

Supplemental Digital Content is available in the text
Abstract
In experimental myocardial infarction (MI), a rise in cell counts of circulating monocyte subsets contributes to impaired myocardial healing and to atherosclerotic plaque destabilization. In humans, the prognostic role of monocyte subsets in patients suffering ST-elevation MI (STEMI) is still unclear. In the present study, we aimed to determine the kinetics of the 3 monocyte subsets (classical CD14++CD16–, intermediate CD14++CD16+, and nonclassical CD14+CD16++ monocytes), as well as the subset-specific monocyte–platelet aggregates (MPA), in acute STEMI followed by primary percutaneous coronary intervention (PCI), and their relationships with cardiovascular outcomes during a 2-year follow-up.
Monocyte subsets and MPA were measured in 100 STEMI patients receiving primary PCI on days 1, 2, 3, 5, and 7 of symptom onset, which were compared with 60 stable coronary heart disease patients and 35 healthy volunteers. From day 1 to day 7, significant increases in the counts of CD14++CD16+ monocytes and CD14++CD16+ MPA were observed, with peak levels on day 2. During a median follow-up of 2.0 years, 28 first cardiovascular events (defined as cardiovascular death, nonfatal ischemic stroke, recurrent MI, need for emergency or repeat revascularization, and rehospitalization for heart failure) were recorded. After adjustment for confounders, CD14++CD16+ monocytosis (day 1 [HR: 3.428; 95% CI: 1.597–7.358; P = 0.002], day 2 [HR: 4.835; 95% CI: 1.106–21.13; P = 0.04], day 3 [HR: 2.734; 95% CI: 1.138–6.564; P = 0.02], and day 7 [HR: 2.647; 95% CI: 1.196–5.861; P = 0.02]), as well as increased levels of CD14++CD16+ MPA measured on all time points (days 1, 2, 3, 5, and 7), had predictive values for adverse cardiovascular events.
In conclusion, our data show the expansion of the CD14++CD16+ monocyte subset during acute phase of STEMI has predictive values for 2-year adverse cardiovascular outcomes in patients treated with primary PCI. Future studies will be warranted to elucidate whether CD14++CD16+ monocytes may become a target cell population for new therapeutic strategies after STEMI.
INTRODUCTION
The past decade has witnessed an explosion of research interest in the roles of innate and adaptive immunity in the healing process after myocardial infarction (MI).1 Specifically, enhanced myocardial inflammatory response mediated by leukocytes is implicated in the pathogenesis of postinfarction remodeling and heart failure.1–3 After experimental MI, circulating monocytes are activated and are undergoing a rapid expansion, which are critical to myocardial injury and repair.4 This MI-induced monocytosis, with enhanced monocytic myocardial and artery wall infiltration, not only impairs post-MI healing but also destabilizes preexisting atherosclerotic lesions.1,5–9 Monocytosis may thereby contribute to heart failure and future ischemic events after MI.
Monocyte heterogeneity is widely acknowledged, and human monocytes can be classified into “classical” CD14++CD16–, “intermediate” CD14++CD16+, and “nonclassical” CD14+CD16++ subsets,10 which differ in their proinflammatory propensities.11,12 Moreover, it has been suggested that approaches targeting the most proinflammatory monocyte subset could improve cardiovascular outcomes after MI.13–15
Although investigated in previous studies,16,17 the dynamics of the 3 monocyte subsets in the acute phase after ST-elevation myocardial infarction (STEMI) necessitates further investigation. Precisely, the prognostic impact of monocyte subsets for post-STEMI cardiovascular events remains unknown. Against this background, in this study we sought to examine the dynamic changes of the 3 monocyte subsets, as well as the subset-specific monocyte–platelet aggregates (MPA) in the acute phase of STEMI, and correlated their changes with long-term adverse cardiovascular outcomes.
METHODS
Study Population
From November 2012 to May 2013, we consecutively enrolled patients with de novo STEMI admitted to Pingjin Hospital Heart Center. The diagnosis and treatment of STEMI was carried out according to recent guideline.18 Exclusion criteria included: patients with factors known to affect monocyte counts (infectious and inflammatory disorders, cancer, decompensated heart failure in the past 6 months, and use of hormone-replacement therapy); patients who were not suitable for primary percutaneous coronary intervention (PCI); and patients who had multivessel coronary heart disease (CHD) and were planned to undergo repeat coronary angiography before discharge. The admission blood samples were taken after initial diagnosis and prior to the 1st antiplatelet medication (clopidogrel and aspirin). Primary PCI was performed only in the infarct-related artery with conventional techniques.
The control groups included an age-matched stable CHD group and a healthy control group. Hypertension was defined as a previous diagnosis of hypertension or antihypertensive medication use, and diabetes was defined as a previous diagnosis of diabetes and use of insulin or oral hypoglycemic agents upon inclusion into the study. This study was performed in accordance with the Helsinki declaration and was approved by the hospital Research Ethics Committee. Written consent was obtained from study participants.
