

Abstract
Positive transcription elongation factor b (P‐TEFb), which comprises cyclin‐dependent kinase 9 (CDK9) kinase and cyclin T subunits, is an essential kinase complex in human cells. Phosphorylation of the negative elongation factors by P‐TEFb is required for productive elongation of transcription of protein‐coding genes by RNA polymerase II (pol II). In addition, P‐TEFb‐mediated phosphorylation of the carboxyl‐terminal domain (CTD) of the largest subunit of pol II mediates the recruitment of transcription and RNA processing factors during the transcription cycle. CDK9 also phosphorylates p53, a tumor suppressor that plays a central role in cellular responses to a range of stress factors. Many viral factors affect transcription by recruiting or modulating the activity of CDK9. In this review, we will focus on how the function of CDK9 is regulated by viral gene products. The central role of CDK9 in viral life cycles suggests that drugs targeting the interaction between viral products and P‐TEFb could be effective anti‐viral agents.
Keywords: CDK9, CTD, EBV, HIV, HSV, HTLV, P‐TEFb
Introduction
Transcription of many human viruses is dependent on host cell factors such as RNA polymerase II (pol II) 1. Pol II is made up of 12 subunits (Rpb1–Rpb12) 2. The carboxyl‐terminal domain (CTD) of the largest subunit of pol II (Rpb1) comprises tandemly repeated heptapeptides with the consensus sequence Tyr1‐Ser2‐Pro3‐Thr4‐Ser5‐Pro6‐Ser7 3, 4. The CTD plays a central role in transcriptional and co‐transcriptional RNA processing 4, 5, 6, 7 by mediating the recruitment of transcription and processing factors at different steps of the transcription cycle through reversible modification of the residues within the heptapeptide repeats 8. CTD modification generates a code that regulates the interaction with transcription and RNA processing factors 9, 10, 11, 12. Among the CTD modifications, the phosphorylation of Ser2 (Ser2P) of pol II CTD, is catalyzed by the cyclin‐dependent kinase 9 (CDK9) subunit of the positive transcription elongation factor b (P‐TEFb). P‐TEFb also phosphorylates two negative transcription elongation factors: DRB sensitivity‐inducing factor (DSIF) and negative elongation factor (NELF), which are recruited to the elongation complex in the promoter proximal region to induce pol II pausing 13, 14. Phosphorylation of NELF promotes its dissociation from the elongation complex, whereas phospho‐DSIF remains associated with pol II as an elongation activator 14. The role of P‐TEFb in stimulating productive elongation was elucidated using 5,6‐dichloro‐1‐β‐d‐ribofuranosylbenzimidazole (DRB), which is a purine nucleoside analog that inhibits CDK9 activity 15. Treatment of cells with DRB results in the production of shortened transcripts, diagnostic of the inability of pol II to transcribe through the early elongation checkpoint in the absence of P‐TEFb activity 16. In addition to Ser2 of the pol II CTD, NELF and DSIF, several serine residues in the key transcription factor p53 are phosphorylated by CDK9, which results in p53 activation 17, 18.
Positive transcription elongation factor b comprises the CDK9 kinase and a cyclin partner 15. CDK9 is an ubiquitously expressed Ser/Thr proline‐directed kinase 19, 20. It was identified in the early 1990s and initially named PITALRE 19, 21. It exists in two isoforms, CDK9‐42 and CDK9‐55, which arise from two transcription start sites 22. The ratio of CDK9‐42/CDK9‐55 is cell type‐dependent 23, and it is unclear if there are functional differences between the two isoforms. CDK9 interacts with T‐type cyclins – T1, T2a, T2b and cyclin K 24, 25. In this heterodimer, the CDK9 subunit provides the enzymatic activity, while the cyclin has a regulatory role 24, 26. However, mass spectrometry has failed to demonstrate that cyclin K is associated with CDK9 27. In addition, a recent study revealed that cyclin K associates with CDK12 and CDK13 28. Therefore, cyclins T1 and T2 may be the major cyclins associated with CDK9. In the cell, P‐TEFb exists in two major forms. A large amount of cellular P‐TEFb is sequestered in an inactive complex with the noncoding 7SK small nuclear RNA (7SK snRNA), hexamethylene bisacetamide‐induced protein 1 (HEXIM1) and/or HEXIM2 and the La‐related protein 7 29, 30, 31, 32, 33, 34, 35. The catalytically active form of P‐TEFb is instead generally associated with bromodomain‐containing protein 4 (Brd4) and the super elongation complex (SEC) 36, 37, 38. The transition between active and inactive forms is dynamic and tightly regulated 37, 38, reflecting the important role that P‐TEFb plays in cellular processes (Fig. 1). Given the importance of P‐TEFb in regulating cellular gene expression, it is perhaps not surprising that this complex is also functionally integrated into the expression of human viruses (Table 1). Several viral factors regulate CTD phosphorylation through recruitment or modulation of the activity of CDK9.
