

Abstract
Background
Cancer is considered the second leading cause of death in the world, and for the treatment of this disease, pharmacological intervention strategies are frequently based on chemotherapy. Doxorubicin (DOX) is one of the most widely used chemotherapeutic agents in clinical practice for treating a number of solid tumours. The treatment with DOX mimics some effects of cancer cachexia, such as anorexia, asthenia, decreases in fat and skeletal muscle mass and fatigue. We observed that treatment with DOX increased the systemic insulin resistance and caused a massive increase in glucose levels in serum. Skeletal muscle is a major tissue responsible for glucose uptake, and the positive role of AMPk protein (AMP‐activated protein kinase) in GLUT‐4 (Glucose Transporter type 4) translocation, is well established. With this, our aim was to assess the insulin sensitivity after treatment with DOX and involvement of AMPk signalling in skeletal muscle in this process.
Methods
We used Wistar rats which received a single dose of doxorubicin (DOX group) or saline (CT group) intraperitoneally at a dose of 15 mg/kg b.w. The expression of proteins involved in insulin sensitivity, glucose uptake, inflammation, and activity of electron transport chain was assessed in extensor digitorum longus muscle, as well as the histological evaluation. In vitro assays were performed in L6 myocytes to assess glucose uptake after treatment with DOX. Agonist of AMPk [5‐aminoimidazole‐4‐carboxamide (AICAR)] and the antioxidant n‐acetyl cysteine were used in L6 cells to evaluate its effect on glucose uptake and cell viability.
Results
The animals showed a significant insulin resistance, hyperglycaemia, and hyperinsulinemia. A decrease in the expression of AMKP and GLUT‐4 was observed in the extensor digitorum longus muscle. Also in L6 cells, DOX leads to a decrease in glucose uptake, which is reversed with AICAR.
Conclusions
DOX leads to conditions similar to cachexia, with severe glucose intolerance both in vivo and in vitro. The decrease of AMPk activity of the protein is modulated negatively with DOX, and treatment with agonist of AMPk (AICAR) has proved to be a possible therapeutic target, which is able to recover glucose sensitivity in skeletal muscle.
Keywords: Doxorubicin, Skeletal muscle, AMPk, Glucose intolerance, Hyperglycaemia, Chemotherapy, Anthracycline
Introduction
Doxorubicin (DOX) is a chemotherapeutic, in the family of anthracyclines, developed in the 1960s.1 It is an antineoplastic antibiotic that is still widely used in the treatment of a variety of malignancies, effective in acute leukaemia, non‐Hodgkin lymphomas, breast cancer, Hodgkin's disease, and sarcomas.2
Doxorubicin exerts antitumor activity by inhibiting DNA Topoisomerase II, thus disrupting DNA replication.3 Moreover, DOX induces the generation of reactive oxygen species (ROS) leading to DNA damage and apoptosis, by stimulation of p53‐DNA binding, which initiates the caspase signalling, and DNA cross‐linking.4, 5
Despite the great antitumuoral efficiency of DOX, the use of this drug in therapy is limited, whereas since the 1970s, its cardiotoxicity was demonstrated in DOX treatment; therefore, this limits its use in cancer treatment.6 Although the deleterious effects of DOX on the heart muscle are the main targets of investigation discussed in the literature, skeletal muscle is also affected. DOX treatment causes severe fatigue7 and muscle weaknesses that reflect a poor quality of life.8 This tissue has important biological functions, and various metabolic disorders are related to changes in metabolism.
It has been shown that the decrease in insulin sensitivity in skeletal muscle may be secondary to a number of pharmacological therapies.9, 10 In humans, skeletal muscle accounts for approximately 50–60% of body weight. In insulin‐stimulated conditions, skeletal muscle is responsible for about 75% of uptake in circulating glucose.11 Insulin resistance favours muscle wasting, a process called sarcopenia.12, 13 Sarcopenia is part of the physiological ageing process, but it is also present in several morbidities, such as diabetes, cancer, and kidney disease, which affect the quality and life expectancy of individuals.14, 15 Clinically, patients usually report severe fatigue during DOX treatment.16 Previous studies have shown that DOX is able to generate muscle dysfunction, leading to lost performance and the physical appearance of debilitating fatigue.17, 18, 19 Worsening in the parameters of maximum strength, maximum relaxation and fatigue were evident and were probably caused by changes in sarcoplasmic calcium metabolism.20, 21
However, few studies have been proposed to investigate a possible association between the use of DOX and insulin resistance, as well as the role of skeletal muscle in the process.
AMPk (AMP‐activated protein kinase) is a protein with serine‐threonine kinase residues,22 which acts as a key sensor of cellular energy levels. The activity of this protein appears to play a pivotal role in increasing glucose uptake in skeletal muscle. In 2005, Tokarska‐Schlattner et al.23 showed that this disruption in AMPk signalling negatively affected the energetic metabolism in cardiomiocytes of the animals treated with DOX. AMPk activation by 5‐aminoimidazole‐4‐carboxamide (AICAR) riboside or genetic approach reduced DOX cardiotoxicity in culture cells.24
The explanation of this condition is extremely important, because insulin resistance alters the pharmacokinetics of the drug, increases cardiotoxicity, and increases oxidative stress.25, 26 Therefore, the skeletal muscle has an important role in glucose and insulin homeostasis. Accordingly, the aim of this study was to evaluate insulin sensitivity after treatment with DOX and involvement of AMPk signalling in skeletal muscle in this process.
Methods
Animals
The Experimental Research Committee of the University of São Paulo approved all procedures for the care of the animals used in this study. A total of 26 male Wistar rats approximately 14 weeks of age were used. They were housed four per cage in an animal room under a 12 h light‐dark cycle at 22 ± 1°C and 60 ± 5% humidity and received a chow diet and water ad libitum. The experiments were carried out after a one‐week acclimation period. Rats were randomly divided into two groups: (i) saline control (CT) (n = 13) and (ii) DOX group (DOX) (n = 13). After the acclimation period, the DOX‐treated group received 15 mg/kg, i.p., DOX chloridrate (Eurofarma Laboratory, Campinas, Brazil); control animals received an equal volume of saline. Food intake and body weight were assessed daily.
