

Abstract
Background
The Muscle‐specific RING‐finger (MuRF) protein family of E3 ubiquitin ligases is important for maintenance of muscular structure and function. MuRF proteins mediate adaptation of striated muscles to stress. MuRF2 and MuRF3 bind to microtubules and are implicated in sarcomere formation with noticeable functional redundancy. However, if this redundancy is important for muscle function in vivo is unknown. Our objective was to investigate cooperative function of MuRF2 and MuRF3 in the skeletal muscle and the heart in vivo.
Methods
MuRF2 and MuRF3 double knockout mice (DKO) were generated and phenotypically characterized. Skeletal muscle and the heart were investigated by morphological measurements, histological analyses, electron microscopy, immunoblotting, and real‐time PCR. Isolated muscles were subjected to in vitro force measurements. Cardiac function was determined by echocardiography and working heart preparations. Function of cardiomyocytes was measured in vitro. Cell culture experiments and mass‐spectrometry were used for mechanistic analyses.
Results
DKO mice showed a protein aggregate myopathy in skeletal muscle. Maximal force development was reduced in DKO soleus and extensor digitorum longus. Additionally, a fibre type shift towards slow/type I fibres occurred in DKO soleus and extensor digitorum longus. MuRF2 and MuRF3‐deficient hearts showed decreased systolic and diastolic function. Further analyses revealed an increased expression of the myosin heavy chain isoform beta/slow and disturbed calcium handling as potential causes for the phenotype in DKO hearts.
Conclusions
The redundant function of MuRF2 and MuRF3 is important for maintenance of skeletal muscle and cardiac structure and function in vivo.
Keywords: Protein homeostasis, Protein surplus myopathy, Heart failure, MuRF2, MuRF3, MAPKAPK
Introduction
Muscle‐specific RING‐finger (MuRF) proteins maintain cardiac and skeletal muscle structure and function.1, 2, 3, 4, 5, 6 MuRF proteins belong to the tripartite motif‐containing (TRIM) family of E3 ubiquitin ligases. All three MuRF family members MuRF1, MuRF2, and MuRF3 are predominantly expressed in the heart and skeletal muscle.7 MuRF1 is involved in skeletal muscle atrophy2 and cardiac hypertrophy.6, 8 MuRF1 knockout mice are resistant to skeletal muscle atrophy2, 9 and when subjected to chronic pressure overload develop exaggerated cardiac hypertrophy.6 MuRF2 is implicated in sarcomere formation because it transiently associates with microtubules, myosin, and titin during sarcomere assembly.10 MuRF2 also mediates signal transduction in cardiomyocytes as it translocates to the nucleus during mechanical inactivity where it decreases the abundance of the serum response transcription factor (SRF).3 MuRF3 binds to and stabilizes microtubules establishing a network resistant to depolymerization.7 MuRF3 knockout mice develop left ventricular dilation, heart failure, and cardiac rupture when subjected to myocardial infarction.1 However, no phenotype has been described for single germ line deletions of MuRF1,2 MuRF2,6 or MuRF3 1 under unstressed conditions, implying partially redundant functions for these highly homologous proteins under physiological circumstances. Earlier, we described a protein‐surplus myosin storage myopathy in skeletal muscle and the heart of MuRF1 and MuRF3 double knockout (DKO) mice that supports this idea.4 Further support for redundancy comes from the phenotype of MuRF1 and MuRF2 DKO mice, because most of these mice die at birth.5 Surviving MuRF1 and MuRF2 DKO mice develop cardiac hypertrophy5 and a decrease in fast‐fibres in skeletal muscle.11 Moreover, in vitro studies also support the notion of redundancy, where MuRF2 compensated for the loss of MuRF3 and vice versa, without involving MuRF1.12 Here, we tested the hypothesis that MuRF2 and MuRF3 have partially redundant functions in striated muscles in vivo. We generated MuRF2 and MuRF3 DKO mice and analysed the skeletal and cardiac muscle for altered structure and function.
To elucidate a potential signalling pathway possibly affected by MuRF2 and MuRF3‐deficiency, we relied on a proteomics‐based approach that identified MAPK‐activated protein kinase 3 (MAPKAPK3) to be enriched in DKO muscle. MAPKAPK3 and its family member MAPKAPK2 are downstream targets of p38 mitogen‐activated protein kinase (MAPK) and mediators of p38 MAPK signalling.13 The p38 MAPK pathway is involved in various striated muscle signalling events.14, 15 In adult skeletal muscle, p38 MAPK and MAPKAPK2 are activated by muscle contraction,16 and MAPKAPK2/3 were reported to modify the skeletal muscle fibre type composition.17, 18 In heart, p38 MAPK and MAPKAPK2/3 are highly abundant, and chronic p38 MAPK activation is involved in cardiac pathologies.19 In addition, MAPKAPK2/3‐deficiency increased contractile activity in cardiomyocytes.18 Therefore, we investigated possible effects of MuRF2 and MuRF3‐deficiency in soleus muscle and left ventricle on MAPKAPK2/3 protein levels and a potential interaction of MuRF2 and MuRF3 with MAPKAPK2/3.
Materials and methods
MuRF2 6 and MuRF3 1 knockout mice have been described previously. Animals were housed under standard conditions at 22 °C with a 12‐hour light/12‐hour dark cycle and maintained on commercial mouse chow and water ad libitum. MuRF2 and MuRF3 double heterozygous mice were interbred, and resulting control and DKO littermates were used for subsequent experiments. The following genotypes were used as controls: MuRF2 +/+/MuRF3 +/+, MuRF2 +/−/MuRF3 +/+, MuRF2 +/+/MuRF3 +/−, and MuRF2 +/−/MuRF3 +/−. The Landesamt für Gesundheit und Soziales approved the studies (LaGeSo, Berlin, Germany; permit number: G 0129/12). The studies followed the ‘Principles of Laboratory Animal Care’ (NIH publication No. 86‐23, revised 1985), as well as the current version of German Law on the Protection of Animals. Muscle dissection, gene expression analyses, and protein purification were performed as recently published;1, 4, 20 for further details, please refer to the online Supporting Information.
Western blot analysis was performed on protein samples from skeletal muscles, hearts, and cells, as previously described.1 The following primary antibodies were used: anti‐MuRF2 (polyclonal, rabbit, own production), anti‐MuRF3 (polyclonal, rabbit, own production), anti‐actin (monoclonal, mouse), anti‐β/slow MyHC (clone NOQ7, monoclonal, mouse), anti‐pan MyHCII (clone My32, monoclonal, mouse; all Sigma–Aldrich, Germany), anti‐MAPKAPK2, anti‐p38 MAPK (all: polyclonal, rabbit, Cell SignalingTechnology Inc., Danvers, USA) antibody. Generation of anti‐MuRF2 and anti‐MuRF3 antibody is described in the online Supporting Information. Loading was controlled by using anti‐glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) antibody (clone 6C5, monoclonal, mouse, Millipore GmbH, Germany). Horseradish peroxidase (HRP)‐linked IgG horse anti‐mouse, goat anti‐rabbit (both Cell Signaling Technology Inc., USA), or rabbit anti‐goat (Abcam, UK) were used as secondary antibody. For further details on Western blot analysis, glutathione S‐transferase (GST)‐pulldowns, and co‐immunoprecipitation please refer to the online data supplement. Analysis of MAPKAPK2, MAPKAPK3, and p38 followed our recently published work.14
Histological and immunohistochemical analyses were performed as described previously.4 Haematoxylin and eosin (H & E), metachromatic ATPase dye, or modified Gomori's trichrome staining were performed as previously described.4, 21 The following primary antibodies were used: anti‐β/slow MyHC (clone NOQ7), anti‐pan MyHCII (clone My32), and anti‐Laminin (all Sigma–Aldrich, Germany). The following secondary antibodies were used: goat anti‐mouse and goat anti‐rabbit IgG (Alexa Fluor 488 nm or 555 nm, Invitrogen). Filamentous actin was stained with phalloidin‐TRITC (Sigma–Aldrich, Germany). Images were acquired with a Leica CTR 6500 HS microscope, and Leica digital camera DFC 425 for histological analyses and Leica digital camera DFC 360 FX for fluorescence pictures. Analyses of images were performed with ImageJ software 1.42c (http://rsb.info.nih.gov/ij/). Myocyte cross‐sectional area (MCSA) was measured from extensor digitorum longus from at least 100 myofibres per genotype and histological section of 9‐ to 22‐week‐old mice. Centralized nuclei were quantified in 60–270 myofibres per genotype and histological section from both soleus and extensor digitorum longus of 21‐ to 22‐week‐old mice. Fibre type composition was determined in 330–970 myofibres per genotype and histological section from both soleus and extensor digitorum longus of 21‐ to 22‐week‐old mice. Electron microscopy (16‐ to 22‐week‐old mice) and measurements of muscle force (14‐ to 27‐week‐old mice) were performed as previously described.20 For further details, please refer to the online Supporting Information. For mass‐spectrometry of muscle proteins and bioinformatics data analysis, please refer to the online Supporting Information.
