoplasm. However, in either case, IR resulted in depressed levels of the encoded full-length protein. A more recent analysis of murine erythropoiesis and megakaryopoiesis similarly revealed alterations in IR during the development of both of these populations as well (Edwards et al. 2016). These studies suggest that IR is a common mechanism used to couple alterations in splicing and gene expression during hematopoietic differentiation. Given that intron-containing transcripts may be subjected to impaired nuclear export as well as cytoplasmic RNA degradation, further efforts to characterize nuclear versus cytoplasmic RNAs during hematopoietic differentiation may identify novel additional features of RNA processing during hematopoiesis. Finally, while increased IR appears to inversely correlate with gene expression in both terminal erythropoiesis (Edwards et al. 2016) and granulopoiesis (Wong et al. 2013), it is possible that a proportion of IR-retained transcripts undergoes translation to generate novel proteins. Use of techniques such as ribosome profiling of purified hematopoietic populations may help to identify unexpected translation of such intron-containing transcripts.
Forms of splicing regulation in addition to IR likely also generate PTCs to regulate gene expression during normal cellular differentiation. As one example, every member of the human serine/arginine-rich (SR) gene family has a PTC-containing isoform that is targeted for degradation by NMD (Lareau et al. 2007; Ni et al. 2007). Indeed, mRNAs encoding several SR proteins are down-regulated during terminal human erythropoiesis, concordant with higher expression of their PTC-containing isoform counterparts (Pimentel et al. 2016). Thus, further efforts to comprehensively characterize changes in splicing beyond IR during hematopoietic differentiation are still needed. In addition, efforts are needed to identify trans-acting splicing regulatory proteins that contribute to stage-specific switches in hematopoietic differentiation (as described above for PTBP1/2 regulation of neuronal development).
The above studies illustrate how global alterations in splicing catalysis can couple splicing to the regulation of total protein produced from particular genes. This connection complements AS's better-known role in regulating the production of specific protein isoforms and indicates how splicing provides an additional mechanism of gene expression regulation in addition to better-studied modes of transcriptional, translational, and post-translational regulation of gene expression in normal hematopoiesis.
In addition to identifying specific splicing events that help define different stages of hematopoiesis, these recent RNA-seq studies also highlighted important deficiencies in our knowledge of normal splicing. For example, the Blueprint consortium's study of splicing in normal human hematopoietic stem and progenitor cells (HSPCs) (Chen et al. 2014b) reported that 26.5% of putative splice junctions identified in their data set were previously unannotated [not found in the Ensemble version 70, University of California at Santa Cruz (UCSC) expressed sequence tag (EST)/mRNA data set or the poly(A)+ RNA-seq data set from Illumina]. Pimentel et al. (2016) similarly reported specific isoforms in proerythroblasts that were not present in the RefSeq transcript set. Thus, further efforts to map the specific isoforms that are present in different stages of normal human hematopoiesis will almost certainly be needed to define how those events are altered in MDSs and other clonal disorders of hematopoiesis.
Discovery of spliceosomal gene mutations and their molecular consequences
As mentioned earlier, in late 2011, frequent mutations in genes encoding spliceosomal proteins were discovered and subsequently shown to be the most common category of mutations in MDSs, related myeloid malignancies, and clonal disorders of hematopoiesis that do not meet the criteria for malignancy (so-called “clonal disorders of hematopoiesis of indeterminate potential” [CHIP]) (Fig. 2A; Papaemmanuil et al. 2011, 2013; Yoshida et al. 2011; Graubert et al. 2012; Haferlach et al. 2014; Jaiswal et al. 2014; Genovese et al. 2015). The vast majority of these mutations is concentrated in one of three genes: SF3B1, U2AF1, or SRSF2. Interestingly, the mutations occur entirely as heterozygous missense mutations at highly restricted residues and also occur in a mutually exclusive manner with one another (Fig. 2B). These genetic features, along with the absence of recurrent nonsense or frameshift mutations in these three genes, suggest that the mutations may confer an alteration of function, such as a neomorphic and/or dominant-negative activity. Given that the roles of these proteins are most established in the context of RNA splicing, a number of studies have recently been performed to evaluate the mechanistic effects of these mutations on RNA splicing. The general findings from these studies are described and appraised below.
Figure 2.

Genetic and bioinformatic characterization of spliceosomal gene mutations and their effects on splicing in myeloid malignancies. (A,B) Frequencies of spliceosomal gene mutations (A) and proportion of mutated residues across myeloid malignancies (B) (references used to make this figure are noted in the Supplemental Material). Known domains of each mutated protein are shown in colored boxes. The height of each bar is proportional to the frequency of mutations affecting each residue. The frequency of mutations represented by each amino acid change is indicated in the parentheses. (HD) HEAT domain repeat; (RRM) RNA recognition motif; (RS) arginine–serine rich domain; (ZF) zinc finger; (UHM) U2AF homology domain. (C–E) Current understanding of the mechanistic effects of mutations in SRSF2 (C), U2AF1 (D), and SF3B1 (E) on RNA splicing. As shown in C, SRSF2 mutations affecting Pro95 alter the recognition of exons containing specific ESE sequences. Wild-type SRSF2 normally binds to and promotes splicing of C- and G-rich ESEs equally well, while mutant SRSF2 has skewed preference for C-rich ESEs (Kim et al. 2015; Zhang et al. 2015). (D) In contrast, U2AF1 mutations promote or repress splice site (SS) recognition based on the identity of the nucleotide at the −3 or +1 sites that flank the AG of the 3′ SS. This occurs in an allele-specific manner in which U2AF1 S34 mutations drive alterations at the −3 position (with a C/A>>T preference), and mutations at Q157 drive alterations at the +1 position (with a G>>A preference) (Ilagan et al. 2015; Okeyo-Owuor et al. 2015; Shirai et al. 2015). Finally, SF3B1 mutations have been shown in three studies to promote use of 3′ cryptic SSs that are 10–30 base pairs (bp) upstream of the canonical 3′ SS (Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016).
