

Abstract
Background
Cancer associated cachexia affects the majority of cancer patients during the course of the disease and thought to be directly responsible for about a quarter of all cancer deaths. Current evidence suggests that a pro‐inflammatory state may be associated with this syndrome although the molecular mechanisms responsible for the development of cachexia are poorly understood. The purpose of this work was the identification of key drivers of cancer cachexia that could provide a potential point of intervention for the treatment and/or prevention of this syndrome.
Methods
Genetically engineered and xenograft tumour models were used to dissect the molecular mechanisms driving cancer cachexia. Cytokine profiling from the plasma of cachectic and non‐cachectic cancer patients and mouse models was utilized to correlate circulating cytokine levels with the cachexia phenotype.
Results
Utilizing engineered tumour models we identified MAP3K11/GDF15 pathway activation as a potent inducer of cancer cachexia. Increased expression and high circulating levels of GDF15 acted as a key mediator of this process. In animal models, tumour‐produced GDF15 was sufficient to trigger the cachexia phenotype. Elevated GDF15 circulating levels correlated with the onset and progression of cachexia in animal models and in patients with cancer. Inhibition of GDF15 biological activity with a specific antibody reversed body weight loss and restored muscle and fat tissue mass in several cachectic animal models regardless of their complex secreted cytokine profile.
Conclusions
The combination of correlative observations, gain of function, and loss of function experiments validated GDF15 as a key driver of cancer cachexia and as a potential therapeutic target for the treatment and/or prevention of this syndrome.
Keywords: Cancer cachexia, GDF15, MAP3K11, MIC‐1, MLK3
Introduction
One of the most debilitating and lethal effects of cancer is the development of cancer‐associated anorexia and cachexia syndrome (also known as wasting syndrome). It affects up to 80% of advanced cancer patients and is thought to be responsible for about 20–30% of all cancer deaths.1, 2, 3 Cancer cachexia is a complex metabolic syndrome defined by the presence of involuntary weight loss that is primarily due to the loss of muscle and fat tissues. The weight loss is thought to result from an energy imbalance caused by reduced food intake and the shift from anabolic to catabolic metabolic processes driven by inflammation.4, 5
The molecular mechanisms responsible for the development of cancer‐associated cachexia are poorly understood. It is postulated that constantly evolving tumours eventually acquire genetic changes that trigger the cachexia phenotype, potentially in combination with host susceptibility factors.6, 7, 8 Key to this phenotype is the increased synthesis of secreted effector molecules (cytokines and/or other pro‐cachectic factors such as IL‐1, IL‐6, TNFα, and IFNγ) that, by acting directly or indirectly on host tissues, elicit metabolic changes that generate an imbalance in energy homeostasis.9 The direct or indirect actions of these pro‐cachectic factors on the central nervous system cause reduced food intake, resulting in anorexia.10, 11 On muscle tissues, these factors suppress the synthesis of muscle proteins and activate the ubiquitin‐mediated proteolytic system, leading to increased muscle protein degradation, hypercatabolism, and ultimately, muscle atrophy. Muscle protein degradation results in the generation of amino acids and oligopeptides, providing building blocks for synthetic processes. The effect on adipose tissues is the reduction of adipogenesis and the induction of lipolysis, leading to elevated free fatty acid levels in circulation.4, 12 Cancer cachexia cannot be reversed by conventional nutritional support.3 Ideally, an early intervention would prevent the cascade of catabolism and worsening of symptoms.13, 14 However, the lack of a detailed molecular understanding of the underlying mechanism(s) of cancer cachexia has thus far prevented the development of effective therapies.
In the past two decades, the pro‐inflammatory molecules tumour necrosis factor‐alpha (TNFα) and interleukin‐6 (IL‐6) have emerged as potential key mediators of cachexia, largely based on their ability to induce cachexia when injected into animals.10, 11, 15, 16 Subsequent clinical trials with inhibitors of these molecules have shown no clinical benefit in preventing or reversing the effect of cachexia on lean body mass, suggesting that neither of these molecules is a critical driver of the syndrome.17 Recently, negative regulators of muscle homeostasis such as myostatin (GDF8) and Activin A have attracted attention based on their ability to induce muscle atrophy.10, 18 Although the exact role of these molecules in inducing cancer‐associated cachexia is not known, several agents targeting this pathway have entered clinical development.19 A related molecule, growth differentiation factor 15 (GDF15), a member of the TGFβ ligand family, has also been implicated in cancer cachexia. Serum levels of GDF15 correlate with body weight loss in patients with prostate cancer, and tumours engineered to overexpress GDF15 can induce cachexia in mice.20, 21
In an effort to identify tumour maintenance genes we tested the ability of members of the mitogen‐activated protein kinase (MAPK) signalling pathway to drive tumour growth in vivo. MAPK pathways play a central role of mediating the effects of a variety of extracellular stimuli and they regulate gene expression, metabolism, cell division, morphology, and survival.22, 23 We demonstrated that MAP3K11/MLK3 is a potent tumour maintenance gene. Surprisingly, we also found that MAP3K11 pathway activation in tumours triggered the induction of a genetic programme that caused severe cachexia of the host. The purpose of the study was to identify the essential mediator(s) of the MAP3K11 induced cachexia phenotype. We identified GDF15 as a key driver of this genetic programme. Furthermore, we also demonstrated that GDF15 acted as the key driver of cachexia in a variety of naturally occurring cachectic tumour models, and its plasma levels strongly correlated with cachexia in cancer patients representing different tumour types.
Results
MAP3K11 overexpression in tumours leads to weight loss in the host
We assessed the ability of potential tumour maintenance genes to drive tumour growth in vivo using a directed complementation (DC) approach. This assay involves testing the ability of an introduced gene to maintain tumour growth when expression of the original driving oncogene is turned off. We tested the ability of candidate tumour maintenance genes, MAP3K7/TAK1 or MAP3K11/MLK3, to form tumours in vivo in a genetically engineered model originally driven by a doxycycline‐inducible HER2(V659E) oncogene. In this system, the expression and activity of HER2 were suppressed by doxycycline withdrawal and followed by the introduction of genes encoding MAP3K7, MAP3K11, or EGFR(L858R) and HER2(V659E) as positive controls. We implanted the transduced tumour cells in mice and assessed the ability of the introduced genes to drive tumour growth in the absence of the original HER2(V659E) oncogene. As shown in Figure 1A, cells infected with the empty vector were not able to form tumours, while 100% of the animals implanted with tumour cells expressing the positive control HER2(V659E) or EGFR(L858R) oncogenes developed tumours by day 18 post‐implantation. All animals implanted with MAP3K11‐expressing tumour cells developed tumours by day 25 post‐injection, while only 40% of the animals injected with MAP3K7‐expressing tumour cells developed tumours by day 30. Thus, the expression of MAP3K11 and to a lesser extent, MAP3K7, was able to complement the missing oncogenic function of HER2(V659E) and drive tumour growth.
Figure 1.
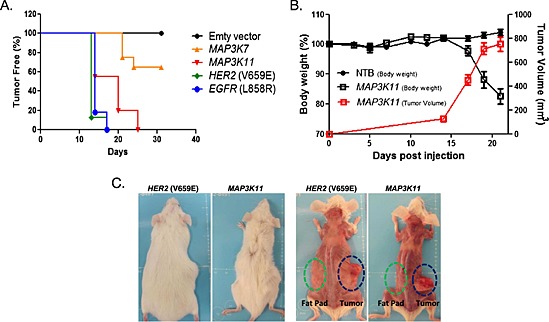
MAP3K11‐driven tumors induce cachexia in mice. (A). MAP3K11 and MAP3K7 expression drive tumor growth in vivo. Inducible HER2(V659E)‐driven murine mammary tumor cells were transduced with either empty vector (negative control) or HER2(V659E) or EGFR(L858R) (positive controls), MAP3K7 or MAP3K11 in the absence of doxycycline and implanted in mice (n = 10/group). (B). Induction of body weight loss by MAP3K11‐driven tumors. Mice (n = 10/group) were implanted with MAP3K11‐driven tumor cells in matrigel (1:1) or sham injected with matrigel and phosphate buffered saline (PBS Buffer) (1:1). Total body weight and tumor growth were monitored three times a week. Mean +/− SEM are represented in the graph. (C). Loss of muscle and fat tissue in mice carrying MAP3K11‐ and HER2‐driven tumors. Mice were sacrificed when tumor volume reached 800 mm3.
Close inspection of mice bearing MAP3K11‐driven tumours revealed severe weight loss‐(Figure 1B and 1C). Body weight loss was only observed in mice bearing with MAP3K11 but not with MAP3K7 or the positive control HER2(V659E) or EGFR(L858R)‐complemented tumours (data not shown). This phenotype was reminiscent of cancer cachexia and was maintained upon the propagation of the MAP3K11‐driven tumours in naïve animals, validating that the phenotype was triggered by the tumour and not by the host.
Identification of secreted proteins induced by MAP3K11 overexpression
Cancer cachexia is thought to be a complex syndrome triggered, at least in part, by tumour‐derived secreted factors that alter the metabolic balance of the host, resulting in severe body weight loss primarily because of the loss of skeletal muscle and adipose tissues.10, 11, 17 We hypothesized that activation of the MAP3K11 signalling pathway results in the elevated expression and increased secretion of effector proteins whose action on host tissues elicits metabolic changes resulting in body weight loss.
To identify secreted proteins whose expression was selectively elevated in the cachexia‐inducing MAP3K11‐driven DC tumours, we took advantage of a panel of closely related control, non‐cachectic DC tumours generated through DC in the same tumour background with the overexpression of MAP3K7, HER3, FAK1, FGFR2αIIIb, FGFR2αIIIc, HER2(V659E), PIP5K1, RON, RON plus MSP, RONΔ160 variant, RSK2, and TNK2 genes. We compared the gene expression profile of 70 secreted proteins in these different tumour models. Also, we analysed the levels of 25 murine cytokines in the plasma of the animals carrying the non‐cachectic HER2(V659E) and EGFR(L858R) and cachexia‐inducing MAP3K11‐driven tumours.