Isolation of adult mouse ventricular cardiomyocytes from 15‐week‐old animals and measurements of single cell contraction and calcium transients were performed as described previously.22 Body composition was measured in 5‐month‐old male (n = 4) and female (n = 3) DKO mice and their control (n = 8 and n = 4, respectively) littermates using the LF90 II time domain NMR analyzer (6.5 mHz, Bruker Optics, USA).23 The mice were placed into the restraint tube, which was adjusted to minimize movements of the animal without impairing respiration. The tube together with the animal was placed into the LF90 II instrument, and fat mass, fat‐free mass, and fluid were measured in triplicate.
Echocardiography was performed on 8‐week‐old male DKO (n = 5) mice and their control littermates (n = 7) as previously described.1, 24 The mice were anesthetized with 2% isoflurane and kept warm on a heated platform. Temperature and electrocardiogram were continuously monitored. Cardiac function and morphology were assessed with a VisualSonics Vevo 2100 High‐Resolution Imaging System (VisualSonics, Toronto, Canada) with the use of a high‐resolution (38 MHz) transducer. Left ventricular (LV) end‐diastolic dimension (EDD) and end‐systolic dimension (ESD), thickness of the left ventricular posterior wall (LVPW) in diastole and systole, left ventricular ejection fraction (EF), and cardiac output were measured. Langendorff heart experiments were performed as recently described.25 For further details, please refer to the online Supporting Information.
Statistics
Values are presented as mean ± SEM. Gene expression was normalized to stably expressed Hprt mRNA and calculated as relative change. Differences in morphologic, physiologic, and biochemical parameters between groups were analysed by Mann–Whitney U test or 2‐sided Student's t‐test. Statistics were calculated with Microsoft Excel 2002 and Sigma Plot software 11.0. A P‐value of less than 0.05 was considered as statistically significant.
Results
General characteristics of muscle ring‐finger 2 and muscle ring‐finger 3 double knock‐out mice
MuRF2 6 and MuRF3 1 knockout mice were used to generate MuRF2 and MuRF3 double heterozygous mice, which were interbred to obtain control and MuRF2 and MuRF3 DKO mice. DKO and control littermates were used for subsequent experiments. The absence of MuRF2 and MuRF3 protein in soleus, extensor digitorum longus, tibialis anterior, and gastrocnemius/plantaris of DKO mice was confirmed by Western blot analyses (Figure 1A, Figure S1a). MuRF1 mRNA was not increased in the skeletal muscle of DKO mice (Figure S2). DKO mice were born in the expected Mendelian ratios, and no differences in behaviour and motion were observed (data not shown). However, male DKO mice had a decreased body weight (Figure 1B), increased muscle mass, and decreased fat content (Figure 1C). Weights of individual skeletal muscles were not different in male and female mice compared with respective controls (Figure 1D, Figure S1b).
Figure 1.
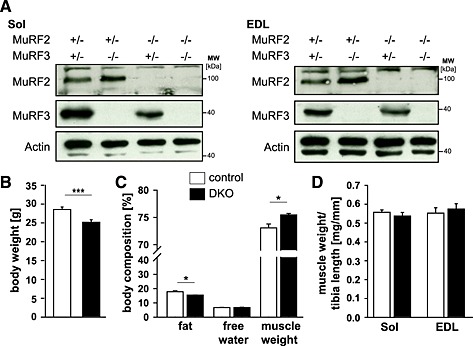
An increase in muscle mass was found in muscle‐specific RING‐finger (MuRF)2 and MuRF3 double knockout (DKO) mice. (A) Immunoblotting of proteins from soleus (Sol) and extensor digitorum longus (EDL) from control and DKO mice using anti‐MuRF2 and anti‐MuRF3 antibody, as indicated, confirmed absence of MuRF2 and/or MuRF3 proteins in the respective single and double knockout mice. Actin served as loading control. (B) Quantification of body weight of male 7‐ to 22‐week‐old control (n = 30) and DKO (n = 26) mice. ***P < 0.001. (C) Body composition of 5‐month‐old male control (n = 8) and DKO (n = 4) mice was analysed by nuclear magnetic resonance spectroscopy. The amount of body fat, free water, and total muscle weight is shown and expressed in percent of body weight. *P < 0.05. (D) Mass of Sol and EDL normalized to tibia length of control (n = 30) and DKO (n = 26) mice. Data are shown as mean ± SEM.
MuRF2 and MuRF3 DKO mice display a protein aggregate myopathy in skeletal muscle
The soleus, extensor digitorum longus, gastrocnemius/plantaris, and tibialis anterior revealed pathological changes in DKO mice. Haematoxylin and eosin staining of histological sections showed intracellular protein aggregates surrounding an inner laying myofibre and adjacent to the membrane of the myofibres in all DKO skeletal muscles (Figure 2A, Figure S3a). Centralized nuclei were found in DKO myofibres of all muscles from male and female mice indicative for regeneration of the skeletal muscle (Figure 2A–C, Figure S3a). Quantitation of MCSA using H & E staining of histological cross‐sections showed a higher amount of smaller myofibres in DKO extensor digitorum longus compared with littermate controls as an unspecific sign for myopathy (Figure 2D). Modified Gomori's trichrome staining showed no increase in fibrotic tissue in skeletal muscle of DKO mice (Figure 2B, Figure S3b). In both H & E and modified Gomori's staining, a demarcation between the inner laying myofibre and the accumulating protein was observed (Figure 2, Figure S3). We excluded the possibility that this structure was a membrane. The accumulations were contained within individual myofibres rather than the interstitium (Figure S4). By electron microscopy, the overall structure of the inner‐laying myofibre core was normal in control and DKO soleus and extensor digitorum longus muscle (Figure 3A α–δ). However, the myofibre core showed Z‐line streaming and myofibrils degenerating into the amorphous mass in DKO soleus and extensor digitorum longus (Figure 3A ε, ζ). This core structure was surrounded by mitochondria and an amorphous mass localized between the myofibre core and the sarcolemma (Figure 3A δ, ε). These observations reveal a protein storage myopathy affecting myofibres in DKO skeletal muscle with a preference for soleus and extensor digitorum longus.
Figure 2.
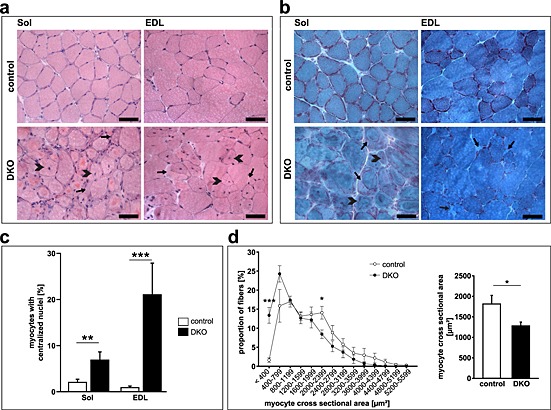
Absence of muscle‐specific RING‐finger (MuRF)2 and MuRF3 resulted in protein surplus myopathy. (A) Haematoxyline and eosin stain of cross‐sections from soleus (Sol) and extensor digitorum longus (EDL) from 21‐week‐old control and DKO mice. Subsarcolemmal accumulations of eosinophilic material occurred around a central core of myofibres in DKO muscles (arrow). Heterogeneity of fibre size and centrally localized nuclei (arrowhead) were found in DKO muscles. Scale bar, 50 µm. (B) Gomori's trichrome stain of cross‐sections from Sol and EDL from 18‐ to 21‐week‐old control and DKO mice. Protein aggregates (arrow) and myofibres with centralized nuclei are depicted (arrowhead). (C) Quantification of myofibres containing centralized nuclei in Sol and EDL of 20‐ to 22‐week‐old control (n = 13) and DKO (n = 8) mice. **P < 0.01, ***P < 0.001. (D) Quantification of myocyte cross‐sectional area (MCSA) from control (n = 7) and DKO (n = 7) mice on cross‐sections from EDL. An increased number of smaller fibres were found in DKO. *P < 0.05, ***P < 0.001.