Figure 1.
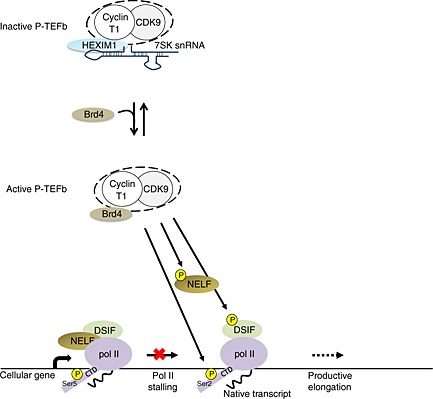
The role of positive transcription elongation factor b (P‐TEFb) in a regulation of pol II transcription. P‐TEFb, comprising cyclin‐dependent kinase 9 (CDK9) and a cyclin T1 subunit, exists in a catalytically inactive small nuclear ribonucleic particle with 7SK snRNA and HEXIM1. Active P‐TEFb is bound instead to bromodomain‐containing protein 4 (Brd4) and stimulates elongation through phosphorylation of the pol II carboxyl‐terminal domain (CTD) on Ser2 and two negative transcription elongation factors: DRB sensitivity‐inducing factor (DSIF) and negative elongation factor (NELF). Phosphorylation converts DSIF from a repressor to an activator, whereas phosphorylated NELF leaves the elongation complex.
Table 1.
Viral factors that interact with P‐TEFb
| Virus | Viral factors that interact with P‐TEFb | References |
|---|---|---|
| Herpes simplex virus | ICP22 | 50, 142 |
| Human cytomegalovirus | pUL69 | 69, 70, 143 |
| IE2 86 | ||
| pUL97 | ||
| Epstein–Barr virus | EBNA2 | 75 |
| Human immunodeficiency virus | Tat | 144, 145 |
| Human T‐lymphotropic virus | Tax | 110, 111 |
| Human adenovirus | E1A | 117 |
| Influenza A virus | vRNP | 121 |
| Dengue virus | DENV core protein | 129 |
| Kaposi's sarcoma‐associated virus | K‐cyclin | 59 |
P‐TEFb, positive transcription elongation factor b; ICP22, immediate‐early protein 22; EBNA2, Epstein–Barr nuclear antigen 2; vRNP, viral RNA‐dependent RNA polymerases.
The studies reviewed here present examples of DNA and RNA viruses that subvert the host cell CTD kinase CDK9 for their own needs. Understanding the requirements of CDK9 for viral infection and how viral infection alters pol II CTD phosphorylation patterns not only advances our knowledge of viral pathogenesis but also provides potential new anti‐viral drug targets.
The α‐herpesvirus ICP22 protein has an intimate relationship with P‐TEFb
After infection, herpes simplex virus (HSV‐1 and HSV‐2) goes through a lytic replicative phase and causes localized lesions. This is followed by a latent phase where viruses reside in sensory neurons and can be frequently reactivated by a range of stresses, including high temperature and ultraviolet light 39. Although HSV infections are generally self‐limiting in healthy individuals, they can cause cancer, inflammation of the brain and the eye, and pose a significant mortality risk in infants and immuno‐compromised adults 40, 41. The HSV immediate‐early protein 22 (ICP22) is encoded by the US1 gene and comprises 420 amino acid residues. Viral deletion mutants without the US1 gene have a reduced capacity to establish latency in animal models 42, 43. In addition, deletion of this gene causes viral growth defects in some cell lines 42. During HSV‐1 productive infection, three classes of genes are sequentially expressed; first, the immediate‐early genes (α‐genes), next, the early genes (β‐genes), and finally, the late genes (γ‐genes) 44, 45. It has been demonstrated that ICP22 represses transcription of the viral α‐genes, β‐genes and γ‐genes by blocking the recruitment of P‐TEFb to their promoters. In contrast, the herpes viral protein 16 (VP16) overcomes the inhibitory effects of ICP22 on α‐gene transcription 46. It has been demonstrated that ICP22 alters the CTD phosphorylation state of pol II 47. ICP22 mediates two distinct effects on pol II: the induction of an intermediately migrating form of pol II (pol IIi) and the loss of pol II forms phosphorylated on Ser2 47, 48. Pol IIi replaces the normal hyperphosphorylated form of pol II in HSV‐1 infected cells. The induction