Mutations in SRSF2 alter RNA splicing in a sequence-specific manner and are distinct from loss-of-function mutations
Two recent studies provided independent and consistent data showing that mutations in SRSF2 alter the protein's role in RNA splicing in a manner completely distinct from loss of function (Kim et al. 2015; Zhang et al. 2015). Although not a member of the core spliceosome, SRSF2 is a member of the SR protein family that facilitates spliceosome assembly by binding to exonic splicing enhancer (ESE) sequences within pre-mRNAs through its RNA recognition motif (RRM) domain (Graveley and Maniatis 1998; Liu et al. 2000). SRSF2 normally recognizes ESE elements within the pre-mRNA with a consensus motif of SSNG (where S represents C or G) to promote exon recognition (Daubner et al. 2012). In the wild-type state, SRSF2 binds ESEs with the motif sequence of CCNG and GGNG equally well to promote inclusion of the exons bearing these motifs.
Mutations in SRSF2 consistently affect the proline 95 (Pro95) residue and occur as heterozygous missense mutations or, rarely, in-frame insertion or deletion mutations that also affect the Pro95 residue. Unbiased genome-wide analysis of RNA-seq data from isogenic genetically modified murine hematopoietic cells as well as K562 cells bearing a substitution of histidine for Pro95 (P95H) in SRSF2 revealed that mutant SRSF2's ability to promote splicing varied based on the exonic SSNG motif. More specifically, cells expressing mutant SRSF2 exhibit enrichment for CCNG ESE motifs in cassette exons with increased inclusion, whereas GGNG ESE motifs are enriched in repressed cassette exons (Fig. 2C). Notably, the sequence-specific requirement of mutant SRSF2 to promote or repress splicing was verified by mutagenizing the ESE motifs in a series of minigene experiments modeling mutant SRSF2-responsive splicing events in both studies (Kim et al. 2015; Zhang et al. 2015). As further evidence of the sequence-specific effects of mutant SRSF2 on splicing, overexpression of wild-type SRSF2 in hematopoietic cells was associated with increased inclusion of cassette exons bearing either CCNG or GGNG ESE motifs. In contrast, depletion of SRSF2 was associated with decreased inclusion of cassette exons bearing either ESE motif. Moreover, when the spatial distribution of CCNG and GGNG motifs across genomic loci containing cassette exons promoted or repressed in association with SRSF2 mutations was examined, these motifs were found enriched specifically over cassette exons and not over the flanking introns or exons (Kim et al. 2015). This spatially restricted enrichment of specific ESEs in association with mutant SRSF2-driven splicing changes further reinforced the concept that SRSF2 mutations alter SRSF2's normal ESE recognition activity.
Mutations in SRSF2 alter RNA-binding preferences, consistent with altered exon recognition
The above data establishing that SRSF2 mutations change the protein's normal ESE preferences led to the question of whether this was due to direct alterations in RNA-binding affinity created by Pro95 mutations. Here again, distinct methods used by two independent groups led to a similar conclusion: The enrichment/depletion of specific ESEs in cassette exons in the presence of SRSF2P95H mutations is due to altered SRSF2:RNA interactions (Kim et al. 2015; Zhang et al. 2015). Isothermal calorimetry and RNA gel shift assays of the interactions between wild-type or mutant SRSF2 peptide (amino acids 1–101, consisting of the RRM and linker domain of SRSF2) and RNA ligands bearing various SSNG sequences revealed that mutant SRSF2 binds RNA oligonucleotides with C-rich ESEs threefold to fourfold more avidly than wild-type SRSF2. In contrast, mutant SRSF2 exhibited a 1.2-fold to 2.1-fold decrease in binding affinity to RNA oligonucleotides with G-rich ESE sequences.
Both direct RNA-binding affinity measurements and informatic computation of ESE motif preferences revealed the same hierarchy of motif preferences for mutant SRSF2 (CC>GC>CG>GG). However, exactly how Pro95 mutations confer this change in RNA-binding preference is still not clear. Although the Pro95 residue is not part of the canonical RRM, recent nuclear magnetic resonance (NMR) data suggest that the Pro95 residue plays a role in SRSF2's RNA-binding activity. Interestingly, NMR titration experiments using the RRM domain of wild-type and mutant forms of SRSF2 with RNA oligos suggested that mutations at Pro95 alter the conformation of the termini of the RRM without affecting the RNA-binding surface of the SRSF2 RRM (Kim et al. 2015). Future efforts to resolve the structure of SRSF2 will be important to elucidate the structural effects of Pro95 mutations on interactions with RNA. In addition, although both studies described above indicate that SRSF2 mutations affect its direct interactions with RNA, these mutations might also directly or indirectly alter SRSF2 interactions with other spliceosomal proteins. SRSF2 mutations reportedly do not affect interactions between SRSF2 and several select spliceosomal proteins (SF3B1, U2AF1, or snRNP70) (Zhang et al. 2015), but further work to understand the potential effects of SRSF2 mutations on a broader network of interacting proteins is needed.
Mutations in U2AF1 alter RNA splicing in a sequence-specific manner and are allele-specific
The earliest attempts to understand the functional implications of spliceosomal mutations on RNA splicing focused on U2AF1 (Yoshida et al. 2011). U2AF1 is the small subunit of the U2AF heterodimer that normally recognizes the consensus motif yAG|r (y = pyrimidine and r = purine) at the intron|exon boundary (Merendino et al. 1999; Wu et al. 1999; Zorio and Blumenthal 1999; for a very recent review of the role of the U2AF heterodimer in splicing, see Wu and Fu 2015). More than 10 distinct mutations have been identified in U2AF1, with nearly all occurring at residues S34 or Q157, each of which is present in one of the zinc finger domains of U2AF1 (Fig. 2B).
Several studies using unbiased genomic analysis of isogenic human and murine cells expressing ectopic mutant or wild-type U2AF1 have now revealed that mutations in U2AF1 consistently alter 3′ splice site (SS) preference in a sequence-specific manner based on the identity of the nucleotide surrounding the AG dinucleotide that forms the 3′ SS's core consensus motif (Przychodzen et al. 2013; Brooks et al. 2014; Ilagan et al. 2015; Okeyo-Owuor et al. 2015; Shirai et al. 2015). Specifically, expression of U2AF1S34F/Y promotes recognition of the 3′ SS bearing a C or A immediately preceding the AG and represses those bearing a T at this position (Fig. 2D). As with SRSF2, this sequence-specific alteration driven by mutant U2AF1 is distinct from splicing alterations that occur with U2AF1 depletion, indicating that U2AF1 mutations cause alteration, not loss, of function (Ilagan et al. 2015). Interestingly, analysis of the effects of Q157P/R mutations revealed that these mutations also affected 3′ SS recognition but in a distinct manner. Q157P/R mutations promoted recognition of the 3′ SS bearing a G immediately following (rather than preceding) the AG and repressed those bearing an A at this position (Fig. 2D; Ilagan et al. 2015). As with SRSF2, the exact structural basis for how disease-associated mutations in U2AF1 alter RNA-binding affinity needs further clarification.