Transcript profiling revealed the selective upregulation of cytokines eotaxin/CCL11, CCL17, CCL24, the chemokine CXCL14, interleukins IL‐11 and IL‐1β, and the TGFβ family members INHBA/Activin A, BMP2, and GDF15 in the cachectic MAP3K11‐driven tumour models (Figure 2A).
Figure 2.
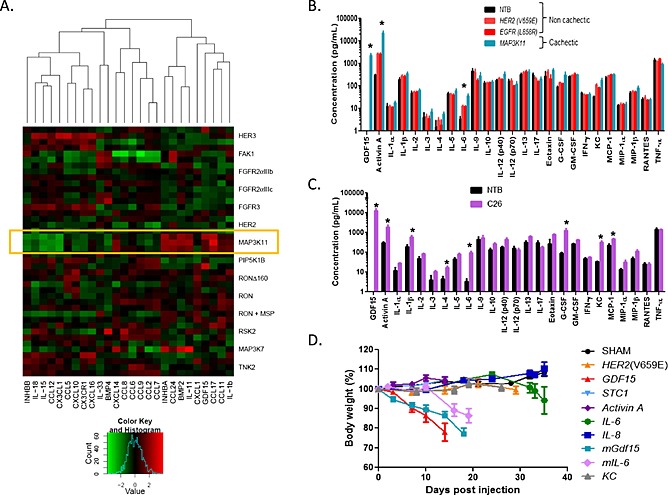
Identification of secreted proteins selectively overexpressed by cachectic MAP3K11‐driven tumors. (A). Microarray profile of secreted proteins in a panel of tumor models. Microarray prolife of a panel of closely related murine DC tumor models generated by the overexpression of the genes indicated on the side of the graph. The expression patterns of 70 secreted factors were analysed and 26 with the most variable expression profile (bottom of the graph) were compared between the tumor models. (B). Plasma protein profile of cachectic MAP3K11 and non‐cachectic HER2(V659E) and EGFR(L858R) DC models. Murine cytokines were measured as described in Materials and methods (n = 3–4/model). Asterisks represent statistically significant elevation in MAP3K11 vs. HER2(V569E) or EGFR(L858R) tumor‐bearing mice (non‐parametric t‐test). (C). Plasma protein profile of cachectic C26 model and non‐tumor‐bearing control mice. Murine cytokines were measured as described in Materials and methods (n = 3–4/model). Asterisks represent statistically significant elevation in C26 tumor‐bearing mice compared with NTB mice (non‐parametric t‐test). (D). Testing the ability of cytokines to induce cachexia in mice. Murine mammary tumor cells driven by the HER2(V659E) oncogene were transduced either with empty vector (sham, which did not develop tumors), hSCT1‐HER2(V659E) (negative controls), human and murine GDF15‐HER2(V659E), Activin A‐HER2(V659E), human and murine IL‐6‐HER2(V659E), human IL‐8‐HER2(V659E), or murine KC‐HER2(V659E). Transduced cells were implanted in mice (n = 10/group) subcutaneously, and the animals were kept without doxycycline. Body weight and tumor growth were monitored three times a week.
Protein profiling identified elevated levels of GDF15 and Activin A in the plasma of the cachectic animals. GDF15 was not detectable in the plasma of the naïve or non‐cachectic hosts but significantly elevated in the plasma of cachectic animals (average plasma concentration 2 ng/mL). Activin A levels were also significantly higher in the cachectic animals (23 ng/mL, p = 0.05) compared with the levels seen in the control non‐cachectic (3 ng/mL) or naïve animals (0.3 ng/mL) (Figure 2B). Eotaxin and IL‐1β plasma levels were not elevated in the cachectic animals while BMP2 was undetectable (data not shown). Assays specific to CCL17, CCL24, CXCL14, and IL‐11 were not included in the panel. In addition, we detected elevated plasma levels of IL‐6 (35 pg/mL vs. 12 pg/ml, p = 0.045) in the tumour bearing animals vs. non‐tumour bearing controls, but its circulating levels were not significantly higher in the MAP3K11 vs. the control EGFR(L858R) and HER2(V69E) models (Figure 2B).
We extended the blood protein analysis to animals carrying the well‐known cachectic murine tumour model, the C26 colorectal carcinoma. Of the proteins analysed, GDF15 was the most significantly increased in the circulation of animals carrying C26 tumours (13 ng/mL), compared with non‐tumour‐bearing (NTB) controls (undetectable). In addition to GFD15, Activin A, granulocyte‐colony stimulating factor (G‐CSF), IL‐1β, IL‐4, IL‐6, MCP‐1/CCL2, and keratinocyte chemoattractant (KC) levels were also significantly increased (Figure 2C).
Taken together, we identified a number of secreted proteins with increased intratumoural expression (eotaxin/CCL11, CCL17, CCL24, CXCL14, IL‐11, IL‐1β, INHBA/Activin A, BMP‐2, and GDF15) and elevated plasma levels (GDF15, Activin A, G‐CSF, IL‐1β, IL‐4, IL‐6, KC, and MCP‐1) in cachectic tumour models as potential effectors of cancer‐induced cachexia. Several of these proteins have already been implicated in the development of cachexia (IL‐6, Activin A, and GDF15).12, 20 Of the proteins examined, GDF15 and Activin A demonstrated a similar profile: their intratumoural expression and circulating levels were selectively upregulated in the cachectic MAP3K11‐driven system and also in the plasma of animals carrying the cachectic C26 tumours.
Identification of secreted plasma proteins associated with cachexia in cancer patients
The experiments described above prompted us to examine if any of the cytokines whose elevated circulating levels correlated with the cachexia phenotype in mouse tumour models are also elevated in the plasma of cachectic cancer patients. To this end, we profiled the levels of 29 circulating human proteins using the Luminex human cytokine panel and ELISA assays (see Materials and methods and Supplementary Table SI) in the plasma of 43 non‐cachectic and 142 cachectic cancer patients representing different tumour types. The cachectic patients were further classified according to the stage of cachexia, using the classification proposed by Fearon and colleagues14 as described by Vigano et al. 24, 25, 26: body mass (weight change, lean, and fat mass), food intake, changes in metabolism and inflammatory markers, and disease progression were among some of the variable analysed for the classification. Twenty eight patients were classified as pre‐cachectic, 49 as cachectic, and 65 as suffering from refractory cachexia.
As shown in Table 1, the analysis revealed that the plasma levels of GDF15, Activin A, IL‐6, IL‐8, and interferon gamma‐induced protein 10 (IP‐10) were significantly elevated in cachectic cancer patients as compared with non‐cachectic controls. The levels of GDF15 and Activin A were increased over two‐fold in the cachectic patients vs. controls (5.42 ng/mL vs. 2.67 ng/mL, p < 0.0001 for GDF15; 1.27 ng/mL vs. 0.62 ng/mL, p = 0.0002 for Activin A). Smaller but highly significant increases in IL‐6 (32.2 pg/mL vs. 21.9 pg/mL, p = 0.0002), IL‐8 (55.6 pg/mL vs. 41.7 pg/mL, p = 0.007), and IP‐10 (1.2 ng/mL vs. 1.0 ng/mL, p = 0.04) were detected in cachectic patients.
Table 1.