Laboratory Measurements
For STEMI patients, blood samples for flow cytometry (FCM) analyses of monocyte subsets and subset-specific MPA were collected on admission (day 1), day 2, day 3, day 5, and day 7. FCM analysis of circulating monocyte subsets and MPA was performed according to our previous work:19–21 briefly, blood samples taken at admission were collected via the antecubital vein in ethylenediaminetetraacetic acid (EDTA) anticoagulated tubes; then, 50 μL whole blood was incubated with 10 μL FITC-labeled antihuman CD14 (clone M5E2), 10 μL PE-labeled antihuman CD16 (clone 3G8), 10 μL PE-Cy5-labeled antihuman CD86 (clone IT2.2), and 10 μL PE-Cy7 labeled antihuman CD41 (clone HIP8) for another 15 minutes at room temperature in dark; then 1 mL red blood cell lysis buffer was added for 10 minutes. We used the following isotype controls: IgG2a-FITC (clone MOPC-173), IgG1-PE (clone MOPC-27), IgG2b-PE-Cy5 (clone MPC-11), and IgG1-PE-Cy7 (clone MPOC-21), which were all obtained from BioLegend (San Diego, CA). Compensation and gating boundaries were adjusted using unstained, single stained, and Fluorescence Minus One (FMO) controls. For absolute counting, 50 μL Flow-Count fluorescent microbeads (Beckman-Coulter, Miami, FL) were added. Analysis was performed using Cytomics FC500 cytometer (Beckman-Coulter) and FlowJo software (Treestar, Ashland, OR). In each sample, we collected a total of 100,000 events. FCM analysis was performed within 1 hour after blood was drawn, which is in accordance with a recent recommendation.22 The gating strategies are shown in Supplemental Figure 1. The coefficients of variation for absolute monocyte counting and for surface makers are 2.0% and less than 5.0%, respectively, in our lab.
For stable CHD and healthy controls, fasting blood samples were used for the above measurements. Baseline blood routine tests and biochemical assays were performed on day 1 using an automated hematology analyzer (XE-5000, Sysmex, Kobe, Japan) and a Hitachi 7180 Clinical Analyzer (Hitachi, Tokyo, Japan), respectively. Estimated glomerular filtration rate was calculated according a modified Modification of Diet in Renal Disease equation.23
Transthoracic echocardiography was performed by a Philips iE33 system (Phillips, Andover, MA) on day 2 and day 7 in STEMI patients. Analyses were carried out by an experienced technician blinded to the clinical and angiographic data, as previously described.24
Coronary lesion severity, as assessed by SYNTAX score, was calculated using the SYNTAX score algorithm.25 Two experienced interventional cardiologists (X.L.L and S.Z.) calculated SYNTAX score by visually assessing all coronary lesions with a diameter stenosis ≥50% in vessels >1.5 mm diameter, using the SYNTAX score algorithm (www.syntaxscore.com). In case of disagreement, the opinion of a 3rd observer (J.X.L) was obtained, and the final decision was made by consensus.
Follow-Up
All patients were followed up for 2 years after STEMI onset via outpatient visits, telephone contact, or hospital records. The follow-up endpoint was defined as the occurrence of a 1st major adverse cardiovascular event (MACE), which was defined as cardiovascular death, nonfatal ischemic stroke, recurrent MI, need for emergency or repeat revascularization, and rehospitalization for heart failure. All deaths were considered cardiovascular deaths if noncardiovascular death could be excluded. All events were adjudicated by the same investigator, blinded to the monocyte data, and were reviewed by an independent committee. After study inclusion, all study participants, or their next of kin, were contacted every 3 months for outcome analysis. We obtained medical records from the treating physicians to verify all events reported by study participants or their next of kin.
Statistical Analysis
Continuous variables with normal distributions are presented as mean ± standard deviation or medians with interquartile ranges. The Δ value of a specific variable in STEMI patients represents a change relative to basal level, defined as the value minus the value measured on day 1 of STEMI onset. Categorical data were compared with Fisher exact test. For comparisons of means between 2 independent groups, an unpaired Student t test or a Mann–Whitney U test was used. For comparison of means between more than 2 groups, one-way analysis of variance with Tukey post hoc analysis or a Kruskal–Wallis test followed by a Dunn test were performed, as appropriate. To test for differences (monocyte subsets and subset-specific MPA) across time, a Friedman test followed by a Dunn test for multiple comparisons was used. Correlation analyses were performed using Pearson coefficient, and data with nonnormal distribution were log-transformed before analysis.
Receiver operator characteristic (ROC) curve was plotted to assess the accuracy and the optimal cut-off value (the best Youden Index: sensitivity + specificity − 1) for each parameter to discriminate between MACE(+) and MACE(−) patients. Parameters with area under curve of P value of P ≤ 0.1 were then used for Kaplan–Meier survival analyses and Cox proportional hazards analyses. For Kaplan–Meier analysis, STEMI patients were stratified by Youden index-derived optimal cut-off values. For Cox proportional hazards analyses, all variables were 1rst transformed into dichotomic variables by using the optimal cut-off values, and then univariate followed by multivariate-adjusted analyses were performed to determine hazard ratio and 95% confidence interval for MACEs. Variables included in multivariate adjustment included those that were significantly different between MACE(+) and MACE(−) patients (age, left ventricular ejection fraction [LVEF], and SYNTAX score; all as dichotomic variables by using the optimal cut-off values derived from ROC curve analyses). Statistical analyses were performed using STATA version 14.1 (STATA Corp., College Station, TX). A 2-tailed P value <0.05 was considered statistically significant.