Many factors are involved in correct SS recognition in complex with U2AF1, including U2AF2, hnRNPA1, and DEK, among others. Thus, future efforts are needed to determine whether S34 and Q157 mutations in U2AF1 alter the extensive protein–protein interaction networks across the exon and the intron that are necessary for spliceosome assembly.
SF3B1 hot spot mutations appear to promote use of cryptic 3′ SSs
Despite being the most common mutations affecting the spliceosome in MDSs, the mechanistic consequences of mutations in SF3B1 have been less clear than those of SRSF2 or U2AF1. SF3B1 is a member of the U2 snRNP complex, which is responsible for 3′ SS recognition. The U2 snRNP is directed to the 3′ SS by short, conserved pre-mRNA sequences, including the branch point sequence (BPS; a degenerate sequence motif usually located 21–34 base pairs [bp]) upstream of the 3′ SS), the Py tract, and the AG dinucleotide at the intron–exon junction (described earlier) (for a recent review of the basic mechanisms of normal splicing, see Wahl and Luhrmann 2015). The mutations in SF3B1 largely cluster within the C-terminal HEAT domains (residues 622–781), with the most commonly mutated residue being K700.
Three recent studies have reported that cells with mutations in SF3B1 are associated with alterations in splicing specifically due to aberrant 3′ SS selection (Fig. 2E; Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016). In each of these reports, SF3B1 mutant cells were found to use cryptic AG dinucleotides lying 10–30 bp upstream of the canonical AG dinucleotide. Although this alteration in splicing was not assayed in myeloid hematopoietic cells, it was seen in patient transcriptomes from a variety of other histologies as well as isogenic Nalm-6 cells (a pre-B lymphoblastic leukemia cell line) bearing SF3B1 mutations in their endogenous locus.
In the study by Darman et al. (2015), the motif sequences associated with cryptic and canonical AGs in mutant SF3B1-expressing cells were studied and revealed an enrichment of pyrimidines upstream of the canonical AG, while the cryptic AGs were associated with a short and weak Py tract and an enrichment of adenines. Darman et al. (2015) generated a minigene of a splicing event that was consistently altered in association with all studied hot spot mutations in SF3B1 (exon 9–exon 10 junction in ZDHHC16) and used a minigene mutagenesis approach to study the contributions of the cryptic 3′ SS, the length/strength of the Py tract, and the potential BPS to splicing in the presence of wild-type versus mutant SF3B1. A series of elegant experiments using this and similar minigenes revealed that (1) the cryptic AG is necessary to induce aberrant splicing by mutant SF3B1, (2) the cryptic AG requires the downstream canonical Py tract for recognition, and (3) distinct BPSs are used in cells expressing wild-type versus mutant SF3B1 (Darman et al. 2015). However, it remains unknown whether these specific mechanisms generalize to the broader spectrum of cryptic 3′ SSs that are activated by SF3B1 mutations.
Normally, the binding of U2 snRNP and other spliceosomal proteins around the BPS prevents 3′ SS selection in a 12- to 18-bp region directly downstream from the BPS due to steric hindrance (Fig. 2E; Smith et al. 1993; Chua and Reed 2001). Informatic characterization of the sequence features around the cryptic 3′ SSs seen by DeBoever et al. (2015) in SF3B1 mutant primary patient transcriptomes indicated that many cryptic 3′ SS used in SF3B1 mutant cells fell within this normally sterically protected region downstream from the BPS. DeBoever et al. (2015) therefore proposed that SF3B1 mutations relieve the steric occlusion of this region, thereby permitting recognition of normally obscured cryptic 3′ SSs.
The study by Darman et al. (2015) highlighted that the majority of cryptic AGs generated in SF3B1 mutant cells were located upstream of the canonical 3′ SSs and induced frameshifts, potentially causing many of the resulting transcripts to be degraded by NMD (Darman et al. 2015). Specific cryptic 3′ SSs in the region downstream from the BPS play roles in some inherited diseases (including those due to disrupted tumor suppressor genes such as ATM, NF1, and TP53) (Kralovicova et al. 2005). However, widespread usage of cryptic 3′ SSs due to a cancer-associated mutation has not been previously described. This enrichment in cryptic 3′ SSs associated with mutant SF3B1 is also distinct from altered splicing in cancer due to somatic mutations at SSs (which appear to most commonly affect 5′ SS recognition) (Jung et al. 2015). Despite this recent progress, however, our understanding of how mutations in SF3B1 result in altered 3′ SS usage is relatively incomplete. Progress is limited by the current lack of understanding of the normal function of SF3B1's HEAT domains, where the majority of SF3B1 mutations are located.
Global splicing changes in spliceosome mutant cells are subtle
Despite the observations made so far regarding global splicing effects of spliceosomal mutations, it is important to note that only a small proportion of splicing events is affected by these mutations. For instance, cryptic 3′ SSs are used in only a small fraction of genes in SF3B1 mutant cells, and they are used relatively infrequently even in that subset of genes (<10% relative to the canonical 3′ SSs for most affected genes) (Darman et al. 2015; DeBoever et al. 2015). Similarly modest global effects were observed for U2AF1 and SRSF2 mutations. Fewer than ∼5% of alternatively spliced events exhibited changes in isoform usage of >10% in isogenic cells bearing these mutations, and most changes in isoform usage were relatively modest (Ilagan et al. 2015; Kim et al. 2015; Zhang et al. 2015). Nonetheless, these observations do not eliminate the potential for functionally important effects of specific individual splicing events and their attendant downstream sequelae on hematopoiesis.
It remains possible that spliceosomal mutations induce more dramatic global splicing changes in specific contexts. Initial efforts to understand the genome-wide effects of the U2AF1S34F mutation reported widespread production of abnormal mRNAs that are substrates for degradation by NMD in association with mutant U2AF1 overexpression (Yoshida et al. 2011). However, this observation was not recapitulated in cells with more modest ectopic overexpression of U2AF1 or in the transcriptomes of patients with U2AF1 mutations (Brooks et al. 2014; Ilagan et al. 2015; Shirai et al. 2015).