Circulating cytokine levels in cachectic cancer patients. Median levels of GDF15 and Activin A are expressed as ng/mL; the concentrations of all other cytokines are expressed as pg/mL. Abbreviations: Non‐Ca = non‐cachectic cancer patients; Ca [all] = all cachectic cancer patients; Pre‐Ca = pre‐cachectic cancer patients; Ca = cachectic cancer patients; Ref‐Ca = cancer‐refractory cachectic patients. Statistical significance between the groups was determined by the non‐parametric t‐test
| Cytokine | Mean levels | P value | |||||||
|---|---|---|---|---|---|---|---|---|---|
| No Ca (n = 43) | Pre‐Ca (n = 28) | Ca (n = 49) | Ref‐Ca (n = 65) | Ca [all] (n = 142) | No‐Ca vs. Pre‐Ca | No‐Ca vs. Ca | No‐Ca vs. Ref‐Ca | No‐Ca vs. Ca [all] | |
| GDF15 a | 2.67 | 5.44 | 5.02 | 5.70 | 5.42 | 0.007 | 0.025 | 0.000 | 0.000 |
| ACTIVIN Aa | 0.62 | 1.03 | 1.05 | 1.53 | 1.27 | 0.007 | 0.028 | 0.000 | 0.000 |
| IL‐6 | 21.89 | 27.29 | 30.39 | 35.61 | 32.23 | 0.024 | 0.117 | 0.000 | 0.000 |
| IL‐8 | 41.74 | 47.56 | 48.94 | 63.77 | 55.58 | 0.206 | 0.764 | 0.000 | 0.007 |
| IP‐10 | 985 | 1832 | 1040 | 1131 | 1232 | 0.070 | 0.152 | 0.067 | 0.041 |
| MIP‐1β | 51.56 | 54.76 | 61.03 | 58.95 | 58.88 | 0.168 | 0.145 | 0.074 | 0.055 |
| VEGF | 36.40 | 50.88 | 49.43 | 51.21 | 50.53 | 0.712 | 0.286 | 0.013 | 0.063 |
| IL‐15 | 3.15 | 4.04 | 10.12 | 5.86 | 6.99 | 0.312 | 0.062 | 0.145 | 0.072 |
| RANTES | 1284 | 1540 | 2265 | 1332 | 1694 | 0.725 | 0.026 | 0.198 | 0.089 |
| IL‐10 | 16.99 | 55.68 | 20.59 | 32.00 | 32.52 | 0.750 | 0.374 | 0.036 | 0.119 |
| PDGF‐BB | 685 | 879 | 1167 | 933 | 1004 | 0.342 | 0.112 | 0.042 | 0.141 |
| IL‐2 | 12.91 | 13.91 | 20.81 | 18.08 | 18.24 | 0.715 | 0.144 | 0.159 | 0.148 |
| IL‐12_P70 | 42.88 | 88.49 | 71.26 | 65.80 | 71.97 | 0.782 | 0.390 | 0.060 | 0.156 |
| GM‐CSF | 6.22 | 1.27 | 14.20 | 8.17 | 8.95 | 0.332 | 0.087 | 0.380 | 0.184 |
| IL‐1Rα | 282.9 | 300.7 | 369.4 | 419.2 | 379.6 | 0.890 | 0.809 | 0.077 | 0.286 |
| TNFα | 105.8 | 108.8 | 126.0 | 150.0 | 133.9 | 0.959 | 0.665 | 0.137 | 0.329 |
| IL‐17 | 46.57 | 44.78 | 62.90 | 51.21 | 54.10 | 0.829 | 0.195 | 0.353 | 0.329 |
| IL‐13 | 14.78 | 51.31 | 16.90 | 14.49 | 22.27 | 0.719 | 0.955 | 0.113 | 0.341 |
| IL‐4 | 8.82 | 8.73 | 9.20 | 10.84 | 9.87 | 0.910 | 0.871 | 0.115 | 0.388 |
| MCP‐1 | 34.43 | 43.35 | 40.80 | 39.82 | 40.82 | 0.735 | 0.523 | 0.658 | 0.554 |
| IL‐1β | 6.82 | 6.83 | 7.13 | 8.79 | 7.85 | 0.992 | 0.913 | 0.238 | 0.565 |
| IFNγ | 211.5 | 206.6 | 217.8 | 296.6 | 252.4 | 0.810 | 0.779 | 0.187 | 0.611 |
| EOTAXIN | 102.8 | 111.7 | 223.1 | 130.7 | 159.1 | 0.297 | 0.274 | 0.654 | 0.618 |
| MIP‐1α | 9.05 | 8.40 | 9.73 | 10.11 | 9.65 | 0.551 | 0.748 | 0.389 | 0.670 |
| IL‐7 | 21.34 | 33.21 | 22.31 | 23.92 | 25.12 | 0.739 | 0.629 | 0.203 | 0.707 |
| G‐CSF | 236.21 | 176.57 | 184.29 | 196.30 | 188.42 | 0.805 | 0.856 | 0.345 | 0.726 |
| IL‐9 | 40.44 | 159.65 | 300.74 | 76.41 | 169.71 | 0.898 | 0.696 | 0.910 | 0.796 |
| bFGF | 50.33 | 43.08 | 62.49 | 52.10 | 53.99 | 0.214 | 0.756 | 0.402 | 0.813 |
| IL‐5 | 8.29 | 18.76 | 8.87 | 8.90 | 10.75 | 0.579 | 1.000 | 0.474 | 0.822 |
Levels of markers expressed as ng/mL of plasma; otherwise pg/mL of plasma.
In patients classified as pre‐cachectic only Activin A, GDF15, and IL‐6 were significantly elevated while in cachectic cancer patients Activin A, GDF15, and RANTES/CCL5 levels were increased with significance. In the plasma of patients classified with refractory cachexia significant elevation of Activin A, GDF15, IL‐6, IL‐8, IP‐10, vascular endothelial growth factor A (VEGFA) and platelet‐derived growth factor BB (PDGF‐BB) levels were detected (Table 1).
Notably, GDF15 plasma levels were significantly increased in pre‐cachectic patients (5.44 ng/mL) and remained relatively flat in patients with a more severe form of cachexia (cachectic 5.03 ng/mL, refractory 5.7 ng/mL), while there was a ~50% increase in Activin A levels in refractory (1.53 ng/mL) vs. pre‐cachectic (1.03 ng/mL) and cachectic (1.05 ng/mL) patients.
Furthermore, high (above median) IL‐8 (hazard ratio [HR]:2.3, p = 1.55E‐6), GDF15 (HR:2.3, p = 4.93E‐6), IL‐6 (HR:2.0, p = 5.56E‐5), Activin A (HR:1.9, p = 1.45E‐4), and IP‐10 (HR:1.4, p = 3.4E‐2) levels correlated with increased probability of death in a multivariate analysis adjusted for patient age, tumour stage, and 6‐month body weight loss (Supplementary Table SII).
Ability of cachexia‐associated secreted proteins to induce cachexia in mice
Transcript and plasma/serum profiling experiments consistently correlated increased GDF15 and Activin A levels with cachexia in human cancer patients and mouse models. To test the ability of these proteins, along with IL‐6 and IL‐8, to induce cachexia in mice we have developed a modification of the DC platform called Double Directed Complementation (DDC), as none of these proteins can drive tumour growth when overexpressed in the DC system (data not shown). In the DDC system, the gene of interest (such as GDF15, Activin A, IL‐6, or IL‐8) is transcriptionally linked to the HER2(V659E) oncogene. The introduction of these fusion constructs into doxycycline‐inducible murine mammary tumour cells ensures the in vivo growth of the transduced tumour cells in the absence of doxycycline because of the expression of HER2(V659E). The complemented tumours also express the gene of interest because its expression is linked to that of HER2(V659E).
Using the DDC approach, we generated tumours programmed to express human and murine GDF15 (hGDF15 and mGDF15), Activin A (human and murine 100% identical), human and murine IL‐6 (hIL‐6 and mIL‐6), human IL‐8, and its murine orthologue KC. Tumours complemented with only HER2(V659E) and the human STC1 gene encoding the secreted stanniocalcin1 protein were used as negative controls. We measured the body weight of tumour‐bearing animals over time until the animals were euthanized either because of large tumour burden (1500–2000 mm3 with the Activin A, human and murine IL‐6, human IL‐8, murine KC, human STC1, and control HER2(V659E) tumour‐bearing mice) or excessive body weight loss observed with the human and murine GDF15‐expressing tumour‐bearing mice (>20%).
No weight loss was detected in the negative control HER2(V659E) and STC1‐expressing tumour‐bearing animals (Figure 2D). Similarly, we did not observe any weight loss in animals with tumours engineered to produce IL‐8 or KC, although average plasma IL‐8 levels reached 1 ng/mL, which is 15–18 times higher than the average plasma concentration seen in cachectic cancer patients (Figure 2D and Table 1). We observed moderate body weight loss (~10–15%) in mice carrying tumours secreting Activin A and the human and murine forms of IL‐6. Average Activin A plasma concentration reached 3–5 ng/mL in the animals, a level that is 3–5 times higher than the average seen in cachectic cancer patients (Table 1). hIL‐6 plasma levels reached an average concentration of 20 ng/mL, which is 500–600 times higher than that observed in cachectic human patients, while mIL‐6 levels reached an average of 7 ng/mL, which is significantly higher (150 to 200 times) than levels observed in cachectic human patients (Table 1).
The overexpression of either human or murine GDF15 resulted in ~25% weight loss of the animals. hGDF15 and mGDF15 serum levels reached 7.5 ng/mL and 6 ng/mL, respectively, which is almost identical to what is seen in cachectic cancer patients (Figure 2D and Table 1).
Inhibition of GFD15 reverses body weight loss induced by human tumours
We identified several human tumour xenograft models with the ability to induce severe body weight loss in mice. These included prostate (LNCaP), ovarian (TOV‐21G, EFO‐21), colon (LS1034), breast (MFM‐223), leukaemia (K562), and fibrosarcoma (HT‐1080) models. We profiled the levels of the same 29 human plasma proteins used for the profiling of human plasma samples (Table 1) in the serum of mice carrying these cachectic tumours as well as in non‐cachectic controls (PC3, RPMI7951, and Chago).
Of the proteins examined, hGDF15 was consistently elevated (>2 ng/mL) in mice carrying cachectic tumours and was very low or undetectable in the non‐cachectic controls (Figure 3A). The cachectic and non‐cachectic tumours also secreted varying amounts of Activin A and hIL‐6; levels of these proteins were particularly elevated in the TOV‐21G model.
Figure 3.
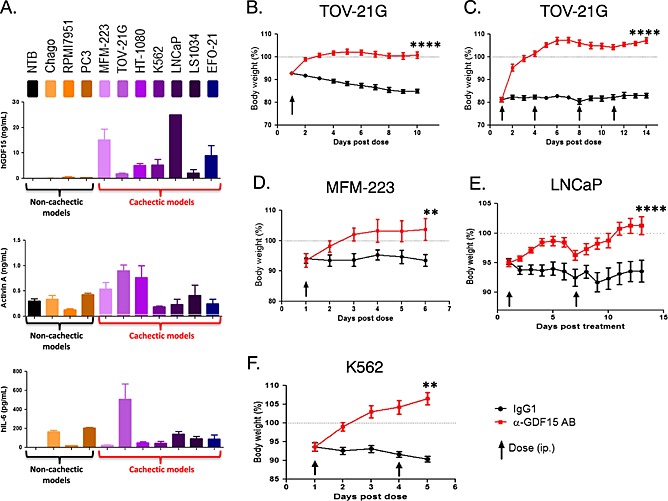
Inhibition of GFD15 reverses body weight loss induced by human tumors. (A). Serum protein profiling of cachectic human xenografts. Human GDF15, Activin A, and human IL‐6 levels of mice carrying cachectic (MFM‐223, TOV‐21G, HT‐1080, K562, LNCaP, LS1034, and EFO‐21) and non‐cachectic (Chago, RPMI7951, PC3) tumor models and NTB naïve mice. Human cytokines were measured as described in Materials and methods (n = 3–4/model), when tumor size reached 500–750 mm3. (B). TOV21‐G ovarian carcinoma model. TOV21‐G tumour‐bearing mice (n = 10/group) were treated with a single dose of the GDF15 inhibitory antibody (mAb01G06) or control IgG1 (2 mg/kg each) upon loss of ~7% of total body weight. (C). TOV21‐G tumour model with severe body weight loss. TOV21‐G tumour‐bearing mice (n = 10/group) were treated with a single dose of the GDF15 inhibitory antibody (mAb01G06) or control IgG1 (4 doses of 10 mg/kg each) upon loss of ~20% of total body weight. (D). MFM‐223 breast adenocarcinoma model. Animals carrying MFM‐223 tumours (n = 10/group) were treated with the GDF15 inhibitory antibody (AF957) or IgG1 control (10 mg/kg each) upon loss of ~7% of total body weight. (E). LNCaP prostate carcinoma model. Animals carrying LNCaP tumours (n = 10/group) were treated with the GDF15 inhibitory antibody (mAb14F11) or IgG1 control (20 mg/kg each) upon loss of ~10% of total body weight. (F). K562 leukaemia model. Animals carrying K562‐tumours (n = 10/group) were treated with the GDF15 inhibitory antibody (mAb01G06) or IgG1 control (10 mg/kg each) upon loss of ~7% of total body weight. In each experiment total weight of the animals was monitored daily. Paired t‐test was applied, **P < 0.01, ****P < 0.0001.