RESULTS
General Characteristics of Healthy Controls, Stable CHD Controls, and STEMI Patients
A total of 100 STEMI patients fulfilled the criteria for eligibility and were prospectively enrolled. All patients finished 2-year follow-up (5 patients lost contact after the 1st MACE was recorded). During a median follow-up of 26.5 months, 28 first MACEs were detected, including 7 cardiovascular deaths, 3 nonfatal ischemic strokes, 1 recurrent MI, 10 emergency or elective repeat revascularizations, and 7 readmissions for heart failure. Additionally, we enrolled 35 healthy and 60 stable CHD controls. As shown in Table 1, compared with healthy controls, stable CHD patients had higher glucose and total cholesterol levels, and lower high-density lipoprotein levels. No statistical difference was observed between stable CHD and healthy controls in terms of other blood tests and monocyte FCM measurements. Compared with stable CHD, STEMI patients presented with higher admission glucose level and compromised LVEF. There was a substantial difference in cell counts of total leukocytes and leukocyte subpopulations between stable CHD and STEMI, as well as in total monocyte counts, CD14++CD16− monocyte counts, CD14++CD16− MPA, and CD14++CD16+ MPA.
TABLE 1.
Comparisons of Baseline Characteristics in STEMI Patients, Healthy and Stable CHD Controls
When comparing STEMI patients suffered MACEs (MACE[+]) to those with event-free survival (MACE[−]), MACE (+) patients were older, with lower LVEF and higher SYNTAX score, and were more likely to have anterior MI (Table 1).
Post-STEMI Dynamics of Monocyte Subsets and Subset-Specific MPA
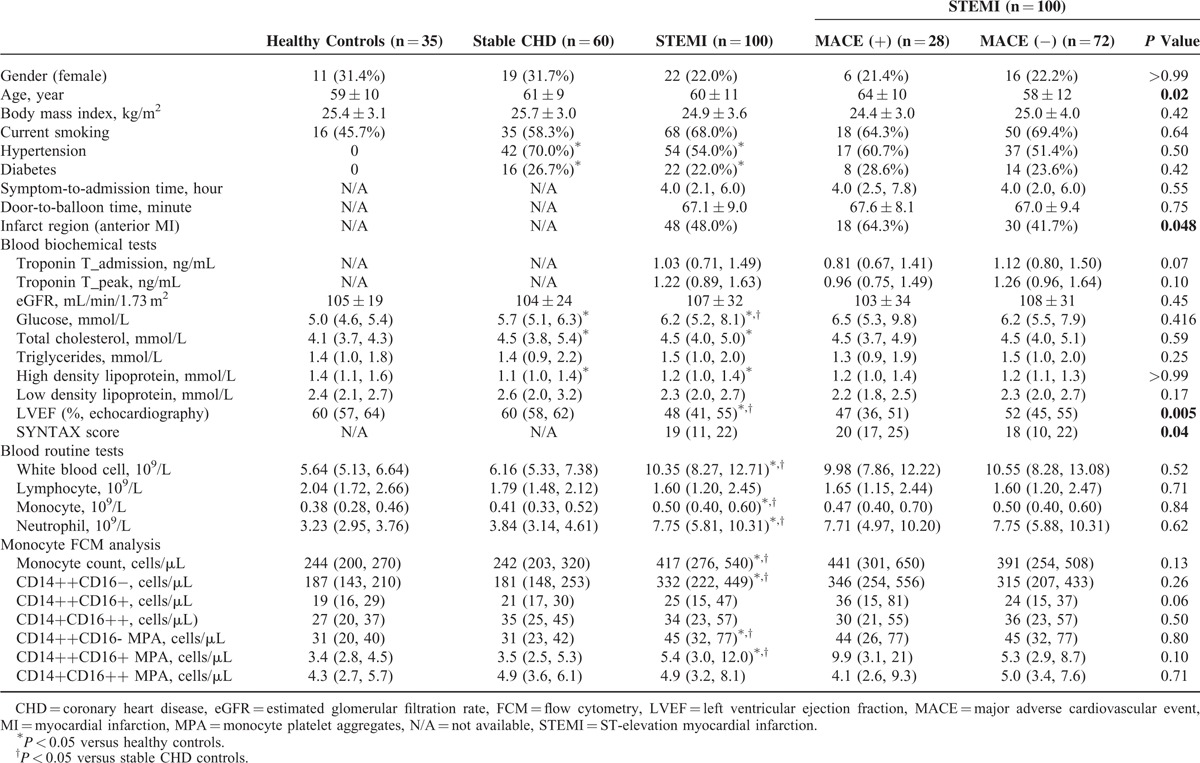
As shown in Figure 1, the longitudinal monitoring of monocyte subsets and subset-specific MPA in STEMI patients revealed coincident changes in CD14++CD16+ monocyte counts (with a 2.26-fold increase compared to stable CHD patients) and subset-specific MPA (a 2.69-fold increase), which all reached their peak levels on day 2, and remained elevated on day 7 (related statistical comparisons are shown in Figure 2). Figure 1 also depicts that throughout the observation period, STEMI patients have higher CD14++CD16− monocyte counts than stable CHD patients. On the contrary, CD14+CD16++ monocyte counts did not increase in STEMI patients compared to stable CHD patients. The concomitant occurrence of peak levels of CD14++CD16− and CD14++CD16+ monocytes promoted us to explore their potential relationships on the 1st 2 days of STEMI onset. As shown in Figure 3, ΔCD14++CD16− monocytes were positively associated with ΔCD14++CD16+ monocytes (Log transformed Δ [day 2–day 1]: r = 0.606, P < 0.001).