Mutations in ZRSR2 appear to be consistent with loss of function and impair splicing of U12-type introns
As noted above, the majority of mutations affecting spliceosomal proteins occurs in SF3B1, SRSF2, and U2AF1. However, the discovery of these mutations by Yoshida et al. (2011) also identified recurrent mutations in ZRSR2 across the entire length of the gene. Yoshida et al. (2011) observed nonsense, frameshift, SS, and missense mutations (Fig. 2B), in stark contrast to the hot spot missense mutations in SF3B1, SRSF2, and U2AF1. Moreover, mutations in ZRSR2, which is encoded in Xp22.1, occur most commonly in males. These data strongly suggest that the mutations cause loss of function.
ZRSR2 is a component of the U12 snRNP and contacts the 3′ SSs of U12-type introns (Shen et al. 2010). U12-type introns are a rare class of introns that are removed by a dedicated, U12-dependent “minor” spliceosome that is distinct from the U2-dependent “major” spliceosome (Hall and Padgett 1994; Tarn et al. 1995; Tarn and Steitz 1996; for a recent review, see Niemela and Frilander 2014). U12-type introns comprise only ∼0.5% of all human introns (Sheth et al. 2006; Alioto 2007). Intriguingly, a subset of U12-type introns is spliced relatively inefficiently relative to their U2-type counterparts. This inefficient splicing can result in nuclear retention of mRNAs containing unspliced U12-type introns, possibly followed by nuclear decay, resulting in a regulatory mechanism analogous to that observed by Pimentel et al. (2016) in erythropoiesis.
Thus far, only one report has studied the mechanistic implications of ZRSR2 loss on splicing in hematopoiesis. Madan et al. (2015) used TF-1 cells with RNAi-mediated down-regulation of ZRSR2 as well as primary MDS patient transcriptomes that were wild type or mutant for ZRSR2. RNA-seq analysis of these cells revealed that IR was enriched in ZRSR2 mutant or knockdown samples relative to ZRSR2 wild-type cells as well as normal BM cells. Categorization of the introns affected as U2- or U12-type revealed that U12-type introns were preferentially affected by ZRSR2 loss. Although prior in vitro splicing assays suggested that ZRSR2 contributes to efficient splicing of both U2- and U12-type introns (Shen et al. 2010), Madan et al. (2015) observed that effects on U2-type introns were mostly restricted to transcripts where U12-type introns were also affected (most genes that depend on the minor spliceosome for processing contain a single U12-type intron surrounded by U2-type introns [Alioto 2007; Chang et al. 2007]). Gene ontology (GO) analysis of genes that were misspliced in association with ZRSR2 mutation or loss revealed enrichment for genes involved in MAPK and ErbB signaling, including predicted IR affecting genes encoding key cytokine signaling intermediates such as PTEN, BRAF, ARAF, and RAF1 as well as E2F transcription factors (Madan et al. 2015). These are intriguing findings that need to be validated at the protein level and functionally linked to MDS disease pathogenesis. Possibly consistent with ZRSR2 mutations predominantly affecting minor intron splicing is the fact that ZRSR2 mutations may coexist with mutations in SF3B1, SRSF2, and/or U2AF1 (which presumably primarily impact splicing of major introns), although this putative co-occurrence needs to be validated using clonal assays (Yoshida et al. 2011).
Biological implications of spliceosome alterations on hematopoiesis
While the observation that mutations in spliceosomal genes present across MDS patients are mutually exclusive might suggest similar effects of each of these mutations on MDS pathogenesis, several facts hint that these mutations might not be simply converging on common downstream effects. First, as described above, it appears that different spliceosomal gene mutations have distinct mechanistic effects on splicing. Moreover, clinical correlative studies have revealed a strong relationship between specific spliceosomal gene mutations and different clinical phenotypes among myeloid malignancies (Fig. 2A). For example, mutations in SF3B1 are highly enriched in MDS patients with ringed sideroblasts (Papaemmanuil et al. 2011; Yoshida et al. 2011), which represents an indolent form of MDS marked by hyperplasia of erythroid precursor cells with aberrant accumulation of mitochondrial ferritin around the nucleus. In contrast, mutations in SRSF2 are highly enriched in the MDS/myeloproliferative syndrome chronic myelomonocytic leukemia (CMML) (Yoshida et al. 2011; Meggendorfer et al. 2012). In addition to these unique phenotypic correlations, mutations in spliceosomal factors are also each associated with particular sets of coexisting and mutually exclusive genetic alterations, again consistent with different pathogenic mechanisms unique to each mutation (Fig. 3A).
Figure 3.