of pol IIi requires ICP22 and another viral factor, UL13 49. A recent study demonstrated that ICP22 interacts directly with cellular CDK9 and that a short segment of ICP22 (residue 193‐256) is sufficient for this interaction 50. However, this short segment does not interact with pol II, unlike the full length of ICP22. Nevertheless, ectopic expression of either full length of ICP22 or amino acids residues 193‐256, leads to the loss of Ser2 CTD phosphorylation and subsequently to the inhibition of transcriptional elongation of host cell genes 50. These findings suggest a model where ICP22 is recruited to the transcriptionally active host genes soon after infection, where its interaction with P‐TEFb inhibits the kinase activity of CDK9, resulting in the loss of productive elongation (Fig. 2). Lytic infection with HSV‐1 also causes a transcription termination defects on host cell genes 51. Termination of transcription of the viral genes is however, efficient, highlighting that HSV‐1 proteins regulate transcription to maximize expression of the viral genome while down‐regulating expression of the host genome. Shutting down host cell gene expression could benefit the virus by, for example, helping to evade anti‐viral responses including production of type I interferon 52. Interaction of ICP22 with the CDK9 target, p53, is also important for efficient HSV‐1 replication 53.
Figure 2.
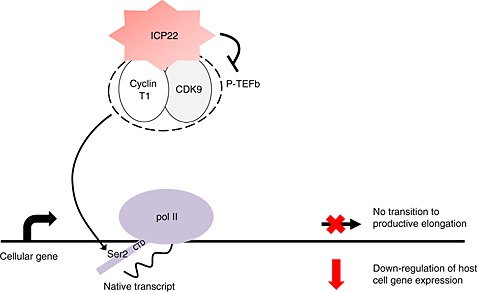
Immediate‐early protein 22 (ICP22) inhibits CDK9. HSV ICP22 interacts with cyclin‐dependent kinase 9 (CDK9). This results in the loss of Ser2 phosphorylation of the carboxyl‐terminal domain (CTD) of pol II and inhibition of transcription elongation of host cell genes. P‐TEFb, positive transcription elongation factor b.
KSHV factors regulate P‐TEFb activity towards pol II and p53
Kaposi sarcoma‐associated herpesvirus (KSHV) is also known as human herpesvirus 8. KSHV causes Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma 54, 55, 56. K‐cyclin, encoded by the virus, can interact and activate CDK6 and can trigger apoptosis in cells with high levels of this cyclin 57, 58. In the search for new K‐cyclin partners, CDK9 was identified as a new interacting CDK. In addition, it was demonstrated that K‐cyclin stimulates CDK9 kinase activity towards p53, and that CDK9 is required for K‐cyclin‐induced p53‐dependent growth suppression 59. However, the role of CDK9 in KSHV replication goes beyond its interaction with K‐cyclin. The replication and transcription activator (RTA) of KSHV, K‐RTA, regulates the reactivation of KSHV from a latent state. As demonstrated by an in vitro kinase assay, RTA is a substrate of CDK9. Furthermore, CDK9 inhibitors suppress the expression of various K‐RTA target genes. This further suggests that CDK9 inhibitors could be used to disturb KSHV replication 60. Interestingly, transcription elongation of KSHV lytic genes is paused during latency and can be reactivated in an RTA‐independent manner. Chip‐on‐chip analysis revealed that during KSHV latent infection, pol II transcription complexes are associated with NELF at the promoters of a group of lytic genes including OriLytL, K5, K6, and K7 (Fig. 3). The CTD of pol II at these promoters is hyperphosphorylated on Ser5 and hypophosphorylated of Ser2. It was hypothesized that hypophosphorylation of Ser2 might be due to the recruitment of enzymatically inactive P‐TEFb to promoters 61. Therefore, the negative control of transcriptional elongation of lytic gene expression by modulating CDK9 activity during KSHV latency could be a key regulatory mechanism.
Figure 3.