The animals performed 6 h of fasting previous to euthanasia, by decapitation, 72 h after the DOX treatment. Following euthanasia, extensour digitorious longus (EDL) and retroperitoneal adipose tissue were removed, weighted, snap frozen in liquid nitrogen, and stored at −80°C. The epididymal adipose tissue and liver were only weighed. Whole blood was drawn, centrifuged at 3000 g for 15 min at 4°C. Serum was removed and kept frozen at −80°C for later determination.
Serum analysed
Fasting blood glucose, uric acid, and aspartate transaminase was assessed using Labtest© kits. Serum insulin, adiponectin, testosterone, and corticosterone were quantified using enzyme‐linked immunosorbent assay (ELISA). For insulin the kit was obtained from Millipore Corp. Bedford, MA, USA, for adiponectin the kit was obtained from R&D Systems, Minneapolis, MN, USA, and for corticosterone and testosterone from Assay Designs, Inc., Ann Arbor, MI, USA. Serum free fatty acid (FFA) levels were analysed in rats using the NEFA‐kit‐U (Wako Chemical GmBH, Neuss, Germany). Homeostatic model assessment of insulin resistance was used to evaluate insulin resistance. The index was determined by calculating: fasting serum insulin (μU/mL) × fasting plasma glucose (mmol l‐1)/22.5.
Histology analysis
The EDL muscle was cut in cryostat sections (10 µm thick) at −25 ° temperature. The sections were incubated with hematoxylin and eosin for the analysis of cross‐sectional area of the fibre (aspartate transaminase). The morphometric analysis was analysed under a microscope (Nikon Eclipse E600, Fukuoka, Japan) equipped with a digital video camera coupled to software to analyse the images (Metamorph, Universal Corporation, Downingtown, USA). The scanned images were analysed using Image‐Pro Plus (Media Cybernetics, Silver Spring, MD) software in a double‐blind manner. For analysis of cross‐sectional area of the fibre, approximately 1000 EDL muscle fibres were analysed per group. Four to five EDL fields per animal were analysed in groups studied.
Enzymatic assays
The EDL muscle was homogenized in SETH buffer, pH 7.4 (250 Mm sucrose, 2 mM EDTA, 10 mM Trizma base, and 50 IU/mL heparin). The enzymatic activity of citrate synthase, malate dehydrogenase, and mitochondrial complexes 1 and 3 were performed as described in27.
Intraperitoneal tolerance test
Forty‐eight hours after starting treatment, some of the animals were subjected to the insulin tolerance test (ITT). After 6 h fasting, insulin (2 IU/kg) was administered by intraperitoneal injection, and blood samples were collected from the tail at 0, 5, 10, 15, 20, 25, and 30 min for measurement of serum glucose.
Quantitative real‐time polymerase chain reaction
Total RNA from the EDL muscle was extracted with Trizol reagent (Invitrogen Life Technologies)28 and reverse transcribed to cDNA using the High‐Capacity cDNA kit (Applied Biosystems). Gene expression was evaluated by real‐time PCR using a Rotor Gene (Qiagen) and SYBR Green as fluorescent dye. Primer sequences are shown in Table S1 . Quantification of gene expression was carried,29 with RPL‐19 gene as an internal control. Primer sequences are shown in Table S1 .
Table 1.
Doxorubicin leads to a severe loss of body weight and anorexia with a disruption in systemic metabolism
| CT | DOX | |
|---|---|---|
| Delta body weight (g) | 12.78 ± 1.26 | −30.05 ± 1.46 *** |
| Food intake (g/day) | 27.10 ± 2.30 | 4.58 ± 0.70 *** |
| Epididymal adipose tissue (g) | 5.99 ± 0.27 | 4.00 ± 0.32 ** |
| Glucose (mg/dl) | 143.2 ± 2.78 | 310.7 ± 11.13 *** |
| NEFA (mEq/L) | 0.62 ± 0.02 | 0.72 ± 0.03 * |
| Insulin (ng/mL) | 1.68 ± 0.12 | 2.46 ± 0.31 * |
Values represent the means and ± SD of the data obtained from analysis of 8–10 animals per group.
P < 0.05.
P < 0.01.
P < 0.001 vs. CT.
Protein analysis by western blotting
The EDL muscle and L6 myocytes were lysed in extraction buffer containing protease and phosphatase inhibitors. The extracts were then centrifuged at 12 000 rpm at 4°C for 40 min to remove insoluble material. Protein determination in the supernatants was performed by the Bradford dye method using the Bio‐Rad reagent (Bio‐Rad Laboratories, Hercules, CA, USA). The proteins were treated with a Laemmli sample buffer containing dithiothreitol and boiled for 5 min before loading into 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis in a Bio‐Rad miniature slab gel apparatus. Electrotransfer of proteins from the gel to nitrocellulose was performed for ~1 h at 15 V (constant) in a Bio‐Rad semi‐dry transfer apparatus. Nonspecific protein binding to the nitrocellulose was reduced by preincubation for 1 h at 22°C in a blocking buffer (5% nonfat dry milk, 10 mM Tris, 150 mM NaCl, and 0.02% Tween 20). The nitrocellulose membranes were incubated overnight at 4°C with antibodies against Akt/PKB, and phospho Akt S473, AMPk α, phospho AMPkα T172, and IR (insulin receptor) were obtained from Cell Signaling Technology® (Danvers, MA, USA). GLUT‐4 (glucose transporter 4), GAPDH, and b‐tubilin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) diluted in blocking buffer added with 1% bovine serum album (BSA) and then washed for 1 h in a blocking buffer without BSA. The blots were subsequently incubated with peroxidase‐conjugated secondary antibody for 1 h.
By evaluation of protein loading, membranes were stripped and reblotted with GAPDH or b‐tubulin antibody, as appropriate. Specific bands were detected by chemiluminescence, and visualization/capture was performed by exposure of the membranes to RX films. Band intensities were quantified by optical densitometry of developed autoradiographs (Scion Image software‐Scion Corporation, Frederick, MD, USA).
L6 cell culture
The cells were maintained in Dulbecco' s modified Eagle' s medium at 37°C in humidified atmosphere containing 5% CO2 and supplemented with 10% fetal bovine serum and 2% antibiotic solution. To differentiate, cells were allowed to reach confluence, and the medium was changed to medium containing 2% fetal bovine serum for 7 days, with medium changes every 2nd day. The serum was removed for 6 h for the determination of glucose uptake ability.