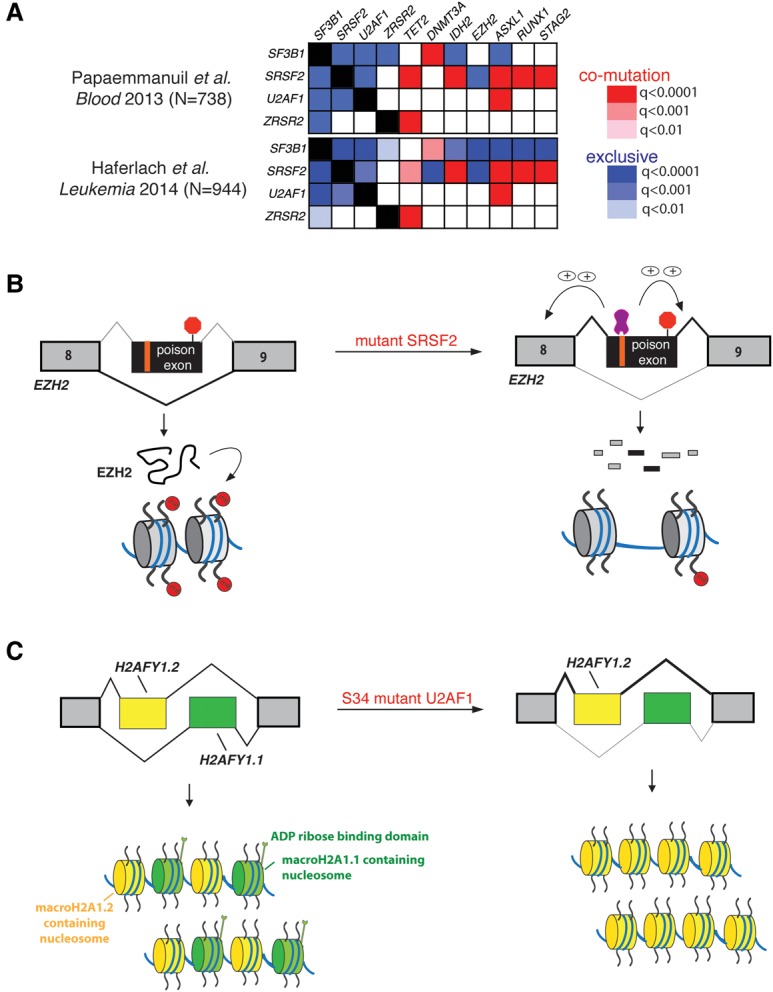
Mechanisms linking spliceosomal gene mutations to myeloid malignancy pathogenesis. (A) The pattern of genetic alterations significantly coexisting with, or mutually exclusive of, individual spliceosomal gene mutations. The q-values testing mutational co-occurrences from two separate MDS sequencing studies involving a total of >1600 MDS patients are shown (Papaemmanuil et al. 2013; Haferlach et al. 2014). The degree of red and blue shading corresponds to the q-value for mutational co-occurrence and exclusivity, respectively, between mutations in each spliceosomal gene and the other genes listed. (B) The effect of SRSF2 mutations on EZH2 expression and consequent histone H3 Lys27 trimethylation. Mutant SRSF2 promotes inclusion of a cassette exon that results in an isoform of EZH2 that undergoes NMD. This event has been validated at the mRNA and protein level, and a mutant SRSF2-responsive ESE that controls the EZH2 splicing change has been identified (Kim et al. 2015). (C) The effects of U2AF1S34 mutations on H2AFY splicing. Although this splicing event has been validated in several reports, the downstream effects on macroH2A1 isoform expression and distribution and consequent biological effects of altered macroH2A1.1/macroH2A1.2 ratios have not been studied in the context of hematopoiesis.
In contrast to the advances made so far in defining the mechanistic implications of spliceosomal mutations for RNA splicing, identifying specific misspliced events that are functionally linked to MDS disease pathogenesis is in its earliest stages. Below, we review published and ongoing efforts to understand how the global effects of each mutated splicing factor on RNA splicing may promote the development of specific subtypes of myeloid malignancies.
SF3B1 mutations and heme synthesis
One of the most interesting findings resulting from the discovery of spliceosomal gene mutations was an unexpected and highly significant association between SF3B1 mutations and MDS subtypes with ringed sideroblasts (Malcovati et al. 2015). Somatic SF3B1 mutations are present in >90% of refractory anemia with ringed sideroblasts (RARS) MDS patients as well as ∼70% of patients with non-RARS forms of MDS with RSs, such as refractory cytopenia with multilineage dysplasias and ringed sideroblasts (RCMD-RS) and RARS associated with marked thrombocytosis (RARS-T).
Ringed sideroblasts are a characteristic feature of a group of disorders known as sideroblastic anemias (SAs), which include congenital, nonclonal disorders as well as acquired clonal disorders such as RARS MDSs. Mutations affecting various stages of iron metabolism, uptake, and trafficking have been found in patients with congenital SA. These include mutations in ALAS2 (5′-aminolevulinate synthase 2) seven-codon in X-linked SA (Cotter et al. 1992), mutations in ABCB7 in X-linked SA with spinocerebellar ataxia (Allikmets et al. 1999), and mutations in GLRX5 (glutaredoxin 5) in congenital SA (Camaschella et al. 2007; Ye et al. 2010), among many others (for a recent review, see Cazzola and Malcovati 2015). Although altered expression of some of these genes, including down-regulation of ABCB7 and up-regulation of ALAS2, had been known to be present in RARS (Boultwood et al. 2008; Nikpour et al. 2010) for some time, mutations in the genes found in congenital SA have not been described in MDSs.
RNA-seq analyses of RARS samples directly from BM or in vitro primary erythroid cultures suggested that aberrant splicing of SLC25A37 (Mitoferrin-1), resulting in an IR event (Visconte et al. 2015), and/or ABCB7, resulting in an NMD substrate, is associated with the presence of SF3B1 mutations (Darman et al. 2015). The putative ABCB7 missplicing event (characterized as use of a cryptic 3′ SS between exons 8 and 9 in ABCB7, causing the addition of a PTC in a seven-codon addition to the protein sequence prior to the start of exon 9) (Darman et al. 2015) is particularly intriguing, as forced down-regulation of ABCB7 in normal CD34+ cells is associated with impaired in vitro erythroid differentiation (Nikpour et al. 2013). Moreover, restoration of ABCB7 levels by cDNA overexpression in CD34+ BM cells from RARS patients appears to functionally rescue impaired in vitro erythroid colony output (although the morphological effect of ABCB7 modulation on ringed sideroblasts themselves was not evaluated in those assays) (Nikpour et al. 2013). However, rigorous confirmation of the consequences of missplicing of ABCB7 and SLC25A37 for protein levels in the proper cell context is needed, as is mechanistic determination of how mutant SF3B1 induces missplicing of these potential target genes. Moreover, despite these potential links between mutant SF3B1 and aberrant heme synthesis, putative targets linking aberrant splicing mediated by SF3B1 mutations and the clonal hematopoiesis that characterizes RARS have not been defined. One early report suggested that constitutive heterozygous Sf3b1 knockout mice had marrow ringed sideroblasts (Visconte et al. 2012), but this feature was not confirmed in two later independent reports using the same murine model (Matsunawa et al. 2014; Wang et al. 2014). It is also important to note that even Abcb7 conditional knockout mice do not exhibit ringed sideroblasts (Pondarre et al. 2007), potentially highlighting a currently unknown difference between human and murine erythroid precursors with respect to mitochondrial iron metabolism.