Figure 3.
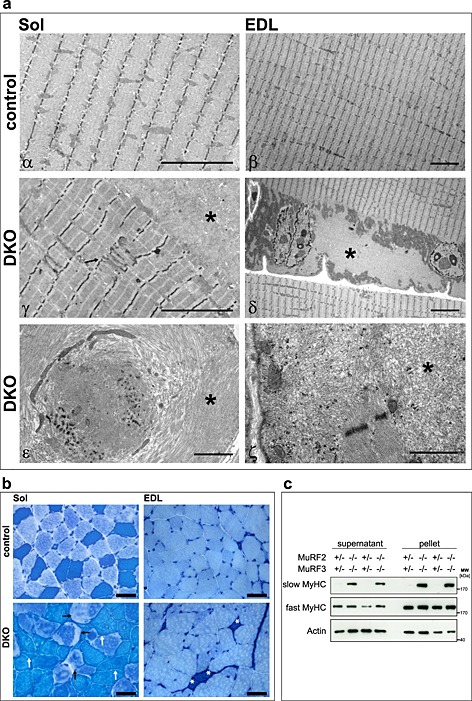
Double knockout (DKO) mice displayed a protein aggregate myopathy and a shift towards slow myofibres in extensor digitorum longus (EDL) muscle. (A) Electron microscopy of soleus (Sol) and EDL sections displayed accumulation of amorphous material in DKO myofibres. Asterisks indicate accumulating aggregates, arrow points to abnormal Z‐line. Scale bar, 5 µm (α–δ), 2 µm (ε, ζ). (B) ATPase stain of Sol and EDL showed accumulations inside of slow/type I fibres (dark blue, white arrows) and fast/type II fibres (bright blue, black arrows) of DKO mice. Slow/type I fibres occurred in EDL of DKO mice (asterisk). Scale bar, 50 µm. (C) Immunoblotting of proteins from the soluble (supernatant) and particulate (pellet) fractions of EDL revealed accumulation of slow/type I myosin heavy chain protein in DKO muscle. Actin was used as a control.
A fibre type shift towards slow/type I fibres occurred in MuRF2 and MuRF3 deficient muscle
We next investigated whether or not protein accumulations were restricted to a specific fibre type. Protein accumulations were found in all types of myofibres and in all DKO but not in control muscles (Figure 3B). Notably, staining of protein accumulations by the ATPase dye resembled the staining of the specific fibre in which the accumulation occurred (Figure 3B). These data show a non‐fibre type specific accumulation of myosin heavy chain (MyHC) protein. Most importantly, we observed slow/type I fibres in extensor digitorum longus of DKO mice where these fibres are usually not present (Figure 3B). Western blot analysis was used to test if myosin heavy chain 7 protein (MyHC‐7), contained in slow/type I fibres, was increased in DKO extensor digitorum longus. Indeed, slow/type I MyHC protein was increased in protein lysates of DKO extensor digitorum longus (Figure 3C). Slow/type I myosin was found in both the supernatant and the pellet fraction indicating increased amounts of soluble and insoluble myosin in DKO muscle (Figure 3C). To confirm suspected accumulation of slow/type I and fast/type II myosin in their respective fibres, immunostaining of histological cross‐sections from soleus and extensor digitorum longus using anti‐slow and anti‐fast myosin antibody was performed. We confirmed that slow and fast myosin accumulated in their respective fibres in soleus and extensor digitorum longus of DKO mice (Figure 4A). Quantification of fibre types showed a decrease in fast/type II and an increase of hybrid fibres (containing both slow and fast myosin) in soleus, and an increase in slow/type I fibres, as well as a decrease in fast/type II fibres in extensor digitorum longus of DKO mice (Figure 4B).
Figure 4.
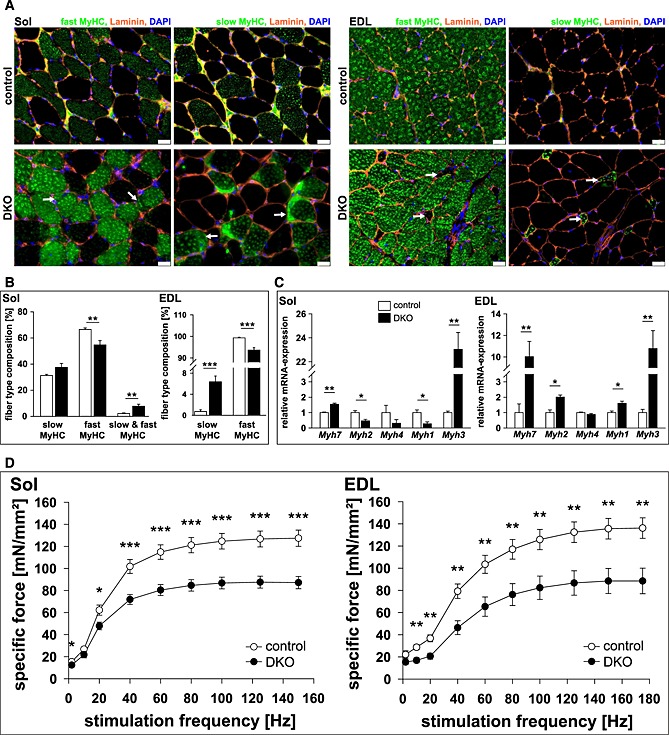
Loss of muscle‐specific RING‐finger (MuRF)2 and MuRF3 leads to an increase in slow/type I fibres in skeletal muscle. (A) Immunohistochemistry of cross‐sections from soleus (Sol, left panel) and extensor digitorum longus (EDL, right panel) using anti‐laminin, anti‐fast/type II myosin heavy chain, or anti‐slow/type I myosin heavy chain antibody. Nuclei were stained with 4',6‐Diamidino‐2‐phenylindol (DAPI). Protein aggregations are indicated (arrows). Slow/type I fibres occurred in EDL of DKO mice (arrow). Scale bar, 25 µm. (B) Slow/type I and fast/type II MyHC containing fibres were quantified in Sol and EDL of control (n = 8–10) and DKO (n = 7–8) mice. Data are shown as mean ± SEM. **P < 0.01, ***P < 0.001. (C) Real‐time RT–PCR analysis of myosin heavy chain (Myh) 1, 2, 3, 4 and 7 gene expression in Sol and EDL from control (n = 8–9) and DKO (n = 4) mice. Hypoxanthine guanine phosphoribosyl transferase (Hprt) expression was used as reference. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01. (D) Maximal force development of Sol and EDL from male 14‐ to 23‐week‐old control (n = 9 each) and DKO (n = 11–12) mice is shown. Specific force [mN/mm2] per stimulation frequency [Hz] is depicted. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Increased expression in Myh7, encoding for slow/type I MyHC, and Myh3, encoding for embryonic MyHC, was found in soleus, extensor digitorum longus, gastrocnemius/plantaris, and tibialis anterior of DKO mice (Figure 4C, Figure S5). In contrast, Myh2 and Myh1 expression (encoding type IIa and IIx MyHC, respectively) decreased in soleus and increased in extensor digitorum longus of DKO mice (Figure 4C), whereas Myh4 expression (encoding type IIb MyHC) remained unchanged in all muscles investigated (Figure 4C, Figure S5). Thus, MuRF2 and MuRF3 are involved in myosin homeostasis in slow/type I and fast/type II fibres.
Contractile function of DKO muscle was reduced
We reasoned that protein accumulations in all myofibres and a shift in fibre type composition towards slow/type I fibres would decrease force development in muscle of DKO mice. Maximum force development was measured on isolated soleus and extensor digitorum longus of male and female DKO mice and compared to littermate controls. As expected, maximum force development of soleus and extensor digitorum longus of male (Figure 4D) and female (Figure S6) DKO mice was reduced compared to controls. These data showed that MuRF2 and MuRF3 are important to regulate the function of skeletal muscle.
Cardiac structure and function were disturbed in DKO mice
We next investigated cardiac structure and function of DKO mice. First, absence of MuRF2 and/or MuRF3 in the heart in single knockout and DKO mice was confirmed by Western blot analyses using anti‐MuRF2 and anti‐MuRF3 antibody (Figure 5A). To analyse if a compensatory up‐regulation of MuRF1 expression occurred in hearts of DKO mice, real‐time RT–PCR was performed. MuRF1 mRNA expression was not increased in DKO animals indicating that no compensatory up‐regulation of MuRF1 occurred in DKO hearts (Figure S7a). Relative heart and lung weights were unchanged in DKO mice compared with littermate controls (Figure 5B). Using H & E and modified Gomori's trichrome staining of histological sections, neither protein accumulations nor interstitial fibrosis were found in hearts of DKO mice (Figure 5C). When using transmission electron microscopy, no ultrastructural changes were observed in DKO hearts (data not shown).