MTT assay
For the assessment of cell viability, MTT assay was performed. 2X104 cells per well in 96‐well plates were cultivated and were differentiated after 24 h. Cells were treated with different concentrations of DOX (0 to 1 μMol) for 48 h, and after this period, we performed the cell viability assay. The cells' medium was replaced with 200 μL fresh medium/well containing 0.125 mg/mL MTT and cultivated for another 3 h darkened in the cells' incubator. The supernatant was removed (until reaching a final volume of 25 uL), and 100 μL isopropanol/HCl (11 M) were added per well. The absorbance at 595 nm was measured. Two wells per plate without cells served as a blank. The effect of the DOX on cell viability was relativized by DMSO group.
Glucose uptake assay
Glucose uptake in L6 cells was measured using the 2‐deoxy‐[C14]‐D‐glucose. The 5.9 × 104 cells per well in 24‐well plates were seeded and were differentiated after 24 h, with a confluence of approximately 80%. Differentiated L6 cells were treated with 100 nMol of DOX or DMSO for 48 h. To evaluate the chronic effect of AICAR, this drug was added together to start treatment with DOX (2 mMol/48 h).
For the glucose uptake assay the cells remained fasting (serum‐free medium) for 6 h. After this, cells were washed in PBS, glucose‐free medium (Hepes, 140 mmol NaCl, 20 mmol Hepes‐Na pH 7.4, 5 mmol KCl, 2.5 mmol MgSO4, and 1 mmol CaCl2) was added for 30 min, with or without insulin (100 nmol) or AICAR (2 mmol/1 h—only for the acute treatment). After this, Hepes medium containing 2‐deoxy‐[C14]‐D‐glucose (1 mCiu/mL) was added for 30 min. Reactions were terminated by washing twice in ice‐cold NaCl (0.9%), cells were digested (50 mmol NaOH), and part of the sample was used for total protein quantification by Bradford, and in part transferred to a scintillation vial with liquid scintillant. The radioactivity was measured by beta counter.
TNF‐α, IL‐10, IL‐6, and adiponectin protein level determination
Frozen tissues (0.1–0.3 g) were homogenized in radio immunoprecipitation assay buffer (0.625% Nonidet P‐40, 0.625% sodium deoxycholate, 6.25 mM sodium phosphate, and 1 mM ethylenediamine tetra acetic acid at pH 7.4) containing 10 µg/mL of a protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, Missouri). Homogenates were centrifuged at 12 000 g for 10 min at 4°C, the supernatant was saved, and protein concentration was determined using the Bradford assay (Bio‐Rad, Hercules, California) with bovine serum albumin as a reference. Quantitative assessment of TNF‐ α, IL‐6, IL‐10, and adiponectin proteins was carried out by ELISA (DuoSet ELISA, R&D Systems, Minneapolis, MN). All samples were run as duplicates, and the mean value was reported.
Statistical analysis
The statistical analysis was performed using the GraphPad Prism statistics software package version 5.0 for Windows (GraphPad Software, San Diego, CA, USA). The data are expressed as the means ± SD. Implementation of the Kolmogorov–Smirnov test revealed that the results of experiments were distributed normally. The data were analysed using a Student's t‐test for comparison between two groups. For comparison of assays in cell culture, the ANOVA one‐way with Tukey post‐test test, or ANOVA two‐way test with Bonferroni post‐test were used. A value of P < 0.05 was considered statistically significant.
Results
Doxorubicin chemotherapy leads to severe anorexia and sarcopenia‐like morbidity
Treatment with DOX causes a marked decrease in body weight and relevant anorectic condition (Table 1). Seventy‐two hours after the initial application of chemotherapy, a severe drop in the weight of epididymal adipose pad (Table 1) and EDL skeletal muscle (Figure 1A) was observed. No change in the weight of the liver, adrenal, and retroperitoneal adipose tissue (Table 2S) was observed. The high levels of aspartate aminotransferase (AST) and uric acid confirm the deleterious effects of chemotherapy with DOX on hepatic and renal function (Table 2S).
Figure 1.
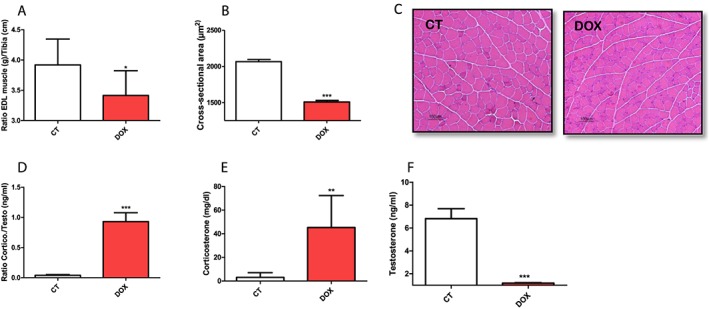
Effect of doxorubicin on muscular atrophy and the electron transport chain activity. (A) Ratio muscle extensor digitorum longus/tibia. (B) Cross‐sectional area in extensor digitorum longus muscle. (C) Histological slices of the extensor digitorum longus muscle stained with H & E. (D) Ratio corticosterone/testosterone. (E) Corticosterone levels. (F) Testosterone levels. The groups were compared by Test T. P < 0.05 was considered statistically significant. * P < 0.5, ** P < 0.01, *** P < 0.001. n = 4–10.
Corroborating with the drop in the weight of the EDL muscle, the application of DOX induced a reduction of its cross‐sectional area (Figure 1B and 1C). To better characterize the systemic change of the use of DOX on the regulation of the control mechanisms of the processes of protein synthesis and degradation, we evaluated the circulating concentration of the two main steroid hormones that regulate these processes. Significantly higher levels of corticosterone/testosterone ratio were found in the DOX group (Figure 1D), resulting from increased levels of corticosterone (Figure 1E) and suppression of circulating testosterone (Figure 1F), thus confirming the high catabolic state of our experimental model.