Effects of SRSF2 and U2AF1 mutations on epigenetic regulatory proteins
In contrast to the association of SF3B1 mutations with ringed sideroblast-containing subtypes of MDS, SRSF2 and U2AF1 mutations appear to be enriched in more aggressive subtypes of MDS, including the WHO subtypes refractory anemia with excess blasts I (RAEB I) and RAEB II. While spliceosomal gene mutations were thought to be rare in de novo acute myeloid leukemia (AML) based on initial reported frequencies of 6.6% by Yoshida et al. (2011) and <5% in The Cancer Genome Atlas (The Cancer Genome Atlas Research Network 2013), subsequent analyses of elderly AML (>60-yr-old) cohorts revealed spliceosomal mutations in 20%–40% of such patients, the majority of which is SRSF2 mutations (Silva et al. 2015).
As is the case for SF3B1 mutations, efforts to link SRSF2 and U2AF1 gene mutations with aberrant HSC self-renewal and myeloid malignancy are still nascent, although one concrete link has been established for SRSF2. Mutations in SRSF2 were found to promote a specific PTC-containing isoform of EZH2, and minigene assays were subsequently used to identify the specific ESE that was responsible for altered EZH2 splicing in the presence of mutant SRSF2 (Fig. 3B; Kim et al. 2015). The PTC-containing isoform of EZH2 was degraded by NMD, resulting in decreased levels of EZH2 protein itself as well as the histone H3 Lys27 trimethylation (H3K27me3) mark that it catalyzes. Finally, restoration of normally spliced EZH2 expression by cDNA rescue partially rescued the hematopoietic defects of Srsf2 mutant HSPCs. These data clearly linked mutations in SRSF2 to aberrant epigenetic regulation through insufficient EZH2 expression and likely underlie the known but unexplained mutual exclusivity between loss-of-function mutations in EZH2 and SRSF2 mutations (Fig. 3A; Papaemmanuil et al. 2013; Haferlach et al. 2014). Further efforts are now needed to define the functional consequences of other splicing events altered by mutant SRSF2 that were identified by Kim et al. (2015) and Zhang et al. (2015).
A number of interesting and potentially functionally relevant misspliced events have also been described in the setting of mutant U2AF1. As with SRSF2-driven missplicing, several of these events also result in altered splicing of epigenetic regulatory genes, some of which are recurrently mutated in myeloid malignancies and/or associated with clonal hematopoiesis. These include altered splicing of BCOR, ASXL1, and DNTM3B (Ilagan et al. 2015; Shirai et al. 2015). The functional consequences of each splicing change needs to be determined in detail, especially given the known statistical enrichment of mutations in ASXL1 and U2AF1 in myeloid malignancies (Fig. 3A; Papaemmanuil et al. 2013; Haferlach et al. 2014).
Perhaps one of the most intriguing potential links between splicing mutations and altered epigenetic regulation is skewed splicing of H2AFY (Ilagan et al. 2015; Shirai et al. 2015), which encodes the histone variant macroH2A1, in association with mutant U2AF1. H2AFY contains two mutually exclusive exons, generating the two isoforms macroH2A1.1 and macroH2A1.2 of macroH2A1. The functions of these two isoforms differ, with important functional and prognostic implications in other forms of cancer (Fig. 3C; Novikov et al. 2011; Dardenne et al. 2012; Chen et al. 2014a). Although these two macroH2A1 isoforms have not been well studied in the context of hematopoiesis, from the study of other tissue types, it appears that the two macroH2A1 isoforms differ in their genome-wide distributions and ability to undergo post-translational modifications to influence gene expression. MacroH2A1.1 tends to localize to transcriptionally active acetylated euchromatin, while macroH2A1.2 tends to be associated with heterochromatin marked by H3K27me3 (Chen et al. 2014a). This association is thought to occur due to the fact that macroH2A1.1 binds ADP-ribose-related molecules, including poly(ADP-ribose) (PAR) (Kustatscher et al. 2005). PAR in turn associates with the histone acetyltransferase CBP, which promotes acetylation of chromatin in the region surrounding macroH2A1.1 (Chen et al. 2014a). In contrast, macroH2A1.2 lacks the ability to bind PAR. Further analysis of the role of H2AFY splicing and macroH2A1 isoforms in malignant hematopoiesis is just one of the many potentially important avenues of investigation highlighted by analyses of target genes downstream from spliceosomal mutations.
Animal models of spliceosomal gene mutations
The study of how spliceosomal alterations affect normal and malignant hematopoiesis will be facilitated by the use of in vivo models that complement primary patient transcriptomes and isogenic cell line models. To this end, conditional knock-in models of the Srsf2P95H (Kim et al. 2015; Kon et al. 2015) and Sf3b1K700E (Obeng et al. 2014; Mupo et al. 2015) mutations from the endogenous loci of these genes have been published or presented in abstract form. In addition, an inducible transgenic model for mutant U2af1S34F expression from the Col1a1 locus has been published (Shirai et al. 2015).
Despite potential differences in sequence conservation of intronic regions and splicing regulatory elements between species, analyses of the transcriptomes of murine hematopoietic cells expressing mutant Srsf2P95H as well as U2af1S34F have already revealed global RNA splicing alterations that are similar to those seen in isogenic human cells and primary patient transcriptomes (Kim et al. 2015; Shirai et al. 2015). Knockout models may prove useful to elucidate the roles of splicing factors in normal hematopoietic development as well as in the context of spliceosomal mutations. For instance, comparison of the hematopoietic effects of conditional deletion of Srsf2 versus expression of Srsf2P95H provided direct evidence that the P95H mutation in Srsf2 is both biologically and transcriptionally distinct from haploinsufficiency or complete loss of function (Kim et al. 2015).
Conclusions
Fewer than 5 years have elapsed since the discovery of spliceosomal gene mutations in myeloid malignancies, yet many insights have already been made about the global effects of these mutations on RNA splicing, and initial murine models of several of these mutations have been described. Further rigorous assessment of the mechanistic effects of the mutations on splicing at both genome-wide and locus-specific levels will be critical for future efforts to therapeutically manipulate splicing in spliceosome mutant malignancies. For example, the finding that mutations in SRSF2 and U2AF1 alter RNA splicing in a sequence-specific manner may be critically important for developing means to manipulate downstream pathologic splicing mediated by these mutant proteins. Furthermore, although several specific splicing alterations of potential importance have been highlighted by published informatics analyses, systematic functional characterization of these and other potentially important splicing events is needed. This effort will hopefully elucidate novel pathways critical for disease pathogenesis as well as potentially elucidate additional specific therapeutic approaches to target particularly important missplicing events. Finally, the generation of additional murine models bearing genetic alterations mimicking those seen in patients, along with alleles for conditional down-regulation of the same genes, will prove useful for defining splicing regulation throughout both normal and impaired hematopoiesis.