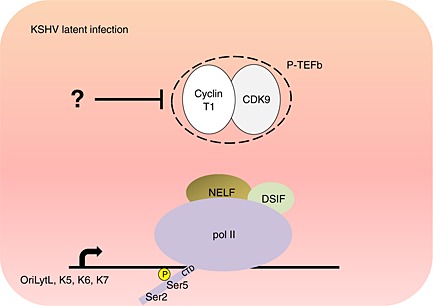
Pol II transcription complexes are paused at the promoters of Kaposi sarcoma‐associated herpesvirus (KSHV) lytic genes during latent infection. The action of negative elongation factor (NELF) and inhibition of cyclin‐dependent kinase 9 (CDK9) prevents expression of KSHV lytic genes OriLytL, K5, K6, and K7 during latency. Inhibition of CDK9 prevents phosphorylation of Ser2 of the pol II carboxyl‐terminal domain (CTD), whereas Ser5 is hyperphosphorylated.
P‐TEFb is a key player in HCMV trancriptosomes
The β‐herpesvirus human cytomegalovirus (HCMV), also known as human herpesvirus 5, infects 50–90% of the human population 62. HCMV infections are the leading viral cause of birth defects and pose a significant mortality risk in immunosuppressed individuals. In addition, HCMV may contribute to atherosclerosis and restenosis after coronary atherectomy 62, 63. HCMV infection results in the formation of viral transcriptosomes in the nucleus 64, 65, 66. These are the sites of transcription of the viral immediate‐early (IE) genes to which various viral and cellular factors are recruited 67. Cellular transcription regulators present at these sites include pol II, CDK9, cyclin T1, CDK7, and Brd4 65, 66, 68. It is well documented that HCMV infection affects the level and kinase activity of the recruited CDKs 68. In addition, recruitment of CDK9 to transcriptosomes during lytic infection results in hyperphosphorylation of the pol II CTD 66, 68, 69. Increased CTD phosphorylation might provide docking sites for processing factors regulating the alternative splicing of the primary HCMV transcripts 68. In addition, P‐TEFb activity is associated with the regulatory functions of HCMV‐encoded proteins pUL69, IE2‐86, and pUL97 67, 69, 70 and thus appears to be important for HCMV replication.
P‐TEFb is critical for immortalization of EBV‐infected cells
The Epstein–Barr virus (EBV), a γ‐herpesvirus, is another important human pathogen, and seropositivity to EBV is estimated to be >90% in adults 71. EBV is able to infect and immortalize human B‐cells and is the causative agent of infectious mononucleosis 72, 73. It has been also associated with several cancers, including Burkitt lymphoma and subsets of Hodgkin's lymphomas and T‐cell lymphomas 74. One of the viral genes translated during latency and required for B‐cell transformation and proliferation of infected cells is Epstein–Barr nuclear antigen 2 (EBNA2) 75. EBNA2 is an activator of viral and cellular transcription, and P‐TEFb is required for its activity 76, 77, 78. EBNA2 activation is sensitive to inhibition by a dominant negative mutant of CDK9 and the CDK9 inhibitor DRB. In addition, EBNA2 promotes Ser5 phosphorylation of the pol II CTD 78. EBNA2 activates transcription from the viral C promoter (Cp) generating a long primary transcript encoding nuclear antigens necessary for immortalization of the host cells. In addition, EBNA2 activates the promoters of viral latent membrane protein genes (LMP1, LMP2A, and LMP2B) 79, 80, 81. Recently, Palermo et al. (2011) demonstrated that Cp directs the buildup of a high level of stalled pol II together with the pausing factors DSIF and NELF. The stalled pol II maintains a nucleosome‐depleted region as measured by chromatin immunoprecipitation for histone H3. Moreover, stalled pol II increases recruitment of Brd4‐associated P‐TEFb to drive high levels of Ser2 phosphorylation of the pol II CTD and facilitate productive elongation 82. These results highlight a key role for P‐TEFb in EBNA2‐dependent transcription activation and the immortalization of EBV‐infected cells.
HIV Tat effectively hijacks P‐TEFb
The human immunodeficiency virus (HIV) is the causative agent of acquired immunodeficiency syndrome 83. The main target cells for HIV are CD4+ T lymphocytes and macrophages 84, 85. The most extensively studied in vivo latent reservoir is found within memory CD4+ T cells 86. HIV can also infect dendritic cells 87. Activation of latent viruses occurs at the level of elongation of transcription and requires binding of the HIV‐encoded elongation factor (Tat) to the transactivation response element at the start of HIV transcripts. The CDK9/cyclin T1 complex is an important factor for productive elongation of transcription of HIV genomes, and the inhibition of CDK9 can ameliorate HIV‐induced disease in animal models 88. In cultured human cells, dominant negative CDK9 mutants impair HIV‐1 replication 89.