Acute treatment with doxorubicin increases systemic insulin resistance
Doxorubicin groups showed substantial increased levels of insulin, glucose, and FFA (Table 1) in serum, after 72 h of DOX treatment (15 mg/kg). Meanwhile, the protein expression of adiponectin in serum and retroperitoneal adipose tissue was decreased (data in press).
The quantification of insulin resistance by homeostatic model assessment of insulin resistance demonstrates that the DOX group has less sensitivity to insulin in the basal state (Figure 2A). The ITT confirmed the impaired insulin sensitivity, under insulin stimulus, in the group treated with DOX compared with the control group. The intraperitoneal tolerance test of insulin was performed to evaluate the response of DOX treatment 48 hours after administration of the drug. DOX treated animals showed higher glucose blood levels at each time interval (0, 5, 10, 15, 20, 25 and 30 minutes) (Figure 2 B). Serum glucose disappearance rate (Kitt) during the ITTs was dramatically decreased in the group treated with DOX (Figure 2C).
Figure 2.
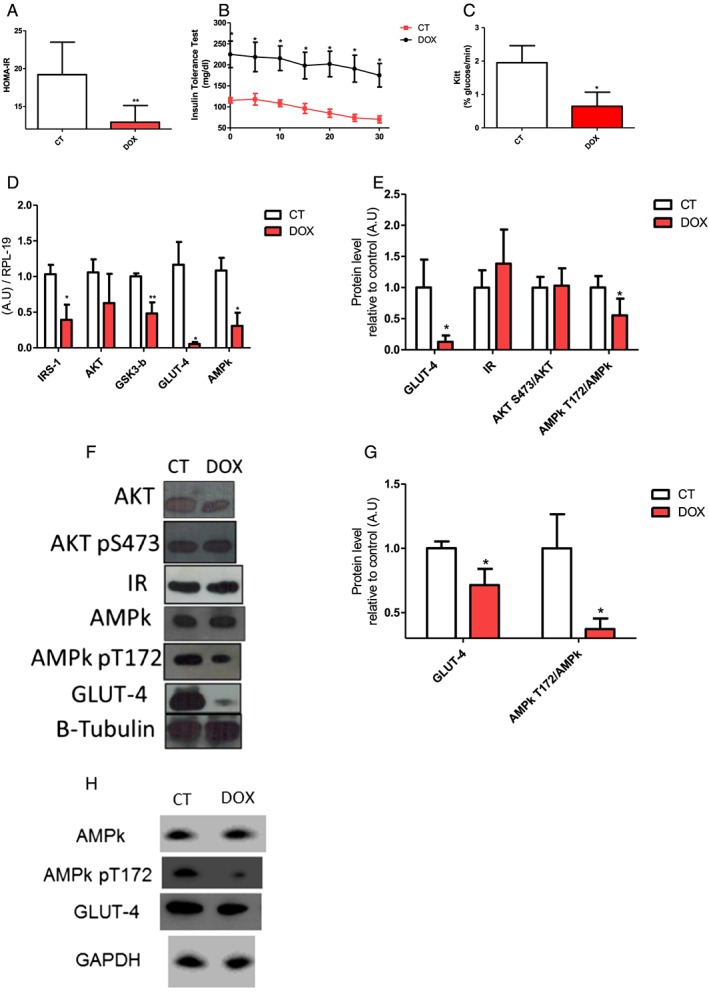
Doxorubicin leads to impaired systemic insulin sensitivity. (A) Homeostatic model assessment of insulin resistance. (B) Curve of the insulin tolerance test C. Kitt. (D)Gene expression involved in glucose metabolism. (E/F) protein expression involved in glucose metabolism in extensor digitorum longus muscle. (G/H) AMPk and GLUT‐4 protein expression in culture L6 myocytes treated with doxorubicin. The groups were compared by Test T. P < 0.05 was considered statistically significant. * P < 0.5, ** P < 0.01, *** P < 0.001. n = 3–8.
However, protein levels of insulin receptor (Figure 2E and 2F) and gene and protein expression of AKT protein downstream of PI3‐K were not altered (Figure 2D, 2E, and 2F) in the EDL muscle after treatment. Nevertheless, other proteins involved in the insulin pathway showed decreased gene expression, such as IRS‐1, GSK3‐b (Figure 2D) in skeletal muscle.
The activity of AMP‐activated protein kinase protein is decreased with treatment with doxorubicin
GLUT4 and AMPk α (pT172) were decreased in mRNA and protein levels (Figure 2D, 2E, and 2F). Our in vitro data corroborate with the decrease of GLUT4 expression and phosphorylation of AMPK α observed in the EDL muscle (Figure 2G and 2H). After 48 h, the L6 myocyte culture treated with DOX presents a decreased phosphorylation of AMPK α in the residue T172, without changing its overall content. The GLUT‐4 expression was also reduced in the treatment. These data suggest that although the insulin signalling cascade has not been completely disrupted, important proteins with effective participation in glucose uptake are less expressed.
Treatment with doxorubicin decreases glucose uptake in L6 myocytes and this effect is reversed by chronic activation of AMP‐activated protein kinase
Our results in isolated myocytes confirm our hypothesis that metabolic chaos generated by this drug on carbohydrate metabolism is due in part to impaired glucose uptake in both the stimulated and basal state (Figure 3A).
Figure 3.