Figure 5.
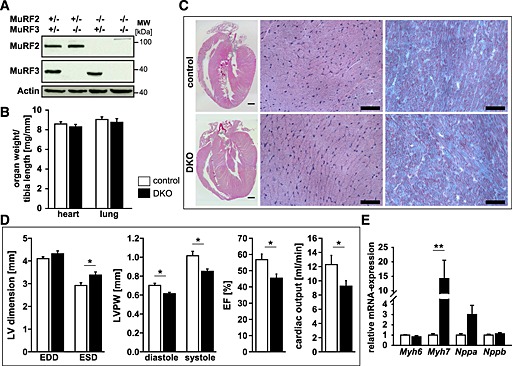
Combined deletion of muscle‐specific RING‐finger (MuRF)2 and MuRF3 leads to decreased cardiac function. (A) Immunoblotting of proteins from the hearts of control and double knockout (DKO) mice using anti‐MuRF2 and anti‐MuRF3 antibody, as indicated, confirmed absence of MuRF2 and/or MuRF3 proteins in the respective single and DKO mice. Actin served as loading control. (B) Quantification of heart and lung weight of 7‐ to 22‐week‐old control (n = 30) and DKO (n = 26) mice. Organ weights were normalized to tibia length. Data are shown as mean ± SEM. (C) Haematoxyline and eosin stain of sections from whole hearts (scale bare, 300 µm) and cross‐sections (middle panel, scale bare, 50 µm), and Gomori's trichrome stain of cross‐sections (right panel, scale bare, 50 µm) of hearts from control and DKO mice. (D) Echocardiography was performed to measure left ventricular (LV) end‐diastolic (EDD) and end‐systolic dimension (ESD), thickness of the left ventricular posterior wall (LVPW) in diastole and systole, left ventricular ejection fraction (EF), and cardiac output in 8‐week‐old control (n = 7) and DKO (n = 5) mice. Data are presented as mean ± SEM. *P < 0.05. (E) Real‐time RT–PCR analysis of myosin heavy chain Myh6, Myh7, Nppa, and Nppb gene expression in the hearts of control (n = 7) and DKO (n = 5) mice. Hprt expression was used as reference. Data are presented as mean ± SEM. **P < 0.01.
Echocardiography revealed an increased left ventricular end‐systolic dimension (LVESD) and a decreased thickness of the left ventricular posterior wall (LVPW) at both systole and diastole in DKO animals. These changes were accompanied by a reduction in systolic function of the left ventricle with a decreased left ventricular ejection fraction and decreased cardiac output of DKO hearts (Figure 5D). With these findings, we conclude that MuRF2 and MuRF3 are important for maintenance of cardiac function. Next, we tested if reduced cardiac function was accompanied by an increased expression of cardiac stress markers. We also tested if genes involved in cardiac remodelling were increased in DKO hearts. We found an increased Myh7 expression in DKO hearts, whereas Myh6, Nppa, Nppb, Ctgf, Col1a1, and Col3a1 remained unchanged (Figure 5E, Figure S7b).
At baseline, systolic left ventricular pressure (LVPsys, Figure 6A) and left ventricular developing pressure (LVPdevp, Figure 6B) were reduced in DKO compared with control hearts. However, following stimulation with the beta‐receptor agonist isoproterenol, LVPsys and LVPdevp increased in both experimental groups (Figure 6A and 6B). We also calculated the derivatives of LVP dLVP/dtmax (Figure 6C) and dLVP/dtmin (Figure 6D) as indices of left ventricular contraction and relaxation velocity, respectively. dLVP/dtmin was decreased whereas dLVP/dtmax remained unchanged in DKO hearts (Figure 6C and 6D). Both indices increased in control and DKO hearts following isoproterenol treatment. However, left ventricular contraction (Figure 6C) and relaxation velocity (Figure 6D) in the response to isoproterenol was reduced in DKO hearts. In summary, DKO hearts showed reduced systolic and diastolic function at baseline and following stress.
Figure 6.
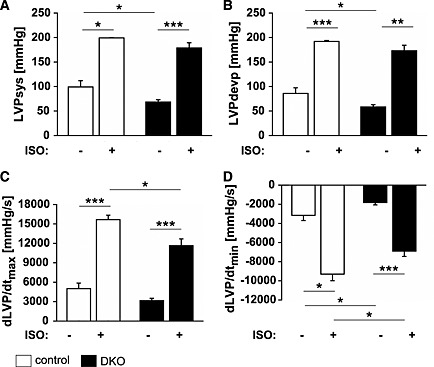
Double knockout (DKO) mice showed a decreased systolic and diastolic cardiac function. (A) Left ventricular systolic pressure (LVPsys) and left ventricular developing pressure (LVPdevp) were measured via an intraventricular balloon. At baseline, LVPsys and LVPdevp were reduced in DKO (n = 6) compared with control (n = 4) animals. Following stimulation with 50 nM isoproterenol (ISO), LVPsys and LVPdevp increased in both experimental groups. (B) Maximal (dLVP/dtmax) and minimal (dLVP/dtmin) derivatives of LVP, indices of contraction and relaxation velocity, respectively, were decreased in DKO (n = 6) compared with control (n = 4) mice. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Systolic and diastolic function was reduced in double knockout cardiomyocytes
We reasoned that the decrease in cardiac function in DKO mice occurred at the level of cardiomyocytes. To test this hypothesis, we analysed contraction and relaxation of adult cardiomyocytes from hearts of DKO and control mice. Cardiomyocytes from DKO mice were longer and wider resulting in an increased surface area compared with controls (Figure 7A). Using sarcomere length tracing, we measured peak sarcomere shortening as amplitude, maximum shortening velocity (+dL/dt), and maximum relaxation velocity (‐dL/dt) of control and DKO myocytes. All these parameters were reduced in DKO cardiomyocytes (Figure 7B) indicative for a diminished systolic and diastolic function of DKO cardiomyocytes.
Figure 7.
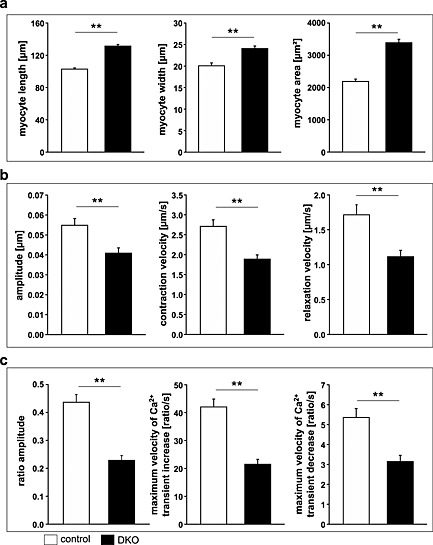
Decreased contraction and relaxation and impaired calcium handling in adult ventricular cardiomyocytes of double knockout (DKO) mice. (A) Quantification of length (left panel), width (middle panel), and area (right panel) of adult cardiomyocytes isolated from hearts of control (n = 100 cells) and DKO (n = 100 cells) animals indicated an increased size of cardiomyocytes in DKO mice. Data are presented as mean ± SEM. **P < 0.01. (B) Peak sarcomere shortening as amplitude (left panel), maximum shortening velocity as + dL/dt (middle panel), and maximum relaxation velocity as ‐dL/dt (right panel) of adult control (n = 135 cells) and DKO (n = 130 cells) myocytes are shown. Cardiomyocytes were stimulated to contract at 5 Hz at 37°C, extracellular Ca2+‐concentration was 1.25 mM. Data are presented as mean ± SEM. **P < 0.01. (C) Intracellular Ca2+‐transients of adult cardiomyocytes from control (n = 76 cells) and DKO (n = 62 cells) animals are shown as ratio transients. Graphs show fura‐2 ratio amplitude (left panel), maximum velocity of ratio increase as + dR/dt (middle panel), and maximum velocity of ratio decay as –dR/dt (right panel). Cardiomyocytes were stimulated to contract at 5 Hz at 37°C, extracellular Ca2+‐concentration was 1.25 mM; cells were fura‐2 loaded. Data are presented as mean ± SEM. **P < 0.01.