As shown on Figure 3B–F, 5A as well as in Supplementary Figure S1, all seven cachectic tumour models were able to induce significant body weight loss. Treatment with hGDF15‐neutralizing antibody reversed the weight loss phenotype in all seven cachectic models at doses as low as 2 mg/kg. hGDF15 inhibition reversed body weight loss in animals carrying TOV‐21G tumours even when they had already lost 20% of their weight prior to treatment (Figure 3C). The reversal of weight loss was complete despite the tumors producing high serum concentrations of hIL‐6 (0.6 ng/mL) and Activin A (1 ng/mL) (Figure 3A) at the start of the experiment (day 1). Likewise, we observed complete reversal of weight loss in the MFM‐223 (Figure 3D) and HT‐1080 (Figure 5A) models although both tumour lines secreted high serum concentrations of Activin A (0.6 ng/mL and 0.8 ng/mL, respectively) at day 1 of the experiment. hGDF15 inhibition also completely reversed body weight loss in the presence of high serum concentrations of hGDF15 (25 ng/mL) in the LNCaP (Figure 3E) tumour model as well as in the K562 tumour model (Figure 3F), LS‐1034, and EFO‐21 tumour models (Supplementary Figure S1).
Figure 5.
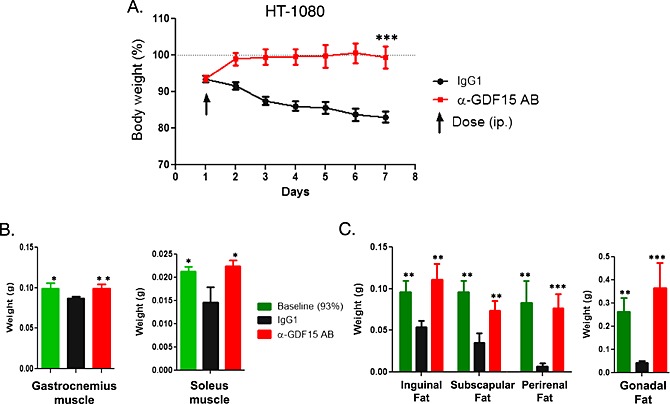
HT‐1080 tumour‐induced muscle and fat loss completely reversed by GDF15 inhibition. (A). HT‐1080 tumour‐bearing mice were treated with a single dose of GDF15 inhibitory antibody (mAb01G06) or IgG1 control (10mg/kg each) when ~7% of their body weight was lost. Body weight was monitored daily. Animals were sacrificed on day 7, and muscle and fat tissues were collected (n = 10/group). Total weight of the animals was monitored daily. Paired t‐test was applied, ***p < 0.001. (B). Weight of isolated gastrocnemius and soleus muscles at day 1 for baseline group (7% weight loss) and day 7 for IgG1 control and GDF15 inhibitory antibody‐treated groups (n = 10/group). Statistical difference vs. IgG1 in post hoc testing is signified as *p < 0.05. (C). Weight of isolated inguinal, perirenal, gonadal, and subscapular white fat pads at day 1 for baseline group (7% body weight loss) and day 7 for IgG1 control and GDF15 inhibitory antibody‐treated groups (n = 10/group). Statistical difference vs. IgG1 in post hoc testing is signified as **p < 0.01, ***p < 0.001.
In none of the models the inhibition of GDF15 function affected tumour growth kinetics or proliferation rate suggesting that GDF15 does not have direct role promoting tumour growth (data not shown).
Inhibition of GDF15 reverses body weight loss induced by MAP3K11‐driven murine breast tumours
The experiments described above demonstrated that GDF15 is an important effector of cancer‐induced body weight loss. In the cachectic, MAP3K11‐driven murine breast DC tumours we identified overexpression of GDF15 along with CCL11, CCL17, CCL24, CXCL14, IL‐11, IL‐1β, INHBA/Activin A, and BMP‐2 (Figure 2A). Plasma levels of GDF15, Activin A, and IL‐6 were also elevated, consistent with their elevated intratumoural expression (Figure 2B).
We utilized a rabbit monoclonal anti‐mGDF15‐specific inhibitory antibody (R‐23) to test if the inhibition of mGDF15 reverses the body weight loss induced by MAP3K11‐driven tumours. Administration of a single dose of the antibody (10 mg/kg) was sufficient to reverse body weight loss in this system, validating GDF15 as the key effector of the cachexia programme triggered by MAP3K11 pathway activation (Figure 4A).
Figure 4.
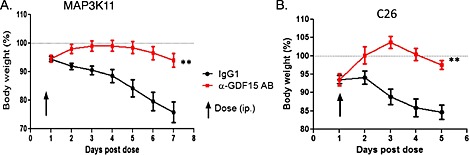
Inhibition of GFD15 reverses body weight loss induced by murine cachectic tumours. (A). MAP3K11‐driven DC tumour model. Mice bearing MAP3K11 DC tumours (n = 10/group) were administered a single dose of murine GDF15‐specific rabbit monoclonal antibody (R‐23) or IgG1 control (10 mg/kg each) at the time of ~7% body weight loss. Total weight of the animals was monitored daily. Paired t‐test was applied, **p < 0.01. (B). C26 colorectal carcinoma model. Mice bearing C26 (n = 10/group) were administered a single dose of murine GDF15‐specific rabbit monoclonal antibody (R‐23) or IgG1 control (10 mg/kg each) at the time of ~7% body weight loss. Total weight of the animals was monitored daily. Paired t‐test was applied, **p < 0.01.
Similarly, the inhibition of GDF15 reversed body weight loss induced by the murine colorectal carcinoma model C26 (Figure 4B), which produces high plasma levels of Activin A, IL‐6, G‐CSF, and KC.
The effect of cancer‐associated cachexia on body composition
As described above, we identified a number of human and murine tumour models capable of inducing body weight loss in mice. Although these tumours express and secrete a diverse set of cytokines, the inhibition of GDF15 alone was sufficient to reverse body weight loss. We further characterized the effect of cancer‐induced cachexia on host tissues and organs as well as the effect of GDF15 inhibition on body composition.
In the experiment shown in Figure 5, HT‐1080 tumour‐bearing mice were treated with a single dose of the hGDF15 inhibitory antibody (mAb01G06, 10 mg/kg) or control murine immunoglobulin G1 (mIgG1, 10 mg/kg) when the animals lost an average of 7% of their weight. Treatment with the GDF15 inhibitory antibody rapidly reversed body weight loss, while the mIgG1‐treated animals continued to lose weight. The experiment was terminated 7 days post‐dosing when the control mIgG1‐treated animals lost ~17% of their total body weight. A separate cohort of animals with ~7% of body weight loss on day 1 (baseline) were also euthanized; muscle and fat tissues were collected and weighted from both treatments and baseline groups (Figure 5A).
The weights of the gonadal, perirenal, and inguinal subscapular white fat tissues, as well as of the gastrocnemius and soleus muscles, were significantly reduced in the mIgG1 control group, compared with those in the baseline controls (Figures 5B and 5C). The weights of the fat and muscle tissues in the mAb01G06‐treated group were similar to those in the baseline group, suggesting that treatment with the GDF15 inhibitory antibody either arrests fat and muscle tissue loss or stimulates de novo fat and muscle tissue creation.
To distinguish between these possibilities we performed an identical experiment in which the body fat and muscle weights of the treated animals were compared with both baseline controls and age‐matched, NTB sham control animals. Animals treated with the GDF15 inhibitory antibody regained their normal weight to levels slightly above the weight of the sham control animals. The marginally higher weight of these animals over the NTB sham controls is attributed to tumour weight. Gonadal fat and gastrocnemius muscle mass recovered to the levels seen in the NTB sham control animals and above the baseline controls, demonstrating that GDF15 inhibition not only prevents additional fat and muscle tissue loss in cachectic animals but results in de novo muscle and adipose tissue synthesis (Figure 6). In HT‐1080 tumour‐bearing cachectic animals there was also a statistically significant reduction in the size of the liver, heart, and spleen, an effect reversed by the GDF15 inhibitory antibody. Moreover, the reversal of body composition changes also correlated with significant prolongation of survival (measured as time to euthanasia) compared with mice treated with IgG control that experienced significant body weight loss (>20%) and were sacrificed (Figure 6D).
Figure 6.