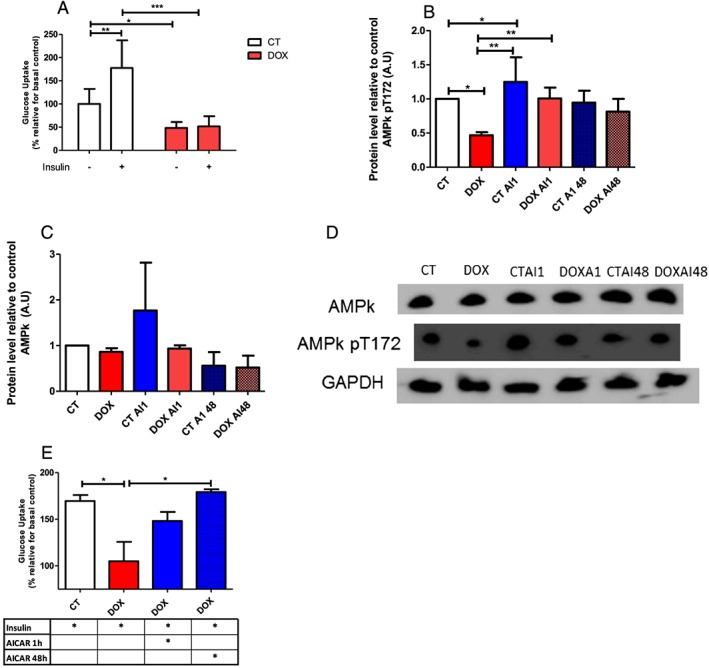
Doxorubicin decrease glucose uptake in L6 cells, which is recovered with chronic treatment with AICAR. (A) The L6 cells were differentiated and submitted to the glucose uptake assay, with (plus sign) and without (minus sign) insulin stimulation (100 nM), after 48 h the treatment with doxorubicin (100 nM). (B–D). Effect of acute and chronic treatment with AICAR on the phosphorylation of AMPK in myocytes. (E) 2‐Deoxy‐[C14]‐D‐glucose uptake in L6 cells. This cells were subjected to treatment with doxorubicin, associated with insulin, and submitted to acute (1 h) or chronic (48 h) AICAR (2 mmol) treatment; The results were relativized by control group without insulin stimulation. For the glucose uptake assay the groups were compared using ANOVA two‐way test with Bonferroni post‐test to compare the groups. P < 0.05 was considered statistically significant. * P < 0.5, ** P < 0.01, *** P < 0.001. n = 4–6.
Accordingly, myocytes were treated with DOX and AICAR acutely (1 h) or chronically (48 h). As previously demonstrated, in vitro treatment with DOX decreased the phosphorylation of AMPK. The acute intervention (1 h) with AICAR is effective in phosphorylation of this protein in control and treated group with DOX compared with the group treated only with DOX. We did not find an increase in the phosphorylation of AMPK α after chronic treatment (48 h) with AICAR (Figure 3B, 3C, and 3D).
Intervention with AICAR recovers glucose uptake in myocytes treated with DOX. By 48 h, the reestablishment of glucose uptake was complete (Figure 3E). Despite the fact that treatment with AICAR for 1 h increased uptake, it was not enough to be statistically significant compared with the group treated with DOX.
Cell viability was increased by n‐acetyl cysteine treatment, but the glucose uptake was not restored by n‐acetyl cysteine
The deleterious role of oxidative stress in chemotherapy treatment, especially with DOX has been well established in cardiac and skeletal muscle tissue. Indeed, our results suggest a potential increase in production of ERO's, because treated animals had increased activity of mitochondrial complex 1 and a decrease in activity of mitochondrial Complex 3 (Figure 4A).
Figure 4.
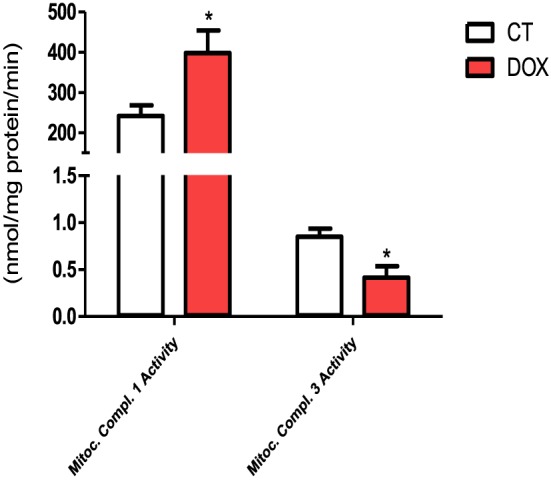
Mitochondrial complex 1 and 3 activity. The groups were compared by Test T. P < 0.05 was considered statistically significant. * P < 0.5. n = 4–6.
To assess whether oxidative stress could be involved in the decrease in cell viability we treated L6 myocytes with DOX for 48 h at a concentration of 1 mmol in the presence or absence of NAC (20 mmol/high concentration and 2 mmol/low concentration). As shown in Figure S2A, the concentration of DOX 1 mmol leads to a decrease in cell viability, whereas the chronic treatment with high concentrations of NAC (20 mmol) reverses this effect (Figure S2B). Low concentrations of NAC were not able to reverse the cell viability.
We investigated whether treatment with the antioxidant NAC reverses glucose uptake in the myocytes treated with this chemotherapeutic agent. Our data show that the treatment with NAC was unable to reverse the reduction in glucose uptake resulting from treatment with DOX (Figure S2C).
Doxorubicin‐induced decreased the proinflammatory cytokines protein content
Pro‐inflammatory cytokine levels (IL‐6/TNF‐α) were not elevated compared with the control group (Figure S3A), including decreased levels of antiinflammatory cytokine IL‐10. Still, the content of IL‐6 released into the medium of myocytes treated with DOX in the presence of LPS is decreased compared with the control group treated with LPS (Figure S3B).
The pathway of inflammasome also seems to be not involved in this process, because the expression of IL‐1β, as well as proteins involved in the maturation of this protein, was not modulated by treatment (Figure S3C).
Discussion
Our main results suggest that treatment with DOX caused hyperglycaemia and insulin resistance mediated by inhibition in AMPk, moreover, the treatment leads to atrophy, weight loss, and anorexia mimicking similar conditions to cachexia. In addition, we observed that rats treated with DOX showed hyperglycaemia, insulin resistance (kITT), increases in FFA, and corticosterone serum concentration. To our knowledge, this is the first study to demonstrate an association between treatment with DOX and insulin resistance in skeletal muscle metabolism.
Chemotherapy can alter insulin sensitivity in clinical practice.30, 31 The treatment with DOX causes the deterioration in glucose metabolism both in vivo and in vitro. Recently, Arunachalam and collegues32 review that DOX may mimic type 2 diabetes, whereas the adipose tissue metabolism was negatively affected. We showed that skeletal muscle plays a significant role in this condition, with dramatically reduced GLUT‐4 protein and mRNA expression in the animal model, and DOX treatment decreased the glucose uptake in the basal and stimulated condition in L6 myocytes.