Multiple levels of regulation of P‐TEFb impact HIV transcription. For example, P‐TEFb activity can be regulated by phosphorylation/dephosphorylation of the CDK9 subunit 90, and phosphatases involved in this process have been implicated in regulating HIV transcription 91, 92. HIV Tat targets CDK9/cyclin T1 to transactivation response element to stimulate transcription elongation through P‐TEFb‐dependent phosphorylation of NELF, DSIF, and Ser2 of pol II CTD 93, 94, 95. Tat competes with HEXIM1 for binding to cyclin T1 to promote the dissociation of P‐TEFb from the large inactive complex 96. In order to further stimulate viral gene expression, Tat recruits additional host factors including the human SEC (Fig. 4). SEC contains a set of factors implicated in regulation of pol II transcription elongation, and its core components include the scaffold proteins AFF1 or AFF4, P‐TEFb, elongation stimulatory factors ELL1 or ELL2, and transcription factors ENL and AF9 97, 98, 99, 100, 101. The function of the SEC is to stimulate elongation of transcription by increasing the processivity of pol II 98. A recent study established that the AFF1‐containing form of the SEC is more potent in supporting HIV‐1 transactivation than AFF4‐SEC 101. The ELL1/ELL2 components of the SEC promote Tat transactivation by suppressing pol II pausing and ELL2 is more effective than ELL1 in Tat transactivation 98.
Figure 4.
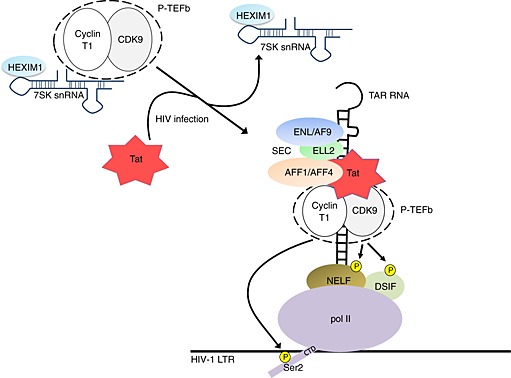
Human immunodeficiency virus type 1 (HIV‐1) Transactivator of transcription (Tat) stimulates transcription elongation by recruiting positive transcription elongation factor b (P‐TEFb). The HIV‐encoded Tat extracts the P‐TEFb from the inactive complex containing the 7SK small nuclear ribonucleic particle (7SK snRNP), targets cyclin‐dependent kinase 9 (CDK9) and a cyclin T1 to the transactivation response element (TAR) and assembles the super elongation complex (SEC) to activate HIV transcription. P‐TEFb triggers productive elongation by phosphorylating negative elongation factor (NELF), DRB sensitivity‐inducing factor (DSIF), and Ser2 of carboxyl‐terminal domain (CTD) of pol II. LTR, long terminal repeat.
The crystal structure of Tat‐P‐TEFb 102 and the recently published Tat‐AFF4‐P‐TEFb crystal structure 103 provide insights into the mechanism of Tat activation. Tat interacts with both cyclin T and CDK9 and induces conformational changes in P‐TEFb upon binding. These structural studies can help in designing small‐molecule compounds that disrupt the Tat‐P‐TEFb and the Tat‐AFF4 interactions to specifically inhibit HIV transcription without affecting normal P‐TEFb function. The central role that P‐TEFb plays in Tat‐dependent activation of latent HIV genomes makes CDK9 an attractive target for the development of novel HIV therapeutics.
More P‐TEFb please for HTLV‐1 too
Human T‐lymphotropic virus type 1 (HTLV‐1) infections cause several human diseases such as adult T‐cell leukemia, the neurological disorder HTLV‐1‐associated myelopathy, and uveitis 104, 105, 106, 107. HTLV encodes a transcriptional transactivator protein called Tax, which functions as an activator of the long terminal repeat (LTR) promoter 108, 109. Similarly to HIV‐1 Tat, HTLV‐1 Tax influences transcription by pol II 110. P‐TEFb is essential for Tax transactivation in vivo and in vitro, and siRNA treatment to CDK9 and the CDK9 inhibitor flavopiridol both inhibit transactivation by Tax 110. In vitro binding studies demonstrate that Tax competes with Brd4 for P‐TEFb binding through direct interaction with cyclin T1. In addition, Tax overexpression decreases the amount of 7SK snRNA associated with P‐TEFb and specifically stimulates Ser2 phosphorylation of the pol II CTD 111. These events give rise to a functional Tax/P‐TEFb complex for viral LTR activation (Fig. 5). As Brd4 competes with Tax for P‐TEFb, LTR promoter activity is higher in HTLV‐1‐transformed Brd4‐deficient cells 112. Interestingly, a recent study showed that in common with HIV‐1 Tat transactivation, the ELL component of the pol II SEC is essential for Tax‐mediated transactivation. Tax enhances ELL incorporation into the histone acetyltransferase p300 and P‐TEFb transcriptional complexes. Depletion of ELL using an shRNA‐mediated approach abrogates Tax transactivation 113. Thus, the HTLV‐1 virus uses similar strategies to HIV‐1 to hijack cellular P‐TEFb.