To evaluate whether or not diminished cardiomyocyte function was due to altered intracellular Ca2+‐availability, we measured Ca2+‐transients simultaneously to sarcomere shortening. Ca2+‐transient amplitudes were significantly decreased in DKO myocytes (Figure 7C). More specifically, diastolic Ca2+‐concentration was reduced in DKO cardiomyocytes compared with controls (Figure 7C). When we calculated the velocities of increase and decay of Ca2+‐transients in cardiomyocytes as maximum velocity of ratio increase (+dR/dt) and maximum velocity of ratio decay (–dR/dt), we found these parameters to be significantly decreased in DKO cardiomyocytes (Figure 7C). In summary, these data are indicative for a disturbed Ca2+‐handling that might at least partially be responsible for decreased systolic and diastolic function of DKO hearts.
MuRF family members physically interact, co‐localize and stabilize each other in cardiac and skeletal muscle cell lines in vitro
The cooperative function of MuRF proteins might be related to their ability to homodimerize and heterodimerize and to be localized to the same subcellular compartment or structure where they might fulfil similar functions. To investigate the molecular basis of functional redundancy of the MuRF family, we performed cell culture analyses. First, we visualized subcellular localization of MuRF proteins in the cardiac myoblasts cell line H9c2 transiently transfected with cDNA expression plasmids encoding MuRF1, MuRF2, and/or MuRF3. When solely expressed, MuRF1 and MuRF2 were predominantly cytoplasmic, and MuRF3 resided at microtubules. However, when MuRF1 and MuRF2 were co‐expressed, both proteins occurred in thick tubule‐like structures. Co‐expression of MuRF1 or MuRF2 with MuRF3 led to co‐localization of MuRF1 and MuRF2 with MuRF3 at microtubules, similar to what was seen for MuRF3 alone (Figure S8a). These data implicated that MuRF3 physically interacts with MuRF2 leading to recruitment of MuRF2 to microtubules. Second, to test if physical interaction was the cause for co‐localization of MuRF2 and MuRF3, co‐immunoprecipitation experiments followed by Western blot analyses were performed. These experiments showed that all MuRF proteins interact with each other (Figure S8b). Because MuRF proteins function as E3 ubiquitin ligases, we tested if co‐expression of MuRF family members would decrease their protein content. First, expression plasmids encoding MuRF1, MuRF2, or MuRF3 were transfected in mouse skeletal myoblasts (C2C12‐cells). Western blot analysis showed a weak signal of MuRF proteins when expressed individually (Figure S8c). However, co‐expression of different MuRFs led to an increase in their protein amount indicating that they do not execute E3 ubiquitin ligase function to degrade their respective family members, but rather stabilize them (Figure S8c). In concert, these data demonstrated that physical interaction of MuRF2 and MuRF3 in myocytes mediates their co‐localization and prevents them from being degraded. Our data also indicate that MuRF1 localizes to the same subcellular compartments as MuRF2 and MuRF3. However, if this subcellular localization enables MuRF1 to compensate for the loss of MuRF2 and MuRF3 remains to be proven.
Proteomic analysis indicated a reduction of proteins important for mitochondrial function and energy supply in DKO muscle.
To search for a potential mechanism by which the absence of MuRF2 and MuRF3 decreased muscle function, we performed mass‐spectrometry on tissue lysates from DKO and control muscle. In total, we detected 37 710 peptides belonging to 3565 proteins in both control (n = 3) and DKO (n = 3) soleus. There were 2024 proteins identified in all six samples. We expected to identify an accumulation of proteins, which are potentially targeted by MuRF2 and MuRF3 for proteasomal degradation. Bioinformatics analyses of mass‐spectrometry data identified proteins which are enriched and diminished in DKO muscle (Figure 8). As expected, GO term analysis of proteins increased in DKO muscle showed an enrichment of proteins predominantly involved in proteolysis, ubiquitin proteasome dependent protein degradation, and regulation of muscle function (Figure 8, Figure S9, Table S2). Importantly, among those proteins that were reduced in DKO muscle, we identified proteins important for mitochondrial function and cellular energy supply (Figure S9 and S10, Table S3).
Figure 8.
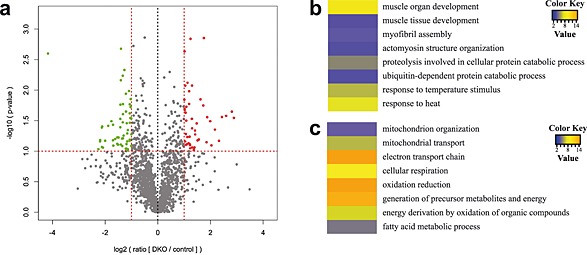
Proteomic analysis of lysates from double knockout (DKO) compared with control soleus muscle. (A) Volcano‐plot. For each of the data sets, a t‐test was used to calculate the − log10 (P‐value), which was plotted vs. the log‐transformed means of the DKO/control ratios. Vertical lines indicate the top or bottom 30% most regulated proteins, while the horizontal line indicates the cut‐off for the significance of the P‐value. Proteins increased and decreased in DKO muscle are shown in red and green, respectively. (B) and (C) Heat‐map of the Z‐transformed P‐values obtained by Gene Ontology (GO)‐analysis of proteins upregulated in DKO muscle. The 30% upregulated (B) and 30% downregulated (C) proteins in DKO soleus were used for the GO analysis with the DAVID online tool 3. The obtained P‐values of GO‐term biological process that were significantly enriched (P < 0.05) were log‐transformed, Z‐transformed, hierarchically clustered, and plotted as a heat map. Colour key indicates P‐values.
Mitogen‐activated protein kinase‐activated protein kinase 2 and mitogen‐activated protein kinase‐activated protein kinase 3 are enriched in DKO muscle
For further analysis, we focussed on MAPKAPK2 and 3, two major final mediators of the p38 MAPK signalling cascade,14, 15 which were enriched in DKO muscle. Western blot analysis from total tissue lysates revealed an increase in the MAPKAPK2 protein content in soleus and left ventricle of DKO mice (Figure 9A and 9B; left panels). A known interaction partner of MAPKAPK2 and MAPKAPK3 is p38 MAPK.14, 15 To avoid unspecific background for the detection of MAPKAPK3 protein levels, we first performed p38‐GST pulldown assays in total tissue lysates of soleus and left ventricle followed by immunoblotting using an anti‐MAPKAPK3 antiserum as described previously.14, 15 MAPKAPK3 protein levels were increased in DKO soleus and left ventricle compared with controls (Figure 9A and 9B; right panels). The anti‐MAPKAPK2 antibody was used in this experiment as a positive control for equal enrichment of p38 MAPK interacting proteins and showed a similar increase in the amount of MAPKAPK2 protein after the p38‐GST pulldown as detected by Western blot analysis in total tissue lysates (Figure 9A and 9B; left panels). In summary, MAPKAPK2 and MAPKAPK3 protein contents are increased in DKO soleus and left ventricle compared with control mice. The amount of total p38 MAPK protein remained unchanged in soleus and left ventricle in control compared with DKO mice (Figure 9A and 9B). In addition, real‐time RT–PCR showed that MAPKAPK2 and MAPKAPK3 mRNA expression was not elevated in DKO soleus and left ventricle indicating that increased MAPKAPK2 and MAPKAPK3 protein contents were not due to their increased gene expression in the absence of MuRF2 and MuRF3 (Figure 9C). Further analyses using co‐immunoprecipitation and co‐localization experiments could not conclusively demonstrate that MAPKAPK2 and MAPKAPK3 interact with or co‐localize with MuRF2 and MuRF3 (data not shown). In concert, our data suggest that MuRF2 and MuRF3 are coordinately involved in maintenance of protein homeostasis in striated muscles primarily regulating protein degradation pathways and energy supply.
Figure 9.