Insulin is a key hormone in glucose uptake in skeletal muscle. The activation of proteins involved in signalling of this hormone is essential for the translocation of GLUT4 from intracellular vesicles to the sarcolemma. Dysfunction of proteins in this pathway causes a strong insulin resistance.33, 34 Our data showed a decrease in insulin sensitivity, whereas in the kITT test, the DOX group showed decreases in the kITT constant. However, in molecular levels our results suggest that the signalling cascade of insulin is not completely disrupted, although important proteins with effective participation in glucose uptake are less expressed. The protein levels of insulin receptors and gene and protein expression of AKT protein downstream of PI 3‐kinase were not affected in the EDL muscle after treatment. Nevertheless, other proteins involved in glucose metabolism had decreased expression such IRS‐1 and GS3kb mRNA expression, and GLUT‐4 mRNA and protein expression. Even with a partially competent signalling of insulin, the need for this glucose transporter is essential as demonstrated by studies using a knockout model for GLUT4.35
Several hypotheses were reported to trigger the development of glucose intolerance. It is well established that DOX increases oxidative stress and mitochondrial dysfunction,36, 37 including in skeletal muscle,38, 39 which both mechanisms are important to lead the insulin resistance. DOX leads to increased ROS through different mechanisms. The interaction of this drug with iron III, the perturbation of regulation of nitric oxide (NO), and the redox cycling of this drug, which takes place by reoxidation of radical DOX‐semiquinone to DOX, leads to the formation of ROS.40, 41, 42
Although our data suggest that treatment with DOX alters the activity of mitochondrial complexes, leading to a proton gradient that is favourable to the formation of ROS with increases in activity of the Complex I and inhibition of Complex III, which are important sites where there is formation of ROS.43, 44 Many studies showed the strong association between oxidative stress and a decrease in glucose uptake in skeletal muscle.45, 46, 47, 48 But the treatment with the antioxidant n‐acetylcysteine (NAC) was not able to reverse the decrease in glucose uptake induced by DOX in L6 culture. However, when cells were treated with concentrations of DOX capable of decreasing cell viability, antioxidant treatment (high concentrations of NAC) was able to reverse this effect. Probably, the absence of effect in restored the glucose uptake after NAC treatment occurs because the oxidative stress leads to impair in glucose uptake mediated by insulin response,46 and the contribution of this pathway in glucose intolerance is less effective after the DOX treatment. Whereas, the main pathway disrupted by DOX is the AMPk signalling in skeletal muscle. The AMPk is the other major regulator of glucose transport, and it leads to GLUT‐4 for the sarcolemma independently of insulin.
Additionally, AMPk regulates the gene expression of GLUT‐4 to the activation and translocation of transcription factors into the nucleus to bind to the promoter of the GLUT4 gene region, as the MEF2A (myocytes enhancer factor) and GEF (GLUT4 enhancer factor).49, 50
Our data demonstrated a large inhibition of GLUT‐4 protein expression in EDL in rats treated with DOX. Together with this, the AMPk mRNA expression and phospho AMPk α thr172 follow the decreases in the GLUT‐4 protein. Because of this we hypothesized that AMPk signalling was disrupted in skeletal muscle in the DOX group. We evaluated the treatment with an agonist of AMPk, AICAR, that was able to completely reverse glucose uptake stimulated by insulin in L6 myocytes.
Other important regulator of AMPk pathway and glucose uptake is the adiponectin, an adipokine produced in adipose tissue and responsible by a cross link between it and other tissues, because its circulating concentration positively correlated with an insulin sensitivity. The adiponectin expression in adipose tissue and the circulating levels was decreased (data not shown) in the DOX group and may be explained, at least in part, by the decrease in AMPk activity and mRNA expression, whereas adiponectin is a positive modulator of AMPk activity.51 Beyond the adiponectin, the Interleukin‐6 increased the AMPk activity in myocites.52, 53 In our results, the DOX treatment inhibits the IL‐6 released in L6 myocites after LPS stimulation, although this phenomenon did not occur in vivo.
Our results showed that DOX treatment lead animals to develop a cachexia‐like syndrome. Cachexia is characterized as a multifactorial syndrome, in which there is involuntary weight loss.54 In agreement with other supporting studies, the use of some chemotherapy drugs appears to contribute to the development of symptoms of cachexia, contributing to an unfavourable metabolic state.19, 55, 56 The DOX also caused increases in glucose intolerance, induced extreme atrophy, weight, and adipose tissue mass loss, anorexia, and a catabolic environment, as demonstrated by an increase in corticosterone/testosterone ratio, with a decrease in myocyte area.
The literature showed that inflammatory mediators are a possible mediator of glucose intolerance, sarcopenia, and anorexia. However, our results do not support this hypothesis in DOX treatment. Although TNF‐α was decreased in the DOX group without changes in the protein content of the cytokine receptors assessed, there was no change in gene expression of proteins involved in the inflammassome pathway in muscle.
In conclusion, our results show that treatment with DOX leads to conditions similar to cachexia, with a severe glucose intolerance both in vivo and in vitro. The decrease of AMPk activity of the protein is modulated negatively with treatment and has proved to be a possible therapeutic target, which is able to recover glucose sensitivity in skeletal muscle, and can lead to improvements in quality of life in chemotherapy patients.
Conflict of interest
Edson Alves de Lima Junior, Alex Shimura Yamashita, Gustavo, Pimentel, Luís Gustavo de Sousa, Ronaldo Vagner T. dos Santos, Cinara Gonçalves, Emílio Streck, Fábio Lira, and José Cesar Rosa Neto declare that they have no conflict of interests.
Supporting information
Supporting info item
Acknowledgements
José Cesar Rosa Neto thanks the Fapesp support (2013/09367‐4), and Fabio Santos Lira thanks the CNPq (481231/2012‐9).
The authors certify that they comply with the ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015.57
de Lima Junior, E. A. , Yamashita, A. S. , Pimentel, G. D. , De Sousa, L. G. O. , Santos, R. V. T. , Gonçalves, C. L. , Streck, E. L. , de Lira, F. S. , and Rosa Neto, J. C. (2016) Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPk signalling in skeletal muscle. Journal of Cachexia, Sarcopenia and Muscle, 7: 615–625. doi: 10.1002/jcsm.12104.