FIGURE 1.
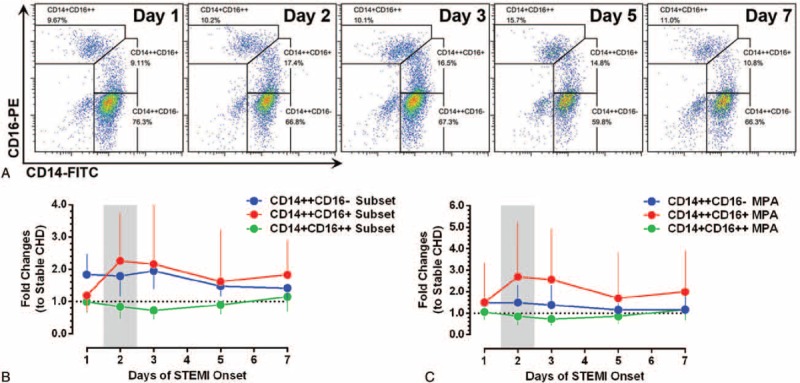
The dynamics of monocyte subsets and subset-specific MPA after STEMI. (A) Illustrate the dynamic changes of monocyte subset in a single patient (FCM analysis; representative example). (B, C) The lower panel shows the temporal profiles of monocyte subsets and subset-specific MPA in STEMI patients (fold changes to stable coronary heart disease patients). Data are presented as medians and interquartile ranges. MPA = monocyte–platelet aggregates, STEMI = ST-elevation myocardial infarction.
FIGURE 2.
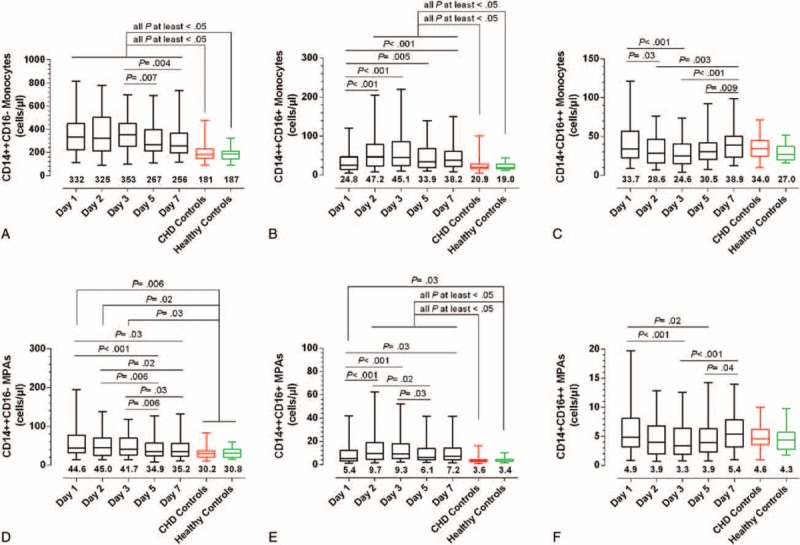
The monocyte subsets and subset-specific MPA in STEMI and coronary heart disease patients, and in healthy controls. (A–C) Changes in CD14++CD16−, CD14++CD16+, and CD14+CD16++ monocytes, respectively. (D–F) Changes in CD14++CD16−, CD14++CD16+, and CD14+CD16++ MPA, respectively. The data are presented by box and whisker plots: the boxes extend from the 25th to the 75th percentile, with a line at the median. The whiskers extend above and below the box to show the 5th to 95th percentiles of values. The number below each box and whisker plot shows the median value. MPA = monocyte–platelet aggregates, STEMI = ST-elevation myocardial infarction.
FIGURE 3.
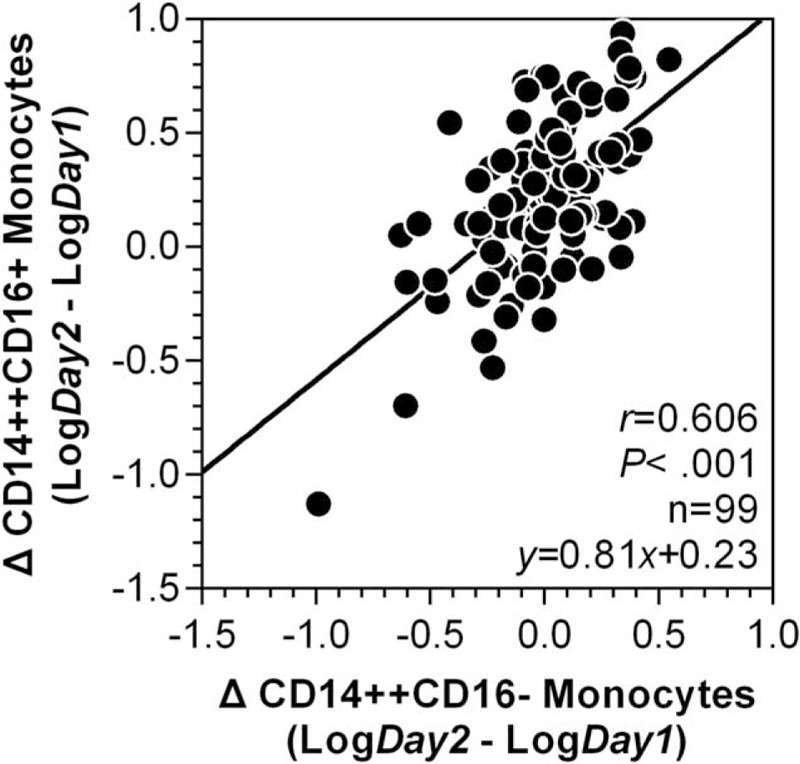
Correlation between ΔCD14++CD16− monocytes and ΔCD14++CD16+ monocytes after ST-elevation myocardial infarction. The Δ value was defined as the value measured on day 2 minus the value measured on day 1 of ST-elevation myocardial infarction onset. The data are presented after log transformation. Correlation coefficients are reported as Pearson linear correlations.