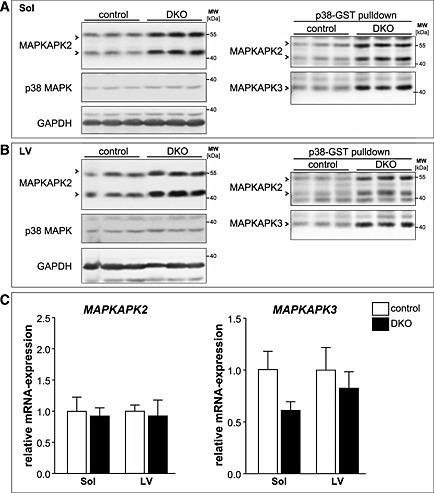
Mitogen‐activated protein kinase‐activated protein kinase (MAPKAPK) 2 and MAPKAPK3 are increased in double knockout (DKO) soleus (Sol) and left ventricle. (A) Immunoblotting of proteins from total tissue lysates of Sol from control and DKO mice using anti‐MAPKAPK2 and anti‐p38 MAPK antibody. GAPDH served as loading control (left panel). GST‐p38 MAPK pulldown assays were performed with tissue lysates from soleus of control and DKO mice and analysed using anti‐MAPKAPK2 antibody and anti‐MAPKAPK3 antiserum (right panel). (B) Immunoblotting of proteins from total tissue lysates of left ventricles (LV) from control and DKO mice using anti‐MAPKAPK2 and anti‐p38 MAPK antibody. GAPDH served as loading control (left panel). GST‐p38 pulldown assays were performed with tissue lysates from hearts of control and DKO mice and analysed using anti‐MAPKAPK2 antibody and anti‐MAPKAPK3 antiserum (right panel). (C) Real‐time RT–PCR analysis of MAPKAPK2 and MAPKAPK3 gene expression in Sol and LV from control (n = 8–9) and DKO (n = 4) mice. Hypoxanthine guanine phosphoribosyl transferase (Hprt) expression was used as reference. Data are presented as mean ± SEM.
Discussion
The main finding of our work is that MuRF2 and MuRF3 are key factors for the maintenance of skeletal muscle and cardiac structure and function, because absence of both leads to myopathy in striated muscles. The observed phenotypes of DKO mice are summarized in Table 1. MuRF2 and MuRF3 avidly interact, co‐localize to microtubules, and stabilize each other in vitro, which could serve as the molecular basis for their redundant function in myocytes. The interactions between MuRF2 and MuRF3 suggest a cooperative activity of both E3 ligases.
Table 1.
Summary of skeletal muscle and cardiac phenotypes of double knockout mice
| Skeletal muscle | |
|---|---|
| Protein aggregate myopathy | |
| Submembraneous accumulations localized inside myofibres containing MyHC and parts of sarcomeres | |
| Protein accumulations are not fibre type restricted | |
| Fibre type shift in soleus and EDL (increased slow‐, decreased fast‐twitch fibres) | |
| Centralized nuclei | |
| Decreased maximal force development of soleus and EDL | |
| Increased Myh3 and Myh7 expression | |
| Mass spectrometry: | |
| Increase in degradation associated proteins | |
| Increase in MAPKAPK3 | |
| Decrease in mitochondrial proteins | |
| Heart | |
|---|---|
| Increased LVESD, decreased wall thickness | |
| Decreased LVEF and cardiac output | |
| Reduced left ventricular systolic and developing pressure | |
| Decreased cardiac contraction and relaxation velocity | |
| Reduced cardiomyocyte contractility and relaxation velocity | |
| Reduced calcium influx and export velocities | |
| Increased Myh7 expression | |
EDL, extensor digitorum longus; LVESD, left ventricular ejection fraction; LVEF, left ventricular end‐systolic dimension; MAPKAPK, mitogen‐activated protein kinase‐activated protein kinase; Myh, myosin heavy chain.
The MuRF2 and MuRF3 DKO mice developed a protein aggregate myopathy of the skeletal muscle. This finding resembles the phenotype of MuRF1 and MuRF3 DKO mice we reported earlier.4 We showed that MuRF1 and MuRF3 associate with, ubiquitinate, and mediate ubiquitin proteasome system (UPS) dependent myosin degradation.4 However, if MuRF2 is involved in myosin turnover and if this leads to myosin aggregates in MuRF2 and MuRF3 DKO mice was unknown. In addition, recent reports suggest E3 ubiquitin ligase independent functions of MuRF proteins.11 Others, and we, reported that MuRF2 10, 26, 27 and MuRF3 7 are important for sarcomere assembly, initiation of myogenesis, and muscle differentiation via their binding to and stabilization of microtubules. These findings together with the observation that knockdown of MuRF2 increased the expression of MuRF3 and vice versa, and that the loss of MuRF2 was partially compensated by MuRF3 12 indicate that both proteins compensate for their respective loss. In addition, combined deletion of MuRF2 and MuRF3 resulted in a myofibrillar phenotype in myocytes in vitro.12 We add to these data that the amount of MuRF2 and MuRF3 is not only balanced at the transcriptional but also at post‐translational level, because MuRF2 and MuRF3 co‐localize to microtubules and prevent their respective degradation. Redundancy of MuRF2 and MuRF3 is one explanation why MuRF2 and MuRF3 single knockout mice have no phenotype at baseline.4, 5 We conclude that MuRF2 and MuRF3 function redundantly during sarcomere formation and that their absence leads to myosin aggregates in vivo. However, normally formed sarcomeres suggest additional factors compensating for the loss of MuRF2 and MuRF3 in DKO muscle during myogenesis.
MuRF1 is mainly expressed in fast/type II fibres,11 MuRF2 is predominantly expressed in slow/type I fibres, and MuRF3 is ubiquitously expressed.28 Up‐regulation of MuRF1 paralleled by a predominant atrophy of fast/type II fibres during muscle atrophy29 underscores a functional relevance of this fibre type specific expression pattern. However, myosin aggregates occurred in both fast/type II and slow/type I myofibres of DKO muscle. These findings argue for a role of MuRF2 and MuRF3 in both fibre types, which seems to be MuRF1 independent. The latter is supported by the fact that myosin aggregates also occurred in fast/type II fibres were MuRF1 is preferentially expressed.11 MuRF1 and MuRF3 DKO,4 and MuRF2 and MuRF3 DKO (shown here), but not MuRF1 and MuRF2 DKO11 mice show a protein aggregate myopathy. Because myosin aggregates only occurred in the skeletal muscle of DKO mice lacking MuRF3, we believe that this phenotype is mediated by the absence of MuRF3. In contrast, based on the phenotype of MuRF1 and MuRF3 DKO4 and MuRF1 and MuRF2 5 DKO mice, MuRF1 appears to play a predominant role in both skeletal muscle and heart. More specifically, we found a severe myosin storage myopathy of both the heart and skeletal muscle in MuRF1 and MuRF3 DKO animals.4 Additionally, three‐fourths of MuRF1 and MuRF2 DKO mice died around birth.5 However, the myopathic phenotype of MuRF2 and MuRF3 DKO mice was less severe. These data suggest that MuRF1 plays a predominant role in muscle and has non‐overlapping functions with MuRF2 and MuRF3.4, 5 Nevertheless, due to the myopathic phenotype of MuRF2 and MuRF3 DKO mice, it appears that MuRF1 cannot fully compensate for the loss of MuRF2 and MuRF3. Furthermore, absent MuRF2 and MuRF3 caused an increase in slow/type I and in hybrid fibres, as well as a decrease in fast/type II fibres. Possibly, this function is related to the regulation of myozenin‐1, as was recently shown in MuRF1 and MuRF2 DKO muscle.11 Of note, recently combined homozygous MuRF1 and heterozygous MuRF3 mutations were shown to cause a protein aggregate myopathy and cardiomyopathy in patients,30 which was reminiscent of the MuRF1 and MuRF3 DKO phenotype described by us.4 Importantly, after comprehensive screening for mutations in several sarcomeric genes, our description of the MuRF1 and MuRF3 DKO mouse phenotype prompted the authors to search for mutations in the MuRF1 and MuRF3 genes.30 This analysis by Olive et al. now led to a new disease entity of skeletal muscle and cardiac protein aggregate myopathy.30 However, if combined MuRF2 and MuRF3 mutations occur in patients and are causative for protein aggregate myopathy and cardiomyopathy needs to be verified. Nevertheless, in those patients suffering from protein aggregate myopathies in whom mutations in sarcomeric genes were excluded, it appears to be reasonable to search for mutations in MuRF1, MuRF2 and MuRF3.