References
- 1. Richardson DS, Johnson SA. Anthracyclines in haematology: preclinical studies, toxicity and delivery systems. Blood Rev 1997;11:201–223. [DOI] [PubMed] [Google Scholar]
- 2. Lai HC, Yeh YC, Ting CT, Lee WL, Lee HW, Wang LC, et al. Doxycycline suppresses doxorubicin‐induced oxidative stress and cellular apoptosis in mouse hearts. Eur J Pharmacol 2010;644:176–187. [DOI] [PubMed] [Google Scholar]
- 3. Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF. Adriamycin‐induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984;226:466–468. [DOI] [PubMed] [Google Scholar]
- 4. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 2004;56:185–229. [DOI] [PubMed] [Google Scholar]
- 5. Fang J, Nakamura H, Iyer AK. Tumor‐targeted induction of oxystress for cancer therapy. J Drug Target 2007;15:475–486. [DOI] [PubMed] [Google Scholar]
- 6. Benjamin RS, Wiernik PH, Bachur NR. Adriamycin chemotherapy–efficacy, safety, and pharmacologic basis of an intermittent single high‐dosage schedule. Cancer 1974;33:19–27. [DOI] [PubMed] [Google Scholar]
- 7. Morrow GR, Andrews PL, Hickok JT, Roscoe JA, Matteson S. Fatigue associated with cancer and its treatment. Support Care Cancer 2002;10:389–398. [DOI] [PubMed] [Google Scholar]
- 8. Gorselink M, Vaessen SF, van der Flier LG, Leenders I, Kegler D, Caldenhoven E, et al. Mass‐dependent decline of skeletal muscle function in cancer cachexia. Muscle Nerve 2006;33:691–693. [DOI] [PubMed] [Google Scholar]
- 9. Jacob S, Rett K, Wicklmayr M, Agrawal B, Augustin HJ, Dietze GJ. Differential effect of chronic treatment with two beta‐blocking agents on insulin sensitivity: the carvedilol‐metoprolol study. J Hypertens 1996;14:489–494. [PubMed] [Google Scholar]
- 10. Pagano G, Cavallo‐Perin P, Cassader M, Bruno A, Ozzello A, Masciola P, et al. An in vivo and in vitro study of the mechanism of prednisone‐induced insulin resistance in healthy subjects. J Clin Invest 1983;72:1814–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin‐dependent (type II) diabetes mellitus. J Clin Invest 1985;76:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Honors MA, Kinzig KP. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J Cachexia Sarcopenia Muscle 2012;3:5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Srikanthan P, Hevener AL, Karlamangla AS. Sarcopenia exacerbates obesity‐associated insulin resistance and dysglycemia: findings from the National Health and Nutrition Examination Survey III. PLoS One 2010;5:e10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L, et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population‐based study. Lancet Oncol 2008;9:629–635. [DOI] [PubMed] [Google Scholar]
- 15. Sakkas GK, Kent‐Braun JA, Doyle JW, Shubert T, Gordon P, Johansen KL. Effect of diabetes mellitus on muscle size and strength in patients receiving dialysis therapy. Am J Kidney Dis 2006;47:862–869. [DOI] [PubMed] [Google Scholar]
- 16. Neidhart JA, Gochnour D, Roach R, Hoth D, Young D. A comparison of mitoxantrone and doxorubicin in breast cancer. J Clin Oncol 1986;4:672–677. [DOI] [PubMed] [Google Scholar]
- 17. Stone P, Hardy J, Broadley K, Tookman AJ, Kurowska A, A'Hern R. Fatigue in advanced cancer: a prospective controlled cross‐sectional study. Br J Cancer 1999;79:1479–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gilliam LA, Ferreira LF, Bruton JD, Moylan JS, Westerblad H, St Clair DK, et al. Doxorubicin acts through tumor necrosis factor receptor subtype 1 to cause dysfunction of murine skeletal muscle. J Appl Physiol (1985) 2009;107:1935–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gilliam LA, Moylan JS, Callahan LA, Sumandea MP, Reid MB. Doxorubicin causes diaphragm weakness in murine models of cancer chemotherapy. Muscle Nerve 2011;43:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Norren K, van Helvoort A, Argiles JM, van Tuijl S, Arts K, Gorselink M, et al. Direct effects of doxorubicin on skeletal muscle contribute to fatigue. Br J Cancer 2009;100:311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Beer EL, Finkle H, Voest EE, Van Heijst BG, Schiereck P. Doxorubicin interacts directly with skinned single skeletal muscle fibres. Eur J Pharmacol 1992;214:97–100. [DOI] [PubMed] [Google Scholar]
- 22. Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev 2009;89:1025–1078. [DOI] [PubMed] [Google Scholar]
- 23. Tokarska‐Schlattner M, Zaugg M, da Silva R, Lucchinetti E, Schaub MC, Wallimann T, et al. Acute toxicity of doxorubicin on isolated perfused heart: response of kinases regulating energy supply. Am J Physiol Heart Circ Physiol 2005;289:H37–H47. [DOI] [PubMed] [Google Scholar]
- 24. Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric restriction mimetic 2‐deoxyglucose antagonizes doxorubicin‐induced cardiomyocyte death by multiple mechanisms. J Biol Chem 2011;286:21993–22006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al‐Shabanah OA, El‐Kashef HA, Badary OA, Al‐Bekairi AM, Elmazar MM. Effect of streptozotocin‐induced hyperglycaemia on intravenous pharmacokinetics and acute cardiotoxicity of doxorubicin in rats. Pharmacol Res 2000;41:31–37. [DOI] [PubMed] [Google Scholar]
- 26. Blasiak J, Arabski M, Krupa R, Wozniak K, Zadrozny M, Kasznicki J, et al. DNA damage and repair in type 2 diabetes mellitus. Mutat Res 2004;554:297–304. [DOI] [PubMed] [Google Scholar]
- 27. Souza CO, Teixeira AA, Lima EA, Batatinha HA, Gomes LM, Carvalho‐Silva M, et al. Palmitoleic acid (n‐7) attenuates the immunometabolic disturbances caused by a high‐fat diet independently of PPARalpha. Mediators Inflamm 2014;2014:582197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem 1987;162:156–159. [DOI] [PubMed] [Google Scholar]
- 29. Liu W, Saint DA. Validation of a quantitative method for real time PCR kinetics. Biochem Biophys Res Commun 2002;294:347–353. [DOI] [PubMed] [Google Scholar]