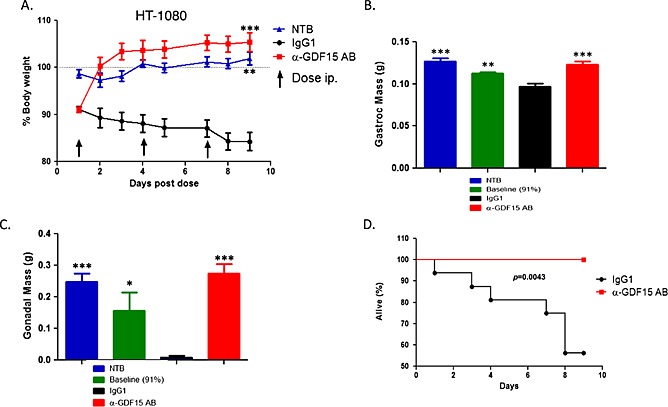
GDF15 inhibition completely reverses cachexia induced by HT‐1080 xenograft tumours. (A). HT‐1080 tumour‐bearing mice with ~9% of body weight loss were treated with the GDF15 inhibitory antibody (HuAb01G06‐127) or hIgG1 control (10 mg/kg each) twice weekly, and the body weight of the animals was measured daily together with non‐tumour‐bearing (NTB) normal controls (n = 10/group). Total weight of the animals was monitored daily. Statistical difference vs. IgG1 in post hoc testing is signified as **p < 0.01, ***p < 0.001. (B). The weight of isolated gastrocnemius muscles (g) was measured on day 1 for baseline (9% body weight loss) and on day 9 (end of study) for treated groups and NTB controls (n = 10/group). Statistical difference vs. IgG1 in post hoc testing is signified as **p < 0.01, ***p < 0.001. (C). The weight of isolated gonadal fat pads (g) was measured on day 1 for baseline (9% body weight loss) and on day 9 (end of study) for treated groups and NTB controls (n = 10/group). Statistical difference vs. IgG1 in post hoc testing is signified as *p < 0.05, ***p < 0.001. (D). Survival of HT‐1080 tumour‐bearing animals following treatment with GDF15 inhibitory antibody combination compared with IgG1 control (n = 10/group). Survival is defined as time to euthanasia (see Materials and methods). Kaplan–Meier plot for vehicle control (IgG1) and α‐GDF15 AB treated tumour‐bearing group.
Taken together, the HT‐1080 tumour‐induced wasting phenotype recapitulates all body composition changes associated with cancer cachexia. Inhibition of GDF15 is not only sufficient to restore normal body composition but also prolong survival. Similar observations were made in the K562 and TOV‐21G models.
Metabolic characterization of the cancer cachexia phenotype
In order to characterize the underlying metabolic changes driving cancer cachexia, we performed whole‐animal indirect calorimetry experiments in a comprehensive laboratory animal monitoring system (CLAMS) using the cachectic HT‐1080 tumour model and NTB sham control mice (Figure 7). By days 9–11 following tumour implantation, the tumour‐bearing animals had significantly reduced food intake, locomotor activity, energy expenditure (EE, measured as oxygen consumption) and resting energy expenditure (REE) during both diurnal and nocturnal periods compared with NTB mice. The combined effect of these metabolic changes was reduction of body weight in the tumour‐bearing animals.
Figure 7.
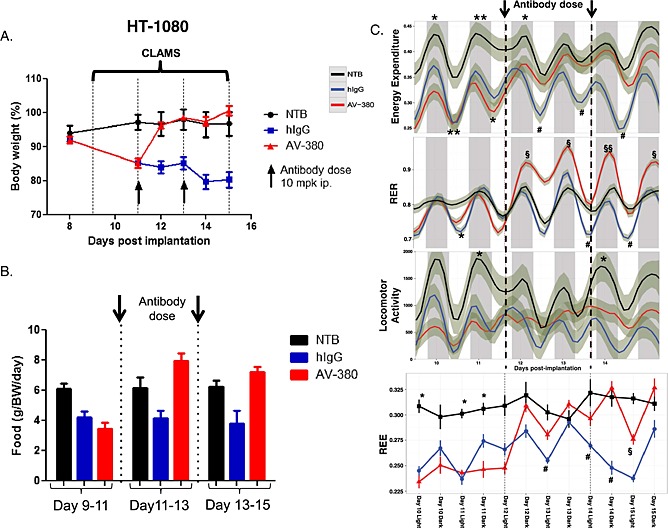
Metabolic characterization of the cancer cachexia phenotype in HT‐1080 model. (A). Animals were housed in CLAMS (at thermoneutrality, 30°C), treated with two doses of the GDF15 inhibitory antibody (AV‐380) or hIgG1 control on day 11 post tumour cell implantation. Body weight was followed up to day 18. NTB: non‐tumour bearing control mice of similar age and weight as the HT‐1080‐injected mice (n = 4/group). The experiment was repeated twice. Total weight of the animals was monitored daily. Statistical difference vs. IgG1 in post hoc testing is signified as *P < 0.05, ***P < 0.001. (B). Food intake (g) shown in two‐day intervals and adjusted for body weight (n = 4 per group) per day. Statistical difference in post hoc testing is signified as *P < 0.05, HT‐1080 groups vs. NTB group; # P < 0.05 IgG treated HT‐1080 group vs. NTB group; §§P < 0.01 AV‐380 treated group vs. IgG treated group. (C). Energy expenditure (EE), respiratory exchange ratio (RER), locomotor activity, and resting energy expenditure (REE) before and after antibody administration (n = 4/group). Shadows represent standard deviation (SD). Statistical difference in post hoc testing is signified as *P < 0.05, **P < 0.01 HT‐1080 groups vs. NTB group; # P < 0.05 IgG treated HT‐1080 group vs. NTB group; §P < 0.05, §§P < 0.01 AV‐380 treated group vs. IgG treated group.
The tumour‐bearing mice received two doses of GDF15 inhibitory antibody or human IgG1 (hIgG1) (10 mg/kg each) on day 11 (when they had lost ~15% of their total body weight) and on day 13. Within two days (days 11–13) following administration of the GDF15 inhibitory antibody, the treated mice increased food intake and regained body weight similar to that of the NTB sham control animals. These animals also displayed increased locomotor activity, EE, and REE, while the IgG1 control‐treated mice continued to experience declines in body weight, locomotor activity, and EE. The respiratory exchange ratio (RER) of the GDF15 inhibitor‐treated mice also dramatically increased (values >1) in this period, indicating increased anabolic activity, with carbohydrates the preferred source of energy. During this period the GDF15 inhibitor‐treated cachectic mice recovered their body weight and body composition.
After the body weight recovery period (days 13–15 after tumour inoculation), the metabolic profile (body weight, EE, REE, RER,) of the GDF15 inhibitor‐treated animals became similar to that of the normal NTB controls. The partial recovery of locomotor activity might be because of the large tumours interfering with the mobility of the animals. The increased EE in these animals might be due increased food intake and anabolic activities (as indicated by shift in RER values). GDF15 inhibition resulted in similar metabolic changes in cachectic animals carrying murine renal cancer cell (RENCA) tumours.
This experiment demonstrated that GDF15 inhibition immediately shifts the metabolic profile of the cachectic animals from catabolism to anabolism, leading to the recovery of normal body weight and restoration of normal body composition (i.e. recovery of muscle, fat, and organ sizes).
Concomitant inhibition of cachexia and tumour growth leads to increased survival
Next we investigated the effect of the combination of tivozanib, a potent angiogenesis inhibitor and anti‐tumour agent, with a GDF15 inhibitory antibody (mAb14F11) on survival in the LNCaP tumour model.27 LNCaP tumours produce high levels of GDF15 (>25 ng/mL) and induce severe body weight loss that can be reversed by GDF15 inhibition (Figure 3E). Tivozanib treatment results in the stabilization of tumour growth. In this experiment, tumours were grown to ~500 mm3 before treatment with either tivozanib alone (5 mg/kg) or the combination of tivozanib and mAb14F11 (20 mg/kg). As compared with controls, tivozanib treatment either alone or in combination prevented further tumour growth and led to the stabilization of tumour size around 500 mm3 while in control treated animals the mean tumour volume exceeded 1500 mm3 (Figure 8A). Despite tivozanib's tumour growth inhibitory activity, animals treated with tivozanib alone continued to lose weight, while animals treated with the combination of tivozanib and the GDF15 inhibitory antibody quickly reversed body weight loss and gained weight (Figure 8B). The inhibition of tumour growth by tivozanib and the reversal of body weight loss by GDF15 inhibition resulted in improved survival over control or animals treated with tivozanib monotherapy (HR:0.13, p = 0.023 and HR:0.14, p = 0.045, respectively) (Figure 8C).
Figure 8.
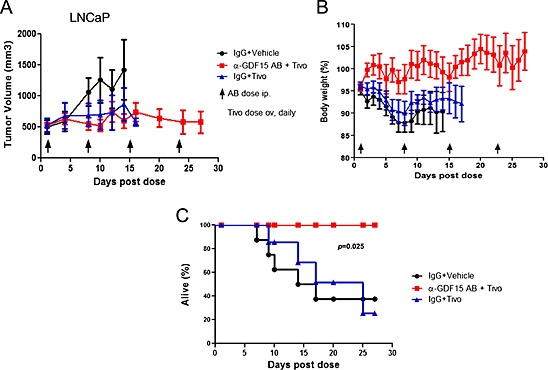
Combination of an anti‐cachectic and anti‐tumour agent extends survival. (A). LNCaP tumour growth inhibition by tivozanib (5 mg/kg, daily) alone or in combination with the GDF15 inhibitory antibody (mAb14F11) as compared with IgG1 controls (20mg/kg each, n = 8/group). Data was plotted until 50% of animals were removed from the study because of either death or euthanasia. (B). Body weight changes in tivozanib or tivozanib + GDF15 inhibitory antibody‐treated animals as compared with controls. Body weight was measured daily. Data was plotted until 50% of animals were removed from the study because of either death or euthanasia. (C). Survival of tumour‐bearing animals following treatment with tivozanib + GDF15 inhibitory antibody combination or tivozanib alone as compared with controls. Survival or time to euthanasia was defined as either the animals having better than moribund health or tumour size <2000 mm3 or body weight loss <20%, according to Institutional Animal Care and Use Committee guidance. Kaplan–Meier plot for control (IgG1 + Vehicle), Tivo treated group (IgG1 + Tivo), and combo group (α‐GDF15 + Tivo).