CD14++CD16+ Monocytosis Predicts 2-Year MACEs
As shown in Figure 4, compared with MACE(−) patients, a consistent increase in CD14++CD16+ subset and CD14++CD16+ MPA was observed in MACE(+) patients, with statistical significances on day 2, day 3, and day 5 (CD14++CD16+ monocytes were increased on day 7 as well). No obvious difference for CD14++CD16−, CD14+CD16++ monocytes, and their associated MPA was observed between MACE(+) and MACE(−), except for an elevation of CD14++CD16− monocytes in MACE(+) patients on day 2. The prognostic capacities of CD14++CD16+ monocytes and CD14++CD16+ MPA on day 2, day 3, and day 5 were further confirmed in ROC curve analyses and Cox regression models (Supplemental Figure 2 and Table 2). Using the optimal cut-off values derived from ROC analyses to generate Kaplan–Meier plots, CD14++CD16+ monocytosis, and increased CD14++CD16+ MPA from day 1 to day 7, all presented prognostic values for MACEs (Figure 5). Moreover, almost all measurements of CD14++CD16+ monocytes and CD14++CD16+ MPA [Table 2; except for day 5 CD14++CD16+ monocytes (P = 0.08)] presented statistical significances in Cox regression models after adjustment for baseline variables (age, LVEF, and SYNTAX score) that significantly differed between MACE(+) and MACE(−). Notably, the discriminative and predictive value for CD14++CD16− monocytes was only observed on day 2 (Supplemental Figures 2 and 3). No association between CD14+CD16++ monocytes and MACEs was observed. Finally, we found that the discriminative and predictive value for the total monocyte counts (the sum of the 3 monocyte subsets) was only observed on day 2 (Table 2 and Supplemental Figures 4).
FIGURE 4.
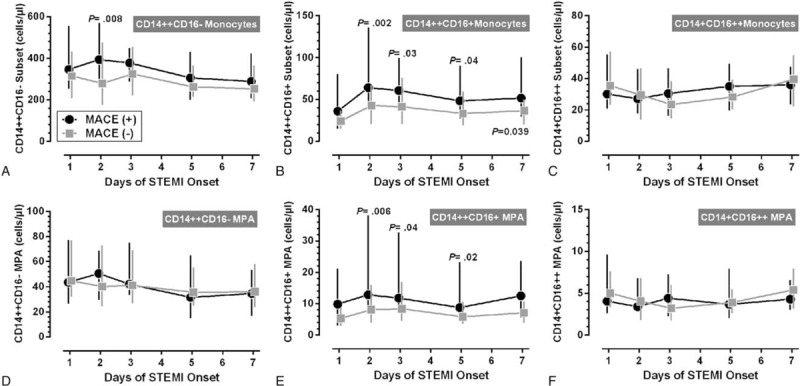
Monocyte subsets and subset-specific MPA between MACE(+) and MACE(−) STEMI patients. (A–C) Changes in CD14++CD16−, CD14++CD16+, and CD14+CD16++ monocytes in MACE(+) and MACE(−) patients, respectively. (D–F) Changes in CD14++CD16−, CD14++CD16+, and CD14+CD16++ MPA in MACE(+) and MACE(−) patients, respectively. Data are presented as medians and interquartile ranges. The P values indicate the results of statistical comparisons between MACE(+) and MACE(−) patients at the same time point. MACE = major adverse cardiovascular events, MPA = monocyte–platelet aggregates, STEMI = ST-elevation myocardial infarction.
TABLE 2.