Figure 5.
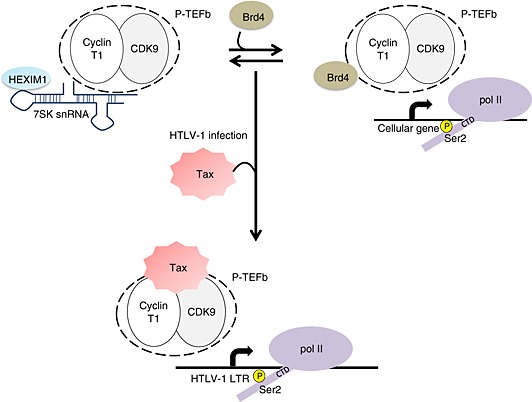
A dual function for Tax in modulating positive transcription elongation factor b (P‐TEFb) activity. Human T‐lymphotropic virus type 1 (HTLV‐1) transcriptional transactivator protein Tax disrupts the inactive 7SK snRNA/HEXIM1/P‐TEFb complex to create a novel Tax/P‐TEFb complex. Tax also acts as an antagonist of bromodomain‐containing the protein 4 (Brd4) and competes with Brd4 for interaction with P‐TEFb. The active Tax/P‐TEFb complex facilitates the phosphorylation of Ser2 of pol II carboxyl‐terminal domain (CTD) and enhances the activation of the HTLV‐1 long terminal repeat (LTR) promoter.
Inhibiting CDK9 blocks adenovirus replication
Human adenovirus (HAdV) is a double‐stranded DNA virus that causes upper and lower respiratory infections 114. The adenovirus large E1A (L‐E1A) protein is a transcriptional activator that functions through the action of an activation domain named “conserved region 3” (CR3). CR3 has been shown to recruit cellular transcription factors including mediator subunit 23 (Med23) 115, 116. Recent proteomic analysis in human cells infected with HAdv5 revealed that L‐E1A is associated with many subunits of the mammalian mediator complex 117. Interestingly, the presence of mediator subunits in the L‐E1A protein complex is greatly reduced in cells depleted of Med23, suggesting that Med23 links the transactivation domain CR3 with the mediator complex. The proteomic analysis also revealed that L‐E1A is associated with super elongation complex components, such as CDK9, cyclin T1, AFF4, ELL, and EAF. The catalytic constituent of the SEC, CDK9, was shown to be critical for the transcription of HAdv5 early genes by L‐E1A and HAdv5 replication 117. Interestingly, a newly developed CDK9 inhibitor, FIT‐039 (N‐[5‐fluoro‐2‐(1‐piperidinyl)phenyl]isonicotinthioamide), suppresses HAdV replication 118. This further suggests that HAdV hijacks P‐TEFb activity for viral‐specific transcription.
P‐TEFb influences influenza
The best‐studied member of the orthomyxovirus RNA virus family is the influenza A virus, which is responsible for acute respiratory diseases in humans. After infection with influenza A, viral RNA‐dependent RNA polymerases (vRNPs) are transported to the nucleus, where viral transcription takes place. vRNPs associate with the promoter region of protein‐coding genes with lower levels in the regions downstream of the promoter 119. This reflects the distribution of pol II in infected cells. In mock‐infected and influenza virus‐infected cells, there are similar levels of pol II associated with the promoter region of genes. However, in infected cells, there are decreased levels of pol II in coding regions suggesting that influenza virus infection inhibits pol II elongation 120. Zhang et al. 121 demonstrated that CDK9/cyclin T1 interacts with the vRNP of influenza A virus and facilitates its association with cellular pol II phosphorylated on Ser2 of the CTD. CDK9 interacts with three subunits of influenza virus vRNP, PB1, PB2, and PA, when they are ectopically expressed in cells. Interestingly, in contrast to HIV transcription, the kinase activity of CDK9 is not crucial for influenza virus transcription as overexpression of the dominant negative form of CDK9 does not reduce influenza virus transcription activity. In addition, the CDK9 inhibitor DRB does not affect viral transcription 120. However, siRNA‐mediated knockdown of cyclin T1 inhibits viral mRNA synthesis and overexpression of cyclin T1 promotes vRNP activity. It was suggested that interaction with P‐TEFb facilitates vRNP association with cellular pol II for cap‐snatching 121. Thus, P‐TEFb, but not its kinase activity, plays an important role in regulating expression of the influenza A virus.