This experiment demonstrated that simultaneous inhibition or stabilization of tumour growth and the suppression of the effects of tumour‐induced cachexia can significantly extend life span.
Discussion
The exact molecular mechanisms responsible for the induction of cancer cachexia are currently unknown. Here we demonstrated that MAP3K11/MLK3 pathway activation in genetically engineered tumour models elicited a genetic programme resulting in the wasting of the host. This effect was specific to MAP3K11 signalling. While both MAP3K11 and MAP3K7/TAK1 overexpression could drive tumour growth, only MAP3K11 pathway activation was able to induce cachexia. This was likely to be the consequence of signalling differences between MAP3K11 and MAP3K7 that are currently not well understood28, 29, 30 but could be identified by careful proteomic mapping of signalling molecules in the closely related MAP3K7‐ and MAP3K11‐driven murine mammary DC tumours. Nevertheless, induction of MAP3K11 signalling represents one mechanism in tumours that can trigger cachexia although the upstream oncogenic events that lead to the activation of the pathway are yet to be identified.
Engineered MAP3K11 driven or naturally occurring cachectic murine and human tumours expressed and secreted a diverse set of cytokines representing potential mediators of cachexia. Of these, IL‐6, Activin A and GDF15 have been implicated in the development of cachexia.10, 20 Elevated circulating levels of GDF15 were most consistently associated with the cachexia phenotype. In line with this observation, elevated circulating levels of GDF15 (along with Activin A, IL‐6, IL‐8, IP‐10, VEGFA, RANTES/CCL5, and PDGF‐BB) were also correlated with cachexia in human patients, where it was also predictive of overall survival. Circulating levels of GDF15 have been shown to be associated with cancer morbidity31, 32 and predict poor survival in endometrial33 and gastric cancers34 but not in esophago‐gastric cancers.35 Circulating levels of GDF15 also robustly correlated with survival in patients with congestive heart failure, in whom it is an independent predictor of mortality,31, 32, 36, 37, 38, 39 as well as in end‐stage renal disease.39, 40
The strong association of GDF15 with the cachexia phenotype in tumour models and in human cancer patients prompted the validation of GDF15 as a potential driver of cachexia in gain of function and loss of function experiments in a variety of tumour models. These experiments distinguished GDF15 as a potent cachexia inducing factor whose inhibition triggered metabolic changes that resulted in the complete reversal of body weight loss and restoration of normal body composition independent of the complex secreted cytokine profile of the tumour.
Recently, it has been shown in preclinical models that inhibition of IL‐6, GDF8, Activin receptor IIB (ActRIIB), and tumour‐derived parathyroid hormone‐related protein (PTHrP) can reverse cachexia.15, 18, 41, 42, 43 These observations suggest that different mechanisms might drive cachexia in different tumour contexts. In our hands, in cachectic tumour models with elevated levels of circulating GDF15 (TOV‐21G, K562, or HT‐1080) the inhibition of tumour‐derived human IL‐6 and PTHrP had no effect on preventing body weight loss. Blockade of ActRIIB prevented further body weight loss but did not induce complete body weight recovery; it reversed muscle loss but had no effect on the loss of adipose tissue (Supplementary Figure S2, manuscript in preparation). In tumours with elevated circulating GDF15 levels, GDF15 inhibition appeared to be the most effective strategy to treat cancer cachexia. Thus, circulating GDF15 could serve as a biomarker to identify patients most likely benefit from this approach.
We further investigated the potential clinical application of anti‐cachexia agents and demonstrated significant increase in overall survival (measured as time to euthanasia) by the combination of a GDF15 inhibitor with an anti‐tumour agent (tivozanib) providing a paradigm for the integration of cachexia treatments into anti‐cancer regimens to enhance therapeutic benefit. In summary, these experiments identified GDF15 as a key driver of cancer cachexia, providing a potential point of intervention for the prevention or treatment of the syndrome.
Material and methods
Cell cultures
HT‐1080, LNCaP, TOV‐21G, Chago, RMPI795, PC3, K562, LS1034, and EFO‐21 cancer cell lines were acquired from the American Type Culture Collection (ATCC). MFM‐223 and endometrial cancer cell lines were obtained from the German Collection of Microorganisms and Cell Cultures. All cancer cell lines were cultured according to the suppliers' recommendations. C26 was a generous gift from Dr. Dario Coletti, University of Rome, Italy.
GDF15 inhibitory antibodies
mAb01G06 and mAb14F11 are mouse monoclonal anti‐human GDF15 inhibitory antibodies. AV‐380 and Hu01G06‐127 are humanized forms of the murine monoclonal antibodies. The generation and characterization of these antibodies will be described elsewhere. Briefly, all these antibodies bind to human GDF15 with high affinity (KD > 10−11) and neutralize GDF15 activity in in vivo cachectic models.
R‐23 is a rabbit anti‐mouse GDF15 monoclonal antibody. It binds (KD > 10‐11) and inhibits the activities of murine GDF15 in vivo. All these antibodies were developed by AVEO Oncology Inc. (Cambridge, MA, USA).
AF957 (R&D Systems, Minneapolis, MN, USA) is a goat anti‐human GDF15 polyclonal antibody that neutralizes human GDF15 activity.
ActRIIB, PTHrP, and IL‐6 neutralizing antibodies
Neutralizing antibodies to ActRIIB, PTHrP, and IL‐6 were produced at AVEO Oncology using sequence information from US Patents No. 20100272734, 7906117, and 8029793.
DC and DDC tumour generation, gene expression profiling
Directed complemented (DC) tumours were engineered as previously described.44 Briefly, tumour cells derived from doxycycline‐inducible HER2(V659)‐driven murine breast tumours were infected in vitro for 48 h with pBDG‐1 replication‐defective retroviral vectors, which directed the expression of either MAP3K7 or MAP3K11 and HER2(V659E), EGFR(L858R) positive controls; or with the empty vector alone as a negative control. The infected cells were then injected subcutaneously into the flank of 9‐week‐old ICR‐SCID female mice in the absence of doxycycline, and tumour latency and penetrance were monitored three times a week. tumours of ~500 mm3 were excised, and the expression of the transduced genes (MAP3K7, MAP3K11, HER2(V659E), and EGFR(L858R)) was verified by real‐time polymerase chain reaction. Tumors expressing the target genes and lacking the expression of the original doxycycline‐controlled HER2(V659E) oncogenes were further propagated in mice.
For the generation of double‐directed complemented (DDC) tumours a modified version of the pBDG‐1 vector was used in which the gene of interest was transcriptionally linked to the HER2(V659E) oncogene via an internal ribosome entry site.
Gene expression profiling
Total RNA isolated from frozen tumour tissues was converted into fluorescence‐labelled cRNA that was hybridized to DNA oligonucleotide microarrays via the GeneChip® Mouse Genome 430 2.0 Array (Affymetrix, Santa Clara, CA, USA). Universal mouse reference RNA from Stratagene was used as the reference and hybridized. Microarray data has been deposited in NCBI Gene Expression Omnibus database (accession no. GSE68162). After Robust Multi‐array Average normalization, p value < 0.05 and the fold‐change threshold > 1.5 were identified to be statistically significant alterations.
Plasma cytokine profiling in tumour‐bearing mice
Bio‐Plex Pro™ Murine Cytokine 23‐Plex Assay from Bio‐Rad® (Hercules, CA, USA) was used to measure the circulating levels of murine cytokines including eotaxin; fibroblast growth factor basic; G‐CSF; GDF15; granulocyte‐macrophage colony‐stimulating factor (GM‐CSF); interferon gamma (IFN‐γ); interleukins IL‐10, IL‐12p40, IL‐12p70, IL‐13, IL‐17, IL‐15, IL‐1α, IL‐1β, interleukin‐1 receptor a (IL‐1Ra), IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐7, IL‐8, and IL‐9; KC; IFN‐γ‐induced protein 10 (IP‐10 or CXCL10); monocyte chemotactic protein 1 (MCP‐1 or CCL2); macrophage inflammatory protein‐1 alpha (MIP‐1α or CCL3); macrophage inflammatory protein‐1 beta (MIP‐1β or CCL4); PDGF‐BB; regulated on activation, normal t cell expressed and secreted (RANTES or CCL5); TNFα; and VEGFA. For the profiling of circulating levels of human cytokines Bio‐Plex Pro™ Human Cytokine 27‐Plex Assay from Bio‐Rad® (Luminex®, magnetic beads, Bio‐Rad, Hercules, CA, USA) was used. Human and murine Activin A and GDF15 were measured using murine or human‐specific ELISA (R&D Systems, Minneapolis, MN, USA).
Plasma cytokine profiling in cancer patients
This retrospective study received ethical approval from the McGill University Institutional Review Board and is in accordance with the Declaration of Helsinki. One hundred and eighty five patients provided written informed consent and were all assessed at the hospital bedside, as described in Vigano AL, et al. 2014.26 Patient recruitment and data collection took place between March 2006 and November 2007. Classification of the cachectic patients was described by Vigano AL, et al. 2012.25 Plasma cytokines were profiled as described above. Patient characteristics and individual cytokine values are summarized in (Supplementary Table SI)
Analysis of cachexia phenotype in vivo
In the TOV‐21G, K562 in vivo studies, 8‐week‐old female C.B‐17‐SCID mice (Taconic Biosciences, Hudson, NY, USA) were inoculated subcutaneously (s.c.) into the flank with 5 × 106 cells in 1:1 RPMI 1640 (Invitrogen, Thermo Fisher Scientific, Grand Island, NY, USA)/Matrigel (BD Biosciences, San Jose, CA, USA). For the MFM‐223 in vivo studies, 5‐week‐old female NCr Nude mice (Taconic) were implanted under isofluerance anaesthesia, with a Troca needle, s.c. on the left flank with 0.72 mg of 90‐day‐release 17β‐estradiol pellets (Innovative Research, Novi, MI, USA) and inoculated s.c. into the right flank with 1 × 107 MFM‐223 cells in 1:1 EMEM (ATCC)/Matrigel. In the HT‐1080, EFO‐21, C26, and MAP3K11 DC in vivo studies, 8‐week‐old female ICR SCID mice (Taconic) were inoculated s.c. into the flank with 5 × 106 cells in 1:1 RPMI 1640 (Invitrogen)/Matrigel (BD Biosciences). For the LS1034 and LNCaP in vivo studies, 8‐week‐old female NCr Nude mice (Taconic) were implanted s.c. into the right flank with 5 × 106 cells in 1:1 EMEM (ATCC)/Matrigel.