Supplementary Material
Acknowledgments
D.I. is supported by Post-doctoral Fellowships for Research Abroad from the Japan Society for the Promotion of Science, the YASUDA Medical Foundation, and the Kanae Foundation for the Promotion of Medical Science. O.A.-W. and R.K.B. are supported by grants from the National Institutes of Health/National Heart, Lung, and Blood Institute (R01 HL128239), the Edward P. Evans Foundation, and the U.S. Department of Defense Bone Marrow Failure Research Program (W81XWH-12-1-0041 and BM150092). R.K.B. is also supported by the Ellison Medical Foundation (AG-NS-1030-13) and National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK103854). O.A.-W. is also supported by grants from the National Institutes of Health/National Cancer Institute (1K08CA160647-01), the Josie Robertson Investigator Program, the Damon Runyon Foundation, the Starr Foundation, the Mr. William H. Goodwin and Mrs. Alice Goodwin Commonwealth Foundation for Cancer Research, and the Center for Experimental Therapeutics at Memorial Sloan Kettering Cancer Center.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.278424.116.
Freely available online through the Genes & Development Open Access option.
References
- Alioto TS. 2007. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 35: D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. 1999. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A). Hum Mol Genet 8: 743–749. [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, Littlewood TJ, Malcovati L, Della Porta MG, Jadersten M, et al. 2008. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts. PLoS One 3: e1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL. 2007. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21: 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. 2014. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9: e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. 2007. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110: 1353–1358. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzola M, Malcovati L. 2015. Diagnosis and treatment of sideroblastic anemias: from defective heme synthesis to abnormal RNA splicing. Hematology Am Soc Hematol Educ Program 2015: 19–25. [DOI] [PubMed] [Google Scholar]
- Chang WC, Chen YC, Lee KM, Tarn WY. 2007. Alternative splicing and bioinformatic analysis of human U12-type introns. Nucleic Acids Res 35: 1833–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, Novikov L, Casill AD, Park JW, Gamble MJ. 2014a. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat Struct Mol Biol 21: 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Kostadima M, Martens JH, Canu G, Garcia SP, Turro E, Downes K, Macaulay IC, Bielczyk-Maczynska E, Coe S, et al. 2014b. Transcriptional diversity during lineage commitment of human blood progenitors. Science 345: 1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Reed R. 2001. An upstream AG determines whether a downstream AG is selected during catalytic step II of splicing. Mol Cell Biol 21: 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy JG, Chan JY, Chasis JA, Kan YW, Mohandas N. 1991. Tissue- and development-specific alternative RNA splicing regulates expression of multiple isoforms of erythroid membrane protein 4.1. J Biol Chem 266: 8273–8280. [PubMed] [Google Scholar]
- Cotter PD, Baumann M, Bishop DF. 1992. Enzymatic defect in ‘X-linked’ sideroblastic anemia: molecular evidence for erythroid δ-aminolevulinate synthase deficiency. Proc Natl Acad Sci 89: 4028–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin JP, et al. 2012. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat Struct Mol Biol 19: 1139–1146. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3? Splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- Daubner GM, Clery A, Jayne S, Stevenin J, Allain FH. 2012. A syn-anti conformational difference allows SRSF2 to recognize guanines and cytosines equally well. EMBO J 31: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguillien M, Huang SC, Moriniere M, Dreumont N, Benz EJ Jr, Baklouti F. 2001. Multiple cis elements regulate an alternative splicing event at 4.1R pre-mRNA during erythroid differentiation. Blood 98: 3809–3816. [DOI] [PubMed] [Google Scholar]
- Edwards CR, Ritchie W, Wong JJ, Schmitz U, Middleton R, An X, Mohandas N, Rasko JE, Blobel GA. 2016. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 10.1182/blood-2016-01-692764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Jaiswal S, Ebert BL, McCarroll SA. 2015. Clonal hematopoiesis and blood-cancer risk. N Engl J Med 372: 1071–1072. [DOI] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. 2012. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Maniatis T. 1998. Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell 1: 765–771. [DOI] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. 2014. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SL, Padgett RA. 1994. Conserved sequences in a class of rare eukaryotic nuclear introns with non-consensus splice sites. J Mol Biol 239: 357–365. [DOI] [PubMed] [Google Scholar]
- Hou VC, Lersch R, Gee SL, Ponthier JL, Lo AJ, Wu M, Turck CW, Koury M, Krainer AR, Mayeda A, et al. 2002. Decrease in hnRNP A/B expression during erythropoiesis mediates a pre-mRNA splicing switch. EMBO J 21: 6195–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JP, Tang CJ, Kou GH, Marchesi VT, Benz EJ Jr, Tang TK. 1993. Genomic structure of the locus encoding protein 4.1. Structural basis for complex combinational patterns of tissue-specific alternative RNA splicing. J Biol Chem 268: 3758–3766. [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, et al. 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371: 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E. 2015. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47: 1242–1248. [DOI] [PubMed] [Google Scholar]
- Keppetipola N, Sharma S, Li Q, Black DL. 2012. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol 47: 360–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kon A, Yamazaki S, Ota Y, Kataoka K, Shiozawa Y, Morita M, Yoshizato T, Sanada M, Yoshida K, Nakayama M, et al. 2015. Srsf2 P95H mutation causes impaired stem cell repopulation and hematopoietic differentiation in mice. Blood 126: 1649. [Google Scholar]
- Kralovicova J, Christensen MB, Vorechovsky I. 2005. Biased exon/intron distribution of cryptic and de novo 3′ splice sites. Nucleic Acids Res 33: 4882–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. 2005. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol 12: 624–625. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929. [DOI] [PubMed] [Google Scholar]
- Lee Y, Rio DC. 2015. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 84: 291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. 2010. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11: 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HX, Chew SL, Cartegni L, Zhang MQ, Krainer AR. 2000. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol 20: 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG, et al. 2015. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 126: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunawa M, Yamamoto R, Sanada M, Sato-Otsubo A, Shiozawa Y, Yoshida K, Otsu M, Shiraishi Y, Miyano S, Isono K, et al. 2014. Haploinsufficiency of Sf3b1 leads to compromised stem cell function but not to myelodysplasia. Leukemia 28: 1844–1850. [DOI] [PubMed] [Google Scholar]
- Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, Kohlmann A, Alpermann T, Yoshida K, Ogawa S, et al. 2012. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 120: 3080–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merendino L, Guth S, Bilbao D, Martinez C, Valcarcel J. 1999. Inhibition of msl-2 splicing by Sex-lethal reveals interaction between U2AF35 and the 3′ splice site AG. Nature 402: 838–841. [DOI] [PubMed] [Google Scholar]
- Mupo A, Sathiaseelan V, Seiler M, Kent D, Peng S, Bautista R, Pacharne S, Rosen B, Koutsourakis M, Manes N, Law F, Papaemmanuil E, Buonamici S, Campbell PJ, Bolli N, Vassiliou GS. 2015. Sf3b1 K700E mutation impairs pre-mRNA splicing and definitive hematopoiesis in a conditional knock-in mouse model. Blood 126: 140. [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr. 2007. Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21: 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela EH, Frilander MJ. 2014. Regulation of gene expression through inefficient splicing of U12-type introns. RNA Biol 11: 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikpour M, Pellagatti A, Liu A, Karimi M, Malcovati L, Gogvadze V, Forsblom AM, Wainscoat JS, Cazzola M, Zhivotovsky B, et al. 2010. Gene expression profiling of erythroblasts from refractory anaemia with ring sideroblasts (RARS) and effects of G-CSF. Br J Haematol 149: 844–854. [DOI] [PubMed] [Google Scholar]
- Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, Giai V, Fernandez-Mercado M, Papaemmanuil E, Hogstrand K, et al. 2013. The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts. Leukemia 27: 889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikov L, Park JW, Chen H, Klerman H, Jalloh AS, Gamble MJ. 2011. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol Cell Biol 31: 4244–4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, McConkey ME, Campagna D, Schneider RK, Chen MC, Schmidt PJ, Mullally A, Fleming MD, Ebert ML. 2014. Mutant splicing factor 3b subunit 1 (SF3B1) causes dysregulated erythropoiesis and a stem cell disadvantage. Blood 124: 828.25104859 [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. 2015. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29: 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122: 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Parra M, Gee SL, Mohandas N, Pachter L, Conboy JG. 2016. A dynamic intron retention program enriched in RNA processing genes regulates gene expression during terminal erythropoiesis. Nucleic Acids Res 44: 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondarre C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. 2007. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis. Blood 109: 3567–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponthier JL, Schluepen C, Chen W, Lersch RA, Gee SL, Hou VC, Lo AJ, Short SA, Chasis JA, Winkelmann JC, et al. 2006. Fox-2 splicing factor binds to a conserved intron motif to promote inclusion of protein 4.1R alternative exon 16. J Biol Chem 281: 12468–12474. [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. 2013. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 122: 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Zheng X, Luecke S, Green MR. 2010. The U2AF35-related protein Urp contacts the 3′ splice site to promote U12-type intron splicing and the second step of U2-type intron splicing. Genes Dev 24: 2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. 2006. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res 34: 3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. 2015. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 27: 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva P, Neumann M, Vosberg S, Schlee C, Isaakidis K, Schroeder MP, Ortiz-Tánchez J, Fransecky LR, Hartung T, Graf A, et al. 2015. Acute myeloid leukemia in the elderly is characterized by a distinct genetic landscape. Blood 125: 1367. [DOI] [PubMed] [Google Scholar]
- Smith CW, Chu TT, Nadal-Ginard B. 1993. Scanning and competition between AGs are involved in 3′ splice site selection in mammalian introns. Mol Cell Biol 13: 4939–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn WY, Steitz JA. 1996. Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 273: 1824–1832. [DOI] [PubMed] [Google Scholar]
- Tarn WY, Yario TA, Steitz JA. 1995. U12 snRNA in vertebrates: evolutionary conservation of 5′ sequences implicated in splicing of pre-mRNAs containing a minor class of introns. RNA 1: 644–656. [PMC free article] [PubMed] [Google Scholar]
- van den Hoogenhof MM, Pinto YM, Creemers EE. 2016. RNA splicing: regulation and dysregulation in the heart. Circ Res 118: 454–468. [DOI] [PubMed] [Google Scholar]
- Visconte V, Rogers HJ, Singh J, Barnard J, Bupathi M, Traina F, McMahon J, Makishima H, Szpurka H, Jankowska A, et al. 2012. SF3B1 haploinsufficiency leads to formation of ring sideroblasts in myelodysplastic syndromes. Blood 120: 3173–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Avishai N, Mahfouz R, Tabarroki A, Cowen J, Sharghi-Moshtaghin R, Hitomi M, Rogers HJ, Hasrouni E, Phillips J, et al. 2015. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 29: 188–195. [DOI] [PubMed] [Google Scholar]
- Wahl MC, Luhrmann R. 2015. SnapShot: spliceosome dynamics I. Cell 161: 1474–e1. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Sashida G, Saraya A, Ishiga R, Koide S, Oshima M, Isono K, Koseki H, Iwama A. 2014. Depletion of Sf3b1 impairs proliferative capacity of hematopoietic stem cells but is not sufficient to induce myelodysplasia. Blood 123: 3336–3343. [DOI] [PubMed] [Google Scholar]
- Wong JJ, Ritchie W, Ebner OA, Selbach M, Wong JW, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. 2013. Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154: 583–595. [DOI] [PubMed] [Google Scholar]
- Wu T, Fu XD. 2015. Genomic functions of U2AF in constitutive and regulated splicing. RNA Biol 12: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR. 1999. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 402: 832–835. [DOI] [PubMed] [Google Scholar]
- Yang G, Huang SC, Wu JY, Benz EJ Jr. 2005. An erythroid differentiation-specific splicing switch in protein 4.1R mediated by the interaction of SF2/ASF with an exonic splicing enhancer. Blood 105: 2146–2153. [DOI] [PubMed] [Google Scholar]
- Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault TA. 2010. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest 120: 1749–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci 112: E4726–E4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Black DL. 2013. Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet 29: 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorio DA, Blumenthal T. 1999. Both subunits of U2AF recognize the 3′ splice site in Caenorhabditis elegans. Nature 402: 835–838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.