P‐TEFb co‐operates with DENV‐C to activate an inflammatory response
Dengue virus (DENV) is a mosquito‐transmitted RNA virus and is one of the most common infectious pathogens worldwide 122. Viral infection results in dengue fever, dengue haemorrhagic fever, or dengue shock syndrome 123, 124. At present, an effective dengue vaccine or anti‐viral drug is not available. A notable issue that impedes dengue vaccine development is the limited understanding of the immunopathology of DENV infection. The DENV single‐stranded RNA molecule is about 11 kb in size. It encodes three structural proteins (core protein, non‐glycosylated membrane protein, and envelope protein) and seven non‐structural proteins 125. DENV infection is associated with increased levels of the inflammatory chemokines IL‐8, IFN‐γ, TNF‐α, and TNF‐β 126, 127, 128. A recent finding has shed new light of the role of P‐TEFb in the activation of IL‐8 gene expression during DENV infection 129. Li et al. 129 demonstrated that during DENV virus infection, P‐TEFb associates with DENV core protein (DENV‐C) to induce IL‐8 expression. DRB treatment or siRNA‐mediated knockdown of cyclin T1 prior to DENV infection abolishes the increase of IL‐8 induction. P‐TEFb and DENV‐C core protein co‐localize in vivo and co‐immunoprecipitate in DENV‐infected cells, and P‐TEFb and DENV core proteins are recruited to the IL‐8 gene. These findings further implicate the association of P‐TEFb with DENV‐C in altering host gene expression. The IL‐8 gene promoter contains sites for the transcriptional activator NF‐κB. Interestingly, in DENV‐infected cells, the DENV‐C protein appears to promote NF‐κB‐dependent activation only in the presence of P‐TEFb 129. The benefit to the virus of activating inflammation is unclear. However, the requirement of P‐TEFb association with DENV‐C to induce IL‐8 expression in DENV‐infected cells emphasizes the importance of P‐TEFb in DENV immunopathogenesis and suggests that P‐TEFb could be a valid drug target to treat some of the symptoms of DENV infection.
CDK9 inhibitors as anti‐viral agents
The high mutation rate of viruses facilitates the emergence of strains resistant to drugs that target the virus directly, which represents a public health risk 130. In addition to combating viral drug resistance, the alternative strategy of targeting cellular proteins used by viruses increases the potential range of targets and could yield drugs active against several viruses. Given the importance of P‐TEFb for the replication of herpesviruses, HIV, HTLV, HAdV, influenza A virus and DENV, CDK9 and cyclin T are obvious potential drug targets. Drugs have already been developed that target P‐TEFb for the treatment of disease. For example, CDK9 inhibitors are currently in clinical trials for the treatment of human malignancies 131.
Given the regulation of CDK9 by viral proteins during HSV, EBV, HCMV, and KSHV infections, the potential use of CDK9 inhibitors as anti‐herpesvirus agents is particularly appealing. Acyclovir and its derivatives are effective, widely available, and have greatly reduced the burden of disease. However, the resistance to these drugs is becoming a major public health problem. The growing resistance is due to their frequent use in suppressive therapy and prophylaxis of HSV infection 132. There is therefore a pressing need for the identification of potential herpesvirus drug targets to facilitate the development of the next generation of anti‐herpesvirus drug. One of the most selective, non‐competitive inhibitors of CDK9 is flavopiridol 133. It was demonstrated that flavopiridol can suppress the replication of HSV, HCMV, HAdV, and HIV 88, 134. In HeLa cells, the anti‐HIV activity of CDK9 inhibitors, including flavopiridol, correlates with a dose‐dependent loss of the large form of P‐TEFb and reduction in HIV‐1 infectivity. Unfortunately, studies in primary cell cultures indicate that cytotoxicity of the drugs used is a major drawback 135. Interestingly, studies by Salerno et al. 136 demonstrated that effects of dominant negative CDK9 and flavopiridol are not equivalent. However, the next generation of small pharmacological compounds targeting CDK9 includes some promising anti‐HIV therapeutic agents.