MuRF2 and MuRF3 DKO mice exhibited cardiomyopathy with decreased systolic and diastolic function. Our data show that this phenotype occurred at the level of cardiomyocytes. First, no interstitial fibrosis was observed in MuRF2 and MuRF3 DKO mice, which often accompanies pathological cardiac remodelling and leads to a decrease in systolic and diastolic function.31, 32 Second, beta/slow myosin expression was increased in DKO hearts. Normally, ventricular myocardium of rodents is mainly composed of alphaMhc/Myh6, accounting for more than 90% of all myosins.33 ATPase activity of alphaMhc/Myh6 is higher compared with beta/slow myosin,34 which allows greater economy in sarcomeric force generation.35, 36 This leads to a higher contractile velocity of hearts expressing alphaMhc/Myh6 compared with hearts expressing beta/slow myosin. Accordingly, a decrease in systolic cardiac function was reported for transgenic mice overexpressing beta/slow myosin.37 Therefore, increased beta/slow myosin expression might have contributed to the reduced systolic and diastolic function of DKO cardiomyocytes and hearts. However, it is unknown why beta/slow myosin expression was increased in DKO hearts. Of note, MuRF1 and MuRF2 were shown to regulate myozenin‐1 and myozenin‐2,11 inhibitors of the calcineurin/nuclear factor of activated T‐cells (NFAT) pathway.38, 39 A downregulation of myozenin‐1 and myozenin‐2 was reported for MuRF1 and MuRF2 DKO mice possibly activating the calcineurin/nuclear factor of activated T‐cells (NFAT) pathway.11 Activation of calcineurin could account for increased beta/slow myosin expression.40 Because calcineurin dephosphorylates calcium channels and phospholamban leading to a reduction in intracellular calcium and calcium uptake by the sarcoplasmic reticulum, its activation could also be responsible for the reduction in systolic and diastolic calcium transients in cardiomyocytes of DKO mice. This could also explain why MuRF2 and MuRF3 DKO hearts and cardiomyocytes did not respond differently when exposed to isoproterenol compared with controls. Normally, isoproterenol mediates phosphorylation of the L‐type calcium channel and phospholamban in a protein kinase‐A dependent manner leading to increased calcium currents and calcium load of the sarcoplasmic reticulum. However, activation of calcineurin opposes the effects of protein kinase‐A.41 Although it is tempting to speculate that MuRF2 and MuRF3 are involved in regulation of calcineurin signalling, this hypothesis needs to be proven. In summary, our data show that MuRF2 and MuRF3 regulate systolic and diastolic cardiac function at the level of cardiomyocytes.
Finally, our proteomic data support a role of MuRF2 and MuRF3 in muscle function and sarcomeric organization. They also provide evidence that proteolytic pathways are activated to possibly compensate for the loss of both E3 ligases. However, the phenotype of DKO mice argues for unique non‐replaceable functions of MuRF2 and MuRF3 in muscle proteolysis. We identified MAPKAPK2 and MAPKAPK3 to be elevated in skeletal muscle and hearts of DKO mice. In addition, proteins involved in mitochondrial function and energy supply were reduced in DKO mice, which could account for reduced muscle function in these mice. Because decreased cardiac function is not explained by protein aggregates in DKO mice, these findings are particularly important. Recently, we reported that cardiomyocytes of MAPKAPK2 and MAPKAPK3 DKO mice showed enhanced contractility and accelerated relaxation.18 Therefore, we assume that an increase in MAPKAPK2 and MAPKAPK3 in DKO hearts leads to an opposing phenotype. Because we also observed improved force parameters in MAPKAPK2/3 DKO soleus muscles,18 increased MAPKAPK2 and MAPKAPK3 protein levels in MuRF2 and MuRF3 DKO skeletal muscle might account for decreased muscle performance. Similarly, because of the observed positive effect of MAPKAPK2/3‐deficiency on the oxidative energy metabolism,18 the increase of MAPKAPK2/3 protein levels in MuRF2 and MuRF3 DKO mice might, at least in part, account for reduced mitochondrial function and energy supply in DKO skeletal muscle. However, it remains to be elucidated how the absence of MuRF2 and MuRF3 leads to an increase in MAPKAPK2 and MAPKAPK3 because we did not observe changes in MAPKAPK2 and MAPKAPK3 gene expression and could not detect physical interaction between MAPKAPK2 and MAPKAPK3 with MuRF2 and MuRF3 in co‐immunoprecipitation and co‐localization experiments. Although physical interaction between MAPKAPK2/3 and their substrates are often difficult to detect, the increase in MAPKAPK2/3 protein amounts could also be caused indirectly because of altered p38 MAPK‐MAPKAPK2/3 signalling in MuRF2 and MuRF3 DKO mice. For example, because activated MAPKAPK2/3 shuttles from the nucleus to the cytoplasm, altered p38 MAPK‐MAPKAPK2/3 signalling in DKO muscle might influence the duration time of MAPKAPK2/3 in one of the two cellular compartments and thereby affect MAPKAPK2/3 protein stabilities. Thus, further studies are needed to elucidate the mechanism of increased MAPKAPK2 and MAPKAPK3 contents in DKO muscle.
The p38 MAPK‐MAPKAPK2/3 cascade not only regulates gene expression at the transcriptional and post‐transcriptional level13 but also causes post‐translational modifications leading to altered protein folding and degradation.42, 43 For example, activated p38 MAPK was shown to interact with and phosphorylate E3 ubiquitin ligases, such as Siah2 and MIB1, resulting in increased ligase activity.42, 43 In addition, MAPKAPK2 directly interacts with and phosphorylates the E3 ubiquitin ligase Hdm2/Mdm244 resulting in increased E3 ligase activity and protein degradation. Although it is tempting to speculate that such a relationship exists for MAPKAPK2/3, MuRF2, and MuRF3, a direct association between these proteins needs to be proven. It would also be interesting to investigate if the observed phenotype in the skeletal muscle and heart of MuRF2 and MuRF3 DKO mice is directly associated with increased MAPKAPK2/3 protein levels, and could be abolished in compound MAPKAPK2/3 and MuRF2/3 quadruple KO mice.
We conclude that MuRF2 and MuRF3 are concomitantly involved in the regulation of structure and performance of the skeletal muscle and the heart. MuRF2 and MuRF3 play a role in fibre type specification of slow/type I fibres in extensor digitorum longus and soleus. Additionally, MuRF2 and MuRF3 affected mitochondrial function and energy supply in striated muscle. Accordingly, absence of MuRF2 and MuRF3 leads to a myopathic phenotype of the skeletal muscle and the heart.
Funding
Jens Fielitz has received research grants from the Deutsche Forschungsgemeinschaft [FI 965/2‐1, FI 965/4‐1], the Experimental and Clinical Research Center, and Ernst und Berta Grimmke‐Stiftung.
Conflict of interest
D. Lodka, A. Pahuja, C. Geers‐Knörr, R. Scheibe, M. Nowak, J. Hamati, C. Köhncke, B. Purfürst, T. Kanashova, S. Schmidt, D. J. Glass, I. Morano, A. Heuser, T. Kraft, R. Bassel‐Duby, E. N. Olson, G. Dittmar, and T. Sommer declare that they have no conflict of interest.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle 2010; 1:7–8 (von Haehling S, Morley JE, Coats AJ, and Anker SD).
We thank Friedrich C. Luft for assistance and advice. We thank Janine Woehlecke and Dorothee Krone for excellent technical assistance. We thank Martin Taube for echocardiography. We thank Monte Willis and Cam Patterson for sharing the MuRF2 mutant mice.
Non‐standard abbreviations
- DKO
MuRF2 and MuRF3 double knockout mice
- +dL/dt
maximum shortening velocity
- ‐dL/dt
maximum relaxation velocity
- EDL
extensor digitorum longus muscle
- GP
gastrocnemius/plantaris muscle
- LVPsys
systolic left ventricular pressure
- LVPdevp
left ventricular developing pressure
- MCSA
myocyte cross‐sectional area
- MAPKAPK
mitogen‐activated protein kinase‐activated protein kinase
- MuRF
Muscle‐specific RING‐finger protein
- MyHC
myosin heavy chain
- Sol
soleus muscle
- TA
tibialis anterior muscle
Lodka, D. , Pahuja, A. , Geers‐Knörr, C. , Scheibe, R. J. , Nowak, M. , Hamati, J. , Köhncke, C. , Purfürst, B. , Kanashova, T. , Schmidt, S. , Glass, D. J. , Morano, I. , Heuser, A. , Kraft, T. , Bassel‐Duby, R. , Olson, E. N. , Dittmar, G. , Sommer, T. , and Fielitz, J. (2016) Muscle RING‐finger 2 and 3 maintain striated‐muscle structure and function. Journal of Cachexia, Sarcopenia and Muscle, 7: 165–180. doi: 10.1007/jcsm.12057.