Body weight measurements were made daily at the same time of the day. When body weight dropped by the desired percentage, mice were randomized based on percentage body weight dropped and tumour volume into groups of 10 animals each and received GDF15 inhibitory antibody (2–20 mg/kg) or a control IgG1 at (2–20 mg/kg) by intraperitoneal injection.
At the end of study, mice were sacrificed and tissues (gastrocnemius and soleus muscle, inguinal, subscapular, perirenal, and gonadal fat pads) were surgically excised and weighed by necropsy. Survival was defined as time to euthanasia, i.e. animals with tumours >2000 mm3 in size or with >20% of body weight loss and/or with moribund health were sacrificed as required by Institutional Animal Care and Use Committee protocol.
Metabolic characterization of cancer‐associated cachexia
Whole‐body energy metabolism was evaluated using a CLAMS (Columbus Instruments, Columbus, OH, USA) according to the manufacturer's instructions. Carbon dioxide and oxygen data were collected every 24 min for each mouse and were normalized to total body weight. Metabolic measurements (oxygen consumption, CO2 production) were measured every 24 min for each mouse. Food intake and locomotor activity were obtained continuously using a CLAMS (OxyMax, Columbus Instruments) open‐circuit indirect calorimetry system. EE and RER were calculated according to the manufacturer's instructions. REE was calculated from metabolic data per subject per light–dark period by considering only EE measurements, which occurred during the lowest 25% of activity for that period.45, 46
Ethics Committee approval
Animal work was carried out in accordance with AVEO's Institutional Animal Care and Use Committee guidelines and with the policies and certification of the Association for Assessment and Accreditation of Laboratory Animal Care. All mice were treated in accordance with the Office of Laboratory Animal Welfare Public Health Service Policy on Human Care and Use of Laboratory Animals and the Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals.
Mice were received from Taconic (Cambridge City, IN) and acclimatized to the animal care facility (ACF) for at least 1 week prior to study start. The room which housed the study animals was maintained on a 12:12 h/h light cycle with lights on at 7:00AM. Room temperature was maintained at 70°F, and humidity was set at 35%. Cage changes and health checks were performed on all animals by animal care staff as per Standard Operating Procedures.
Mice were housed, 5 per cage, on a ventilated Innorack® IVC v. 3.5 mouse caging system (San Diego, CA). Cage bottoms were filled with paper twist enriched corn cob bedding. For additional enrichment, Innodome™ huts were added to each cage. For the duration of the study, mice were maintained on Irradiated Purina Lab Diet 5V75 and Innovive Aquavive® water bottles. Food and water were available ad libitum for the duration of the study.
Statistics
Parameters are expressed as mean ± standard error of the mean. Tests were carried out two‐tailed with α = 0.05 using GraphPad Prism (GraphPad Software, CA, USA). One‐way analysis of variance (ANOVA) followed by Dunnett test adjustments for multiple comparisons and multiple ANOVA followed by Bonferroni post hoc test were used where applicable in group comparisons. Survival analysis was performed using log‐rank Mantel–Cox test.
Author contributions
LL, JL, QL, IC, JT, and JG were in charge with conception, design, analysis, and interpretation of data. JT, BK, LH, SZ, RW, LS, and EM performed the experiments and acquisition of data. AV made available the patient plasma collection, and ZW, SW, and WW provided reagents. RN and BF performed bioinformatics analyses, and LL and JG wrote manuscript.
Conflict of interest
LL, JT, QL, RN, BF, BK, EM, ZS, RW, LH, JL, IC, ZW, WW, SW, and JG are/were employees and/or stockholders of AVEO Oncology.
Supporting information
Figure S1: Reversal of body weight loss induced by cachectic tumour models by GDF15 inhibition. LS1034 colon carcinoma and EFO‐21 ovarian carcinoma models. Mice were dosed at the indicated time with a GDF15 inhibitory antibody or IgG control antibody (10 mg/kg each). Total body weight was monitored daily (n = 10/group). Total weight of the animals was monitored daily. Paired t‐test was applied **p < 0.01, ****p < 0.0001.
Figure S2: Comparing the biological activities of GDF15 (AV‐380), Activin receptor IIB, hIL‐6, and hPTHrP inhibitory antibodies in the cachectic TOV‐21G tumour model. TOV‐21G tumour‐bearing mice were treated with inhibitory antibodies to GDF15 (AV‐380), ActRIIB (α‐ActRIIB), human IL‐6 (α‐IL‐6) and human PTHrP (α‐PTHrP), or control human IgG1 (3 doses of 10 mg/kg each) upon the loss of ~7% of total body weight. The total body weight of the animals was monitored daily (n = 10 animals/group). Total weight of the animals was monitored daily. Statistical difference vs. IgG1 in post hoc testing is signified as **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical difference vs. NTB in post hoc testing is signified as ### p < 0.001.
Table SI: Patient demographics, clinical characteristics, and cytokine levels. Baseline patient characteristics: age, gender, diagnosis, weight, % weight loss in the last 6 months, cachexia classification. Cytokine levels in plasma (mean and SD).
Table SII: Association between IL‐8, GDF15, IL‐6, Activin A, and IP‐10 plasma levels and overall survival in cancer patients. Multivariate analysis using the Cox Proportional Hazard Model adjusted by age, stage (locally advanced, metastatic, recurrent), and 6‐month weight loss. Median was used as a cutoff for each cytokine. Top 5 (ranked by p value) out of 29 cytokines analysed (n = 188 cancer patients).
Supporting info item
Supporting info item
Acknowledgements
We would like to thank Rosemary Mazanet, Heidi Okamura, and Bob Young for comments on the manuscript; Peter Steinberg for editorial assistance; and Sandy Abbot, Jinwei Jiang, Ting Cheng, Laura Poling, Sara Haserlat, Jamie Gifford, Ailin Bai, Lingxin Kong, Angela Bressel, Sireesha Yalavarthi, Yanyu Zhang, Nesreen Ismail, and Alisa Bell for reagents, advice, and helpful discussions. We are especially grateful to Miguel Norden, Lily Yen, Sheryl Perry, and Sumita Roy for providing a productive laboratory and ACF environment.
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle (von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle 2010;1:7–8.).
Lerner, L. , Tao, J. , Liu, Q. , Nicoletti, R. , Feng, B. , Krieger, B. , Mazsa, E. , Siddiquee, Z. , Wang, R. , Huang, L. , Shen, L. , Lin, J. , Vigano, A. , Chiu, M. I. , Weng, Z. , Winston, W. , Weiler, S. , and Gyuris, J. (2016) MAP3K11/GDF15 axis is a critical driver of cancer cachexia. Journal of Cachexia, Sarcopenia and Muscle, 7: 467–482. doi: 10.1002/jcsm.12077.
Contributor Information
Lorena Lerner, Email: llerner@aveooncology.com.
Jeno Gyuris, Email: jenogyuris@gmail.com.