Roscovitine, a purine‐derived CDK inhibitor, inhibits replication of HSV, HCMV, and HIV 137, 138, 139. Roscovitine also reduces tumor size and plasma EBV DNA in patients with nasopharyngeal carcinoma 140. Unfortunately, these inhibitors affect a wide range of CDKs and often negatively affect cell cycle progression 141. It was recently demonstrated that R22, a novel selective CDK9 inhibitor, possess anti‐cytomegaloviral activity 67. In addition, a new CDK9 inhibitor (FIT‐039) that exhibits an anti‐viral effect on DNA and RNA viruses was identified 118. It was shown to inhibit the replication of DNA viruses such as HSV‐1, HSV‐2, HAdV, and HCMV in cultured cells. Furthermore, FIT‐039 also suppresses the replication of influenza virus H1N1 and of HIV. Interestingly, FIT‐039 has a less significant effect on the cellular transcriptome than flavopiridol, and it does not affect cellular proliferation 118. It is therefore a prospective drug that affects a broad spectrum of viruses.
Concluding remarks
P‐TEFb is an important regulator of cellular processes, and it is functionally integrated into the expression of many human viruses. In particular, the activity of CDK9 is crucial for the course of HSV, HCMV, EBV, HIV, HTLV, HAdV, DENV, and KSHV virus infection.
P‐TEFb is therefore a key player not only in cellular processes but also in viral biology (Fig. 6). Either by modifying its activity or regulating the amount of active and inactive complexes of P‐TEFb and/or physical interaction with components of P‐TEFb, viruses have evolved strategies to hijack this key factor via their own regulatory proteins and P‐TEFb functions as a central player in virus‐host interaction to facilitate the viral replication cycle. Importantly, studies of viruses that use P‐TEFb as a specific factor for efficient transcription will also help to elucidate the molecular mechanisms controlling transcription in uninfected cells.
Figure 6.
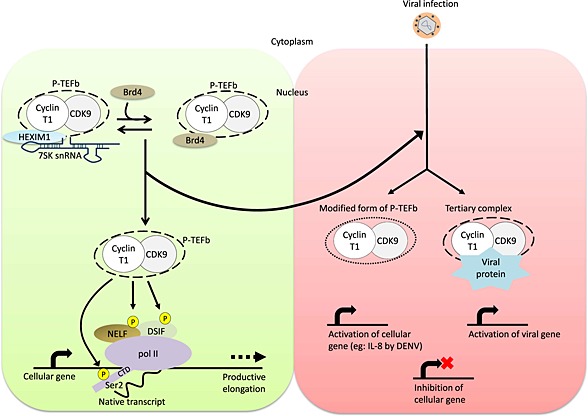
Positive transcription elongation factor b (P‐TEFb) as a master regulator of cellular and viral transcription. In uninfected cells (left panel), the negative elongation factor (NELF) and the DRB sensitivity‐inducing factor (DSIF) enhance pol II stalling. Subsequent recruitment of P‐TEFb allows phosphorylation of DSIF, NELF, and Ser2 of carboxyl‐terminal domain (CTD) of pol II, which leads to productive elongation. In the context of virus‐infected cells (right panel), various viral factors affect transcription by modulating or recruiting the activity of P‐TEFb. Consequently, the cellular genes are inhibited, whereas the viral genes become activated.
Supporting information
Supporting info item
{kind=link}
Supporting info item
Supporting info item
Acknowledgements
We would like to thank Michael Tellier, Ivan Ferrer‐Vicens, and Gabriel Klobusovsky for discussions and critical comments. This work was supported by Wellcome Trust (WT106134A) and Sir Edward Penley Abraham Trust Fund grants (EPA‐RF202) to SM and a scholarship from the Malaysian Government to NFI.
Zaborowska, J. , Isa, N. F. , and Murphy, S. (2016) P‐TEFb goes viral. Inside the Cell, 1: 106–116. doi: 10.1002/icl3.1037.
Footnotes
Thomas et al. 146 have shown that the NSs protein of Bunyamwera virus (BUNV) can also affect phosphorylation of Ser2 of the pol II CTD. It is therefore possible that NSs also interacts with P‐TEFb.
References
- 1. Spencer CA, Dahmus ME, Rice SA. 1997. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J Virol 71: 2031–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cramer P. 2002. Common structural features of nucleic acid polymerases. Bioessays 24: 724–9. [DOI] [