Cox Proportional Hazards Regression Models for Predicting 2-year MACEs
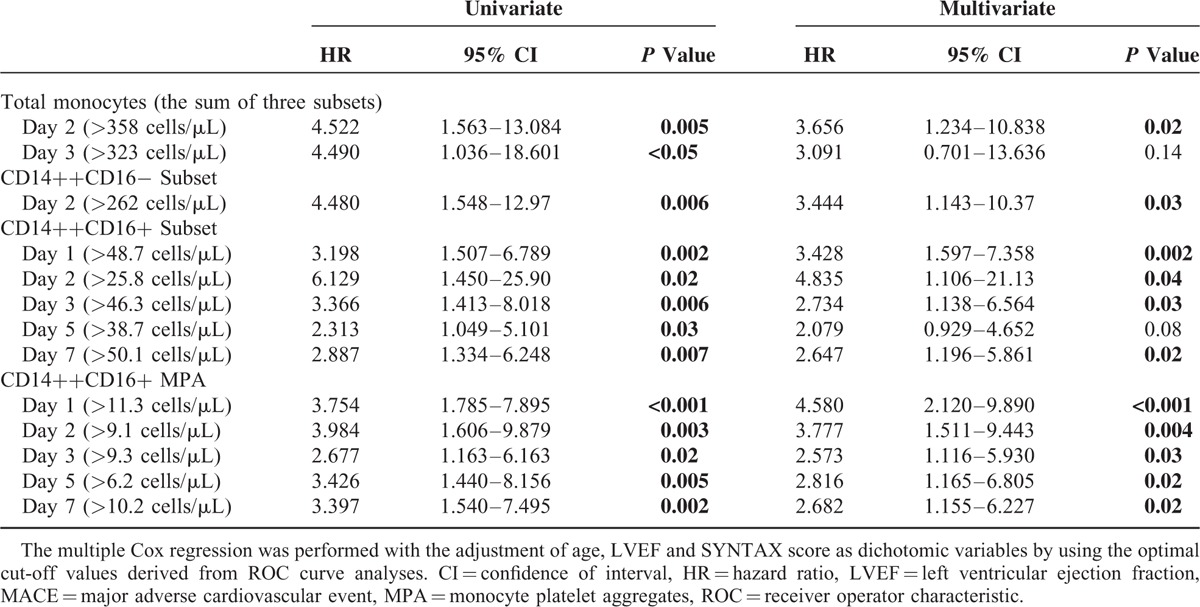
FIGURE 5.
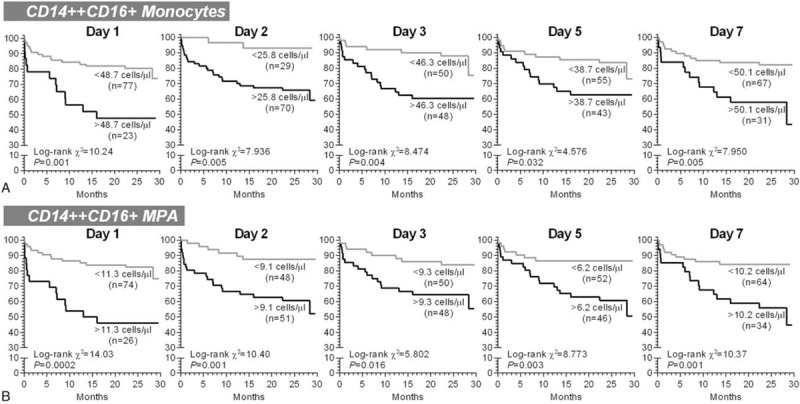
The univariate Kaplan–Meier survival analyses for 2-year major adverse cardiovascular events. The patients were stratified according the optimal cut-off values derived from receiver operator characteristic curve analyses. MPA = monocyte-platelet aggregates.
DISCUSSION
In experimental MI, the expansion of proinflammatory monocyte subset contributes to atherosclerotic plaque destabilization and impaired myocardial healing.5,6 In humans, the prognostic role of monocyte subsets in STEMI remains unexplored. In this work, we demonstrated a substantial expansion of CD14++CD16+ monocytes, as well as CD14++CD16+ MPA on day 2 of STEMI onset. After adjustment for confounders, increased counts of CD14++CD16+ monocytes and CD14++CD16+ MPA had predictive values for 2-year MACEs after STEMI. For clinical perspective, because CD14++CD16+ monocytes and CD14++CD16+ MPA are highly correlated, the measurement of CD14++CD16+ monocytes alone would provide sufficient information for the prediction. To our knowledge, the present study is the 1st report demonstrating a link between circulating monocyte subsets and hard clinical endpoints after STEMI. These data provide the 1st human evidence and strengthen the emerging concept of MI-induced inflammatory monocytosis in the initiation and propagation of adverse cardiovascular outcome.6
Recent studies showed that CD14++CD16+ monocytes represent unique proinflammatory properties associated with atherosclerosis and post-MI healing.9,11,12,26 In our previous reports, CD14++CD16+ monocyte counts independently predict cardiovascular events in patients receiving elective coronary angiography,27 and are associated with high-risk profiles in unstable angina.28 So far, limited information exists regarding whether CD14++CD16+ monocytes have prognostic implication for post-STEMI cardiovascular events. Two previous studies pioneered the investigation of human monocyte subset dynamics after STEMI.16,17 The 1st study explored the relationship between monocyte subset dynamics with the extent of myocardial salvage in 36 cases.16 However, the gating strategy did not allow the discrimination between CD14++CD16− and CD14++CD16+ monocytes. A more recent work by Tapp et al.,17 using the up-to-date classification,10 demonstrated an association between the peak level of CD14++CD16+ monocytes, which occurred within 24 hours of primary PCI (equivalent to day 2 in our study), with worsening of LV function in 50 patients. In comparison with the above work, our analyses include the following strengths: larger sample size (100 vs 36 STEMI in Tsujioka et al16 and 50 in Tapp et al);17 and longer follow-up with hard clinical endpoints (26.5 months vs 6 months in Tsujioka et al16 and 6 weeks in Tapp et al).17 Additionally, we found that CD14++CD16+ MPA closely follows temporal changes of CD14++CD16+ monocyte counts. The interaction of platelets and monocytes is a structural basis for mutual activation, and facilitates the expansion of the CD14++CD16+ monocyte pool,29 and atherosclerotic plaque development and destabilization.2 Thus, these dynamic profiles provide valuable information for future clinical research.