References
- 1. Fielitz J, van Rooij E, Spencer JA, Shelton JM, Latif S, van der Nagel R, et al. Loss of muscle‐specific RING‐finger 3 predisposes the heart to cardiac rupture after myocardial infarction. Proc Natl Acad Sci U S A 2007; 104: 4377–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001; 294: 1704–1708. [DOI] [PubMed] [Google Scholar]
- 3. Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 2005; 308: 1599–1603. [DOI] [PubMed] [Google Scholar]
- 4. Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest 2007; 117: 2486–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. Embo J 2008; 27: 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res 2007; 100: 456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spencer JA, Eliazer S, Ilaria RL Jr, Richardson JA, Olson EN. Regulation of microtubule dynamics and myogenic differentiation by MURF, a striated muscle RING‐finger protein. J Cell Biol 2000; 150: 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle‐specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci U S A 2004; 101: 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. Faseb J 2004; 18: 39–51. [DOI] [PubMed] [Google Scholar]
- 10. Pizon V, Iakovenko A, Van Der Ven PF, Kelly R, Fatu C, Furst DO, et al. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MuRF2 RING‐finger protein. J Cell Science 2002; 115: 4469–4482. [DOI] [PubMed] [Google Scholar]
- 11. Moriscot AS, Baptista IL, Bogomolovas J, Witt C, Hirner S, Granzier H, et al. MuRF1 is a muscle fiber‐type II associated factor and together with MuRF2 regulates type‐II fiber trophicity and maintenance. J Struct Biol 2010; 170: 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perera S, Holt MR, Mankoo BS, Gautel M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev Biol 2011; 351: 46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gaestel M. MAPKAP kinases ‐ MKs ‐ two's company, three's a crowd. Nat Rev Mol Cell Bio 2006; 7: 120–130. [DOI] [PubMed] [Google Scholar]
- 14. Ronkina N, Kotlyarov A, Dittrich‐Breiholz O, Kracht M, Hitti E, Milarski K, et al. The mitogen‐activated protein kinase (MAPK)‐activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol 2007; 27: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moise N, Dingar D, Mamarbachi AM, Villeneuve LR, Farhat N, Gaestel M, et al. Characterization of a novel MK3 splice variant from murine ventricular myocardium. Cell Signal 2010; 22: 1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Widegren U, Ryder JW, Zierath JR. Mitogen‐activated protein kinase signal transduction in skeletal muscle: effects of exercise and muscle contraction. Acta physiol Scandinavica 2001; 172: 227–238. [DOI] [PubMed] [Google Scholar]
- 17. Puigserver P, Spiegelman BM. Peroxisome proliferator‐activated receptor‐gamma coactivator 1 alpha (PGC‐1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 2003; 24: 78–90. [DOI] [PubMed] [Google Scholar]
- 18. Scharf M, Neef S, Freund R, Geers‐Knorr C, Franz‐Wachtel M, Brandis A, et al. Mitogen‐activated protein kinase‐activated protein kinases 2 and 3 regulate SERCA2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol Cell Biol 2013; 33: 2586–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marber MS, Rose B, Wang Y. The p38 mitogen‐activated protein kinase pathway‐‐a potential target for intervention in infarction, hypertrophy, and heart failure. J Mol Cell Cardiol 2011; 51: 485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim MS, Fielitz J, McAnally J, Shelton JM, Lemon DD, McKinsey TA, et al. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle performance. Mol Cell Biol 2008; 28: 3600–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fielitz J, Kim MS, Shelton JM, Qi X, Hill JA, Richardson JA, et al. Requirement of protein kinase D1 for pathological cardiac remodeling. Proc Natl Acad Sci U S A 2008; 105: 3059–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mutig N, Geers‐Knoerr C, Piep B, Pahuja A, Vogt PM, Brenner B, et al. Lipoteichoic acid from Staphylococcus aureus directly affects cardiomyocyte contractility and calcium transients. Mol Immunol 2013; 56: 720–728. [DOI] [PubMed] [Google Scholar]
- 23. Primeaux SD, Tong M, Holmes GM. Effects of chronic spinal cord injury on body weight and body composition in rats fed a standard chow diet. Am J Physiol Regul Integr Comp Physiol 2007; 293: R1102–1109. [DOI] [PubMed] [Google Scholar]
- 24. Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, et al. Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 2008; 40: 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pankonien I, Alvarez JL, Doller A, Kohncke C, Rotte D, Regitz‐Zagrosek V, et al. Ahnak1 is a tuneable modulator of cardiac Ca(v)1.2 calcium channel activity. J Muscle Res Cell Motil 2011; 32: 281–290. [DOI] [PubMed] [Google Scholar]
- 26. McElhinny AS, Perry CN, Witt CC, Labeit S, Gregorio CC. Muscle‐specific RING finger‐2 (MURF‐2) is important for microtubule, intermediate filament and sarcomeric M‐line maintenance in striated muscle development. J Cell Science 2004; 117: 3175–3188. [DOI] [PubMed] [Google Scholar]
- 27. Pizon V, Gerbal F, Diaz CC, Karsenti E. Microtubule‐dependent transport and organization of sarcomeric myosin during skeletal muscle differentiation. Embo J 2005; 24: 3781–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perera S, Mankoo B, Gautel M. Developmental regulation of MURF E3 ubiquitin ligases in skeletal muscle. J Muscle Res Cell Motil 2012; 33: 107–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bierbrauer J, Koch S, Olbricht C, Hamati J, Lodka D, Schneider J, et al. Early type II fiber atrophy in intensive care unit patients with nonexcitable muscle membrane. Crit Care Med 2012; 40: 647–650. [DOI] [PubMed] [Google Scholar]
- 30. Olive M, Abdul‐Hussein S, Oldfors A, Gonzalez‐Costello J, van der Ven PF, Furst DO, et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum Mol Genet 2015; 24: 3638–3650. [DOI] [PubMed] [Google Scholar]
- 31. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med 2008; 358: 1370–1380. [DOI] [PubMed] [Google Scholar]
- 32. Fielitz J, Hein S, Mitrovic V, Pregla R, Zurbrugg HR, Warnecke C, et al. Activation of the cardiac renin‐angiotensin system and increased myocardial collagen expression in human aortic valve disease. J Am Coll Cardiol 2001; 37: 1443–1449. [DOI] [PubMed] [Google Scholar]
- 33. Lompre AM, Schwartz K, d'Albis A, Lacombe G, Van Thiem N, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature 1979; 282: 105–107. [DOI] [PubMed] [Google Scholar]
- 34. Pope B, Hoh JF, Weeds A. The ATPase activities of rat cardiac myosin isoenzymes. FEBS Lett 1980; 118: 205–208. [DOI] [PubMed] [Google Scholar]
- 35. Alpert NR, Mulieri LA. Increased myothermal economy of isometric force generation in compensated cardiac hypertrophy induced by pulmonary artery constriction in the rabbit A characterization of heat liberation in normal and hypertrophied right ventricular papillary muscles. Circ Res 1982; 50: 491–500. [DOI] [PubMed] [Google Scholar]
- 36. Holubarsch C, Goulette RP, Litten RZ, Martin BJ, Mulieri LA, Alpert NR. The economy of isometric force development, myosin isoenzyme pattern and myofibrillar ATPase activity in normal and hypothyroid rat myocardium. Circ Res 1985; 56: 78–86. [DOI] [PubMed] [Google Scholar]
- 37. Tardiff JC, Hewett TE, Factor SM, Vikstrom KL, Robbins J, Leinwand LA. Expression of the beta (slow)‐isoform of MHC in the adult mouse heart causes dominant‐negative functional effects. Am J Physiol Heart Circ Physiol 2000; 278: H412–419. [DOI] [PubMed] [Google Scholar]
- 38. Frey N, Barrientos T, Shelton JM, Frank D, Rutten H, Gehring D, et al. Mice lacking calsarcin‐1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med 2004; 10: 1336–1343. [DOI] [PubMed] [Google Scholar]
- 39. Frey N, Frank D, Lippl S, Kuhn C, Kogler H, Barrientos T, et al. Calsarcin‐2 deficiency increases exercise capacity in mice through calcineurin/NFAT activation. J Clin Invest 2008; 118: 3598–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, et al. A calcineurin‐dependent transcriptional pathway for cardiac hypertrophy. Cell 1998; 93: 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Santana LF, Chase EG, Votaw VS, Nelson MT, Greven R. Functional coupling of calcineurin and protein kinase A in mouse ventricular myocytes. J Physiol 2002; 544: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakayama K, Qi J, Ronai Z. The ubiquitin ligase Siah2 and the hypoxia response. Mol Cancer Res: MCR 2009; 7: 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Papin J, Subramaniam S. Bioinformatics and cellular signaling. Curr Opin Biotech 2004; 15: 78–81. [DOI] [PubMed] [Google Scholar]
- 44. Weber HO, Ludwig RL, Morrison D, Kotlyarov A, Gaestel M, Vousden KH. HDM2 phosphorylation by MAPKAP kinase 2. Oncogene 2005; 24: 1965–1972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item