References
- 1. von Haehling S, Anker SD. Cachexia as a major underestimated and unmet medical need: facts and numbers. J Cachexia Sarcopenia Muscle 2010;1:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muliawati Y, Haroen H, Rotty LW. Cancer anorexia–cachexia syndrome. Acta Med Indones 2012;44:154–62. [PubMed] [Google Scholar]
- 3. Donohoe CL, Ryan AM, Reynolds JV. Cancer cachexia: mechanisms and clinical implications. Gastroenterology research and practice 2011;2011:601434. doi:10.1155/2011/601434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Argiles JM, Fontes‐Oliveira CC, Toledo M, Lopez‐Soriano FJ, Busquets S. Cachexia: a problem of energetic inefficiency. J Cachexia Sarcopenia Muscle 2014. doi: 10.1007/s13539-014-0154-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Vos‐Geelen J, Fearon KC, Schols AM. The energy balance in cancer cachexia revisited. Curr Opin Clin Nutr Metab Care 2014;17:509–14. [DOI] [PubMed] [Google Scholar]
- 6. Seyfried TN, Shelton LM. Cancer as a metabolic disease. Nutrition & metabolism 2010;7:7. doi:10.1186/1743-7075-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arias JI, Aller MA, Arias J. The use of inflammation by tumor cells. Cancer 2005;104:223–8. [DOI] [PubMed] [Google Scholar]
- 8. Tan BH, Fladvad T, Braun TP, Vigano A, Strasser F, Deans DA, et al P‐selectin genotype is associated with the development of cancer cachexia. EMBO Mol Med 2012;4:462–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Laine A, Iyengar P, Pandita TK. The role of inflammatory pathways in cancer‐associated cachexia and radiation resistance. Molecular cancer research: MCR 2013;11:967–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 2012;16:153–66. [DOI] [PubMed] [Google Scholar]
- 11. Onesti JK, Guttridge DC. Inflammation based regulation of cancer cachexia. BioMed research international 2014;2014:168407. doi:10.1155/2014/168407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agustsson T, Ryden M, Hoffstedt J, van Harmelen V, Dicker A, Laurencikiene J, et al Mechanism of increased lipolysis in cancer cachexia. Cancer Res 2007;67:5531–7. [DOI] [PubMed] [Google Scholar]
- 13. Fearon KC, Voss AC, Hustead DS. Definition of cancer cachexia: effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am J Clin Nutr 2006;83:1345–50. [DOI] [PubMed] [Google Scholar]
- 14. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–95. [DOI] [PubMed] [Google Scholar]
- 15. Strassmann G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest 1992;89:1681–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oliff A, Defeo‐Jones D, Boyer M, Martinez D, Kiefer D, Vuocolo G, et al Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 1987;50:555–63. [DOI] [PubMed] [Google Scholar]
- 17. Fearon K, Arends J, Baracos V. Understanding the mechanisms and treatment options in cancer cachexia. Nat Rev Clin Oncol 2013;10:90–9. [DOI] [PubMed] [Google Scholar]
- 18. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–43. [DOI] [PubMed] [Google Scholar]
- 19. Padhi D, Higano CS, Shore ND, Sieber P, Rasmussen E, Smith MR. Pharmacological inhibition of myostatin and changes in lean body mass and lower extremity muscle size in patients receiving androgen deprivation therapy for prostate cancer. J Clin Endocrinol Metab 2014;99:E1967–75. [DOI] [PubMed] [Google Scholar]
- 20. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, et al Tumor‐induced anorexia and weight loss are mediated by the TGF‐beta superfamily cytokine MIC‐1. Nat Med 2007;13:1333–40. [DOI] [PubMed] [Google Scholar]
- 21. Tsai VW, Husaini Y, Manandhar R, Lee‐Ng KK, Zhang HP, Harriott K, et al Anorexia/cachexia of chronic diseases: a role for the TGF‐beta family cytokine MIC‐1/GDF15. J Cachexia Sarcopenia Muscle 2012;3:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vivanco I. Targeting molecular addictions in cancer. Br J Cancer 2014;111:2033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rodriguez‐Berriguete G, Fraile B, Martinez‐Onsurbe P, Olmedilla G, Paniagua R, Royuela M. MAP kinases and prostate cancer. Journal of signal transduction 2012;2012:169170. doi:10.1155/2012/169170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scheede‐Bergdahl C, Watt HL, Trutschnigg B, Kilgour RD, Haggarty A, Lucar E, et al Is IL‐6 the best pro‐inflammatory biomarker of clinical outcomes of cancer cachexia? Clin Nutr 2012;31:85–8. [DOI] [PubMed] [Google Scholar]
- 25. Vigano A, Del Fabbro E, Bruera E, Borod M. The cachexia clinic: from staging to managing nutritional and functional problems in advanced cancer patients. Crit Rev Oncog 2012;17:293–303. [DOI] [PubMed] [Google Scholar]
- 26. Vigano AL, di Tomasso J, Kilgour RD, Trutschnigg B, Lucar E, Morais JA, et al The abridged patient‐generated subjective global assessment is a useful tool for early detection and characterization of cancer cachexia. Journal of the Academy of Nutrition and Dietetics 2014;114:1088–98. [DOI] [PubMed] [Google Scholar]
- 27. Eskens FA, de Jonge MJ, Bhargava P, Isoe T, Cotreau MM, Esteves B, et al Biologic and clinical activity of tivozanib (AV‐951, KRN‐951), a selective inhibitor of VEGF receptor‐1, ‐2, and ‐3 tyrosine kinases, in a 4‐week‐on, 2‐week‐off schedule in patients with advanced solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research 2011;17:7156–63. [DOI] [PubMed] [Google Scholar]
- 28. Mishra P, Senthivinayagam S, Rangasamy V, Sondarva G, Rana B. Mixed lineage kinase‐3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol 2010;24:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muniyappa H, Das KC. Activation of c‐Jun N‐terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: a MLK‐3‐MKK7‐dependent mechanism. Cell Signal 2008;20:675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki S, Singhirunnusorn P, Mori A, Yamaoka S, Kitajima I, Saiki I, et al Constitutive activation of TAK1 by HTLV‐1 tax‐dependent overexpression of TAB2 induces activation of JNK‐ATF2 but not IKK‐NF‐kappaB. J Biol Chem 2007;282:25177–81. [DOI] [PubMed] [Google Scholar]
- 31. Wiklund FE, Bennet AM, Magnusson PK, Eriksson UK, Lindmark F, Wu L, et al Macrophage inhibitory cytokine‐1 (MIC‐1/GDF15): a new marker of all‐cause mortality. Aging Cell 2010;9:1057–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wallentin L, Zethelius B, Berglund L, Eggers KM, Lind L, Lindahl B, et al GDF‐15 for prognostication of cardiovascular and cancer morbidity and mortality in men. PLoS One 2013;8:e78797. doi:10.1371/journal.pone.0078797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Staff AC, Trovik J, Eriksson AG, Wik E, Wollert KC, Kempf T, et al Elevated plasma growth differentiation factor‐15 correlates with lymph node metastases and poor survival in endometrial cancer. Clinical cancer research: an official journal of the American Association for Cancer Research 2011;17:4825–33. [DOI] [PubMed] [Google Scholar]
- 34. Blanco‐Calvo M, Tarrio N, Reboredo M, Haz‐Conde M, Garcia J, Quindos M, et al Circulating levels of GDF15, MMP7 and miR‐200c as a poor prognostic signature in gastric cancer. Future Oncol 2014;10:1187–202. [DOI] [PubMed] [Google Scholar]
- 35. Skipworth RJ, Deans DA, Tan BH, Sangster K, Paterson‐Brown S, Brown DA, et al Plasma MIC‐1 correlates with systemic inflammation but is not an independent determinant of nutritional status or survival in oesophago‐gastric cancer. Br J Cancer 2010;102:665–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wollert KC, Kempf T, Peter T, Olofsson S, James S, Johnston N, et al Prognostic value of growth‐differentiation factor‐15 in patients with non‐ST‐elevation acute coronary syndrome. Circulation 2007;115:962–71. [DOI] [PubMed] [Google Scholar]
- 37. Damman P, Kempf T, Windhausen F, van Straalen JP, Guba‐Quint A, Fischer J, et al Growth‐differentiation factor 15 for long‐term prognostication in patients with non‐ST‐elevation acute coronary syndrome: an Invasive versus Conservative Treatment in Unstable coronary Syndromes (ICTUS) substudy. Int J Cardiol 2014;172:356–63. [DOI] [PubMed] [Google Scholar]
- 38. Rohatgi A, Patel P, Das SR, Ayers CR, Khera A, Martinez‐Rumayor A, et al Association of growth differentiation factor‐15 with coronary atherosclerosis and mortality in a young, multiethnic population: observations from the Dallas Heart Study. Clin Chem 2012;58:172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ho JE, Hwang SJ, Wollert KC, Larson MG, Cheng S, Kempf T, et al Biomarkers of cardiovascular stress and incident chronic kidney disease. Clin Chem 2013;59:1613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Breit SN, Carrero JJ, Tsai VW, Yagoutifam N, Luo W, Kuffner T, et al Macrophage inhibitory cytokine‐1 (MIC‐1/GDF15) and mortality in end‐stage renal disease. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association—European Renal Association 2012;27:70–5. [DOI] [PubMed] [Google Scholar]
- 41. Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, et al JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL‐6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 2012;303:E410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–54. [DOI] [PubMed] [Google Scholar]
- 43. Busquets S, Toledo M, Orpi M, Massa D, Porta M, Capdevila E, et al Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al Compensatory insulin receptor (IR) activation on inhibition of insulin‐like growth factor‐1 receptor (IGF‐1R): rationale for cotargeting IGF‐1R and IR in cancer. Mol Cancer Ther 2010;9:2652–64. [DOI] [PubMed] [Google Scholar]
- 45. Nestoridi E, Kvas S, Kucharczyk J, Stylopoulos N. Resting energy expenditure and energetic cost of feeding are augmented after Roux‐en‐Y gastric bypass in obese mice. Endocrinology 2012;153:2234–44. [DOI] [PubMed] [Google Scholar]
- 46. Stylopoulos N, Hoppin AG, Kaplan LM. Roux‐en‐Y gastric bypass enhances energy expenditure and extends lifespan in diet‐induced obese rats. Obesity 2009;17:1839–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Reversal of body weight loss induced by cachectic tumour models by GDF15 inhibition. LS1034 colon carcinoma and EFO‐21 ovarian carcinoma models. Mice were dosed at the indicated time with a GDF15 inhibitory antibody or IgG control antibody (10 mg/kg each). Total body weight was monitored daily (n = 10/group). Total weight of the animals was monitored daily. Paired t‐test was applied **p < 0.01, ****p < 0.0001.
Figure S2: Comparing the biological activities of GDF15 (AV‐380), Activin receptor IIB, hIL‐6, and hPTHrP inhibitory antibodies in the cachectic TOV‐21G tumour model. TOV‐21G tumour‐bearing mice were treated with inhibitory antibodies to GDF15 (AV‐380), ActRIIB (α‐ActRIIB), human IL‐6 (α‐IL‐6) and human PTHrP (α‐PTHrP), or control human IgG1 (3 doses of 10 mg/kg each) upon the loss of ~7% of total body weight. The total body weight of the animals was monitored daily (n = 10 animals/group). Total weight of the animals was monitored daily. Statistical difference vs. IgG1 in post hoc testing is signified as **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical difference vs. NTB in post hoc testing is signified as ### p < 0.001.
Table SI: Patient demographics, clinical characteristics, and cytokine levels. Baseline patient characteristics: age, gender, diagnosis, weight, % weight loss in the last 6 months, cachexia classification. Cytokine levels in plasma (mean and SD).
Table SII: Association between IL‐8, GDF15, IL‐6, Activin A, and IP‐10 plasma levels and overall survival in cancer patients. Multivariate analysis using the Cox Proportional Hazard Model adjusted by age, stage (locally advanced, metastatic, recurrent), and 6‐month weight loss. Median was used as a cutoff for each cytokine. Top 5 (ranked by p value) out of 29 cytokines analysed (n = 188 cancer patients).
Supporting info item
Supporting info item