The prognostic value of day 2 CD14++CD16− monocyte counts observed in the present study could, at least partly, be explained by the developmental relationship from CD14++CD16− to CD14++CD16+ monocytes after their egression from bone marrow and spleen.10,30 As we found that the expansion of CD14++CD16− monocytes precedes the rise in CD14++CD16+ monocytes, and the sizes of the increases of CD14++CD16− monocytes and CD14++CD16+ monocytes are strongly correlated (Figure 3), which is consistent with a recent study showing that an isolated elevation of CD14++CD16− monocytes occurs 24 hours after transcoronary ablation of septal hypertrophy, a procedure mimicking acute MI.31 Currently, the underlying pathophysiological mechanism triggering post-MI monocytosis in humans remains unexplored. A serial of work by Nahrendorf group has demonstrated a contribution of stress-induced hematopoietic stem cell activation to the increased output of inflammatory monocytes.6,32 Thus, the molecular mechanism underlies STEMI-induced monocytosis and rapid expansion of CD14++CD16+ subset, especially the potential contribution of G protein-coupled downstream molecular cascade as the results of stress response following STEMI and during heart failure33,34 warrants future work. Notably, a recent evidence that human bone marrow also harbors a CD14++CD16+ monocyte pool,35 raises another possibility that a portion of circulating CD14++CD16+ monocytes may be directly mobilized from bone marrow after STEMI, which should be investigated in future work as well.
The present work has the following limitations. First, the consecutive characterization of monocyte heterogeneity among STEMI patients imposes a substantial logistical challenge: FCM analysis requires rapid sample preparations, while patients with STEMI are typically admitted to hospital outside regular working hours. Thus, our sample size is limited, and a relatively small number of subsequent cardiovascular events among study participants precluded a more sophisticated Cox regression analyses with a higher number of independent variables. However, the study was adequately powered to allow adjustment for baseline variables that significantly differed between MACE(+) and MACE(−) in multivariate analyses. Second, the observational design of our study did not allow detailed insight into the mechanisms by which monocytosis induces post-STEMI MACEs. Third, in addition to MI-induced monocytosis, the expansion of granulocytes,31 as well as a reduction in lymphocytes,36 is also observed in the acute phase of MI. Notably, B lymphocytes could trigger the expansion Ly6Chi monocytes (mouse counterpart to human CD14++CD16− monocytes).37 However, the correlations and interactions between monocyte subsets other leukocyte subpopulations warrant future investigations. Fourth, our analysis was based on a Chinese population, in which the prevalence of smoking is higher in males than their counterparts in western countries,38 whereas the prevalence of obesity is lower than western countries.39 As the results of their potential impacts on post-MI inflammatory profiles,40 the extrapolation of our finding to a more general population awaits further studies.
CONCLUSIONS
The present study reveals the expansion of the CD14++CD16+ monocyte subset during acute phase after STEMI has predictive value for adverse cardiovascular outcome following primary PCI in a Chinese population. Future studies will be warranted to elucidate whether CD14++CD16+ monocytes may become a target cell population for new therapeutic strategies after STEMI.
Supplementary Material
Footnotes
Abbreviations: CHD = coronary heart disease, FCM = flow cytometry, LVEF = left ventricular ejection fraction, MACE = major adverse cardiovascular event, MI = myocardial infarction, MPA = monocyte–platelet aggregates, PCI = percutaneous coronary intervention, ROC = receiver operator characteristic, STEMI = ST-elevation myocardial infarction.
XZ and X-LL contributed equally to this work.
This work was supported by research grants from National Natural Science Foundation of China (81070121, 81102088, 81170238, and 81570335), China Scholarship Council, Tianjin Municipal Science and Technology Commission Key Funding (15ZXJZSY00010), and intramural research grants from Pingjin Hospital (FYZ201402 and FYM201421).