- 30. Feng JP, Yuan XL, Li M, Fang J, Xie T, Zhou Y, et al. Secondary diabetes associated with 5‐fluorouracil‐based chemotherapy regimens in non‐diabetic patients with colorectal cancer: results from a single‐centre cohort study. Colorectal Dis 2013;15:27–33. [DOI] [PubMed] [Google Scholar]
- 31. Chala E, Manes C, Iliades H, Skaragkas G, Mouratidou D, Kapantais E. Insulin resistance, growth factors and cytokine levels in overweight women with breast cancer before and after chemotherapy. Hormones (Athens) 2006;5:137–146. [DOI] [PubMed] [Google Scholar]
- 32. Arunachalam S, Tirupathi Pichiah PB, Achiraman S. Doxorubicin treatment inhibits PPARgamma and may induce lipotoxicity by mimicking a type 2 diabetes‐like condition in rodent models. FEBS Lett 2013;587:105–110. [DOI] [PubMed] [Google Scholar]
- 33. Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA‐mediated gene silencing. Proc Natl Acad Sci U S A 2003;100:7569–7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shao J, Yamashita H, Qiao L, Friedman JE. Decreased Akt kinase activity and insulin resistance in C57BL/KsJ‐Leprdb/db mice. J Endocrinol 2000;167:107–115. [DOI] [PubMed] [Google Scholar]
- 35. Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, et al. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med 1997;3:1096–1101. [DOI] [PubMed] [Google Scholar]
- 36. Zhou S, Palmeira CM, Wallace KB. Doxorubicin‐induced persistent oxidative stress to cardiac myocytes. Toxicol Lett 2001;121:151–157. [DOI] [PubMed] [Google Scholar]
- 37. Childs AC, Phaneuf SL, Dirks AJ, Phillips T, Leeuwenburgh C. Doxorubicin treatment in vivo causes cytochrome C release and cardiomyocyte apoptosis, as well as increased mitochondrial efficiency, superoxide dismutase activity, and Bcl‐2:Bax ratio. Cancer Res 2002;62:4592–4598. [PubMed] [Google Scholar]
- 38. Gilliam LA, St Clair DK. Chemotherapy‐induced weakness and fatigue in skeletal muscle: the role of oxidative stress. Antioxid Redox Signal 2011;15:2543–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smuder AJ, Kavazis AN, Min K, Powers SK. Exercise protects against doxorubicin‐induced oxidative stress and proteolysis in skeletal muscle. J Appl Physiol (1985) 2011;110:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davies KJ, Doroshow JH. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 1986;261:3060–3067. [PubMed] [Google Scholar]
- 41. Weinstein DM, Mihm MJ, Bauer JA. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J Pharmacol Exp Ther 2000;294:396–401. [PubMed] [Google Scholar]
- 42. Deavall DG, Martin EA, Horner JM, Roberts R. Drug‐induced oxidative stress and toxicity. J Toxicol 2012;2012:645460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu X, Persson HL, Richardson DR. Molecular pharmacology of the interaction of anthracyclines with iron. Mol Pharmacol 2005;68:261–271. [DOI] [PubMed] [Google Scholar]
- 44. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 2003;278:36027–36031. [DOI] [PubMed] [Google Scholar]
- 45. Boden MJ, Brandon AE, Tid‐Ang JD, Preston E, Wilks D, Stuart E, et al. Overexpression of manganese superoxide dismutase ameliorates high‐fat diet‐induced insulin resistance in rat skeletal muscle. Am J Physiol Endocrinol Metab 2012;303:E798–E805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jaiswal N, Maurya CK, Pandey J, Rai AK, Tamrakar AK. Fructose‐induced ROS generation impairs glucose utilization in L6 skeletal muscle cells. Free Radic Res 2015;49:1055–1068. [DOI] [PubMed] [Google Scholar]
- 47. Aoi W, Naito Y, Yoshikawa T. Role of oxidative stress in impaired insulin signaling associated with exercise‐induced muscle damage. Free Radic Biol Med 2013;65:1265–1272. [DOI] [PubMed] [Google Scholar]
- 48. Pillon NJ, Croze ML, Vella RE, Soulere L, Lagarde M, Soulage CO. The lipid peroxidation by‐product 4‐hydroxy‐2‐nonenal (4‐HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012;153:2099–2111. [DOI] [PubMed] [Google Scholar]
- 49. Holmes BF, Sparling DP, Olson AL, Winder WW, Dohm GL. Regulation of muscle GLUT4 enhancer factor and myocyte enhancer factor 2 by AMP‐activated protein kinase. Am J Physiol Endocrinol Metab 2005;289:E1071–E1076. [DOI] [PubMed] [Google Scholar]
- 50. Zheng D, MacLean PS, Pohnert SC, Knight JB, Olson AL, Winder WW, et al. Regulation of muscle GLUT‐4 transcription by AMP‐activated protein kinase. J Appl Physiol (1985) 2001;91:1073–1083. [DOI] [PubMed] [Google Scholar]
- 51. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003;423:762–769. [DOI] [PubMed] [Google Scholar]
- 52. Ruderman NB, Keller C, Richard AM, Saha AK, Luo Z, Xiang X, et al. Interleukin‐6 regulation of AMP‐activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes 2006;55:S48–S54. [DOI] [PubMed] [Google Scholar]
- 53. Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, et al. Interleukin‐6 increases insulin‐stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP‐activated protein kinase. Diabetes 2006;55:2688–2697. [DOI] [PubMed] [Google Scholar]
- 54. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 55. Sinno MH, Coquerel Q, Boukhettala N, Coeffier M, Gallas S, Terashi M, et al. Chemotherapy‐induced anorexia is accompanied by activation of brain pathways signaling dehydration. Physiol Behav 2010;101:639–648. [DOI] [PubMed] [Google Scholar]
- 56. Sanchez‐Lara K, Ugalde‐Morales E, Motola‐Kuba D, Green D. Gastrointestinal symptoms and weight loss in cancer patients receiving chemotherapy. Br J Nutr 2013;109:894–897. [DOI] [PubMed] [Google Scholar]
- 57. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for Publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2015. J Cachexia Sarcopenia Muscle 2015;6:315–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item