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
REFERENCES
- 1.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 2014; 11:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seropian IM, Toldo S, Van Tassell BW, et al. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol 2014; 63:1593–1603. [DOI] [PubMed] [Google Scholar]
- 3.Zhou X, Yun JL, Han ZQ, et al. Postinfarction healing dynamics in the mechanically unloaded rat left ventricle. Am J Physiol Heart Circ Physiol 2011; 300:H1863–H1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruparelia N, Godec J, Lee R, et al. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J 2015; 36:1923–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013; 339:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature 2012; 487:325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol 2015; 35:1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Libby P, Tabas I, Fredman G, et al. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res 2014; 114:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Laan AM, Ter Horst EN, Delewi R, et al. Monocyte subset accumulation in the human heart following acute myocardial infarction and the role of the spleen as monocyte reservoir. Eur Heart J 2014; 35:376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010; 116:e74–e80. [DOI] [PubMed] [Google Scholar]
- 11.Wong KL, Tai JJ, Wong WC, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011; 118:e16–e31. [DOI] [PubMed] [Google Scholar]
- 12.Zawada AM, Rogacev KS, Rotter B, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011; 118:e50–e61. [DOI] [PubMed] [Google Scholar]
- 13.Majmudar MD, Keliher EJ, Heidt T, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation 2013; 127:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, Luo YC, Ji WJ, et al. Modulation of mononuclear phagocyte inflammatory response by liposome-encapsulated voltage gated sodium channel inhibitor ameliorates myocardial ischemia/reperfusion injury in rats. PLoS One 2013; 8:e74390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leuschner F, Dutta P, Gorbatov R, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol 2011; 29:1005–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsujioka H, Imanishi T, Ikejima H, et al. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol 2009; 54:130–138. [DOI] [PubMed] [Google Scholar]
- 17.Tapp LD, Shantsila E, Wrigley BJ, et al. The CD14++CD16+ monocyte subset and monocyte-platelet interactions in patients with ST-elevation myocardial infarction. J Thromb Haemost 2012; 10:1231–1241. [DOI] [PubMed] [Google Scholar]
- 18.Steg PG, James SK, Atar D, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J 2012; 33:2569–2619. [DOI] [PubMed] [Google Scholar]
- 19.Heine GH, Ulrich C, Seibert E, et al. CD14(++)CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int 2008; 73:622–629. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Zhang L, Ji WJ, et al. Variation in dietary salt intake induces coordinated dynamics of monocyte subsets and monocyte-platelet aggregates in humans: implications in end organ inflammation. PLoS One 2013; 8:e60332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Yuan F, Ji WJ, et al. High-salt intake induced visceral adipose tissue hypoxia and its association with circulating monocyte subsets in humans. Obesity (Silver Spring) 2014; 22:1470–1476. [DOI] [PubMed] [Google Scholar]
- 22.Ji WJ, Lu RY, Liu JX, et al. The influence of different anticoagulants and time-delayed sample processing and measurements on human monocyte subset and monocyte-platelet aggregate analyses. Cytometry B Clin Cytom (Epub Date 2016/02/11) 10.1002/cyto.b.21363. [DOI] [PubMed] [Google Scholar]
- 23.Ma YC, Zuo L, Chen JH, et al. Modified glomerular filtration rate estimating equation for Chinese patients with chronic kidney disease. J Am Soc Nephrol 2006; 17:2937–2944. [DOI] [PubMed] [Google Scholar]
- 24.Cai W, Dong Y, Zhou X, et al. Left ventricular systolic dyssynchrony in patients with isolated symptomatic myocardial bridge. Scand Cardiovasc J 2013; 47:11–19. [DOI] [PubMed] [Google Scholar]
- 25.Sianos G, Morel MA, Kappetein AP, et al. The SYNTAX Score: an angiographic tool grading the complexity of coronary artery disease. EuroIntervention 2005; 1:219–227. [PubMed] [Google Scholar]
- 26.Ghattas A, Griffiths HR, Devitt A, et al. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol 2013; 62:1541–1551. [DOI] [PubMed] [Google Scholar]
- 27.Rogacev KS, Cremers B, Zawada AM, et al. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol 2012; 60:1512–1520. [DOI] [PubMed] [Google Scholar]
- 28.Zeng S, Zhou X, Ge L, et al. Monocyte subsets and monocyte-platelet aggregates in patients with unstable angina. J Thromb Thrombolysis 2014; 38:439–446. [DOI] [PubMed] [Google Scholar]
- 29.Passacquale G, Vamadevan P, Pereira L, et al. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One 2011; 6:e25595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rogacev KS, Zawada AM, Hundsdorfer J, et al. Immunosuppression and monocyte subsets. Nephrol Dial Transplant 2015; 30:143–153. [DOI] [PubMed] [Google Scholar]
- 31.Liebetrau C, Hoffmann J, Dorr O, et al. Release kinetics of inflammatory biomarkers in a clinical model of acute myocardial infarction. Circ Res 2015; 116:867–875. [DOI] [PubMed] [Google Scholar]
- 32.Heidt T, Sager HB, Courties G, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med 2014; 20:754–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santulli G, Campanile A, Spinelli L, et al. G protein-coupled receptor kinase 2 in patients with acute myocardial infarction. Am J Cardiol 2011; 107:1125–1130. [DOI] [PubMed] [Google Scholar]
- 34.Santulli G. Adrenal signaling in heart failure: something more than a distant ship's smoke on the horizon. Hypertension 2014; 63:215–216. [DOI] [PubMed] [Google Scholar]
- 35.Mandl M, Schmitz S, Weber C, et al. Characterization of the CD14++CD16+ monocyte population in human bone marrow. PLoS One 2014; 9:e112140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boag SE, Das R, Shmeleva EV, et al. T lymphocytes and fractalkine contribute to myocardial ischemia/reperfusion injury in patients. J Clin Invest 2015; 125:3063–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zouggari Y, Ait-Oufella H, Bonnin P, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med 2013; 19:1273–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin P, Jiang CQ, Cheng KK, et al. Passive smoking exposure and risk of COPD among adults in China: the Guangzhou Biobank Cohort Study. Lancet 2007; 370:751–757. [DOI] [PubMed] [Google Scholar]
- 39.Das SR, Alexander KP, Chen AY, et al. Impact of body weight and extreme obesity on the presentation, treatment, and in-hospital outcomes of 50,149 patients with ST-Segment elevation myocardial infarction results from the NCDR (National Cardiovascular Data Registry). J Am Coll Cardiol 2011; 58:2642–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruff CT, Braunwald E. The evolving epidemiology of acute coronary syndromes. Nat Rev Cardiol 2011; 8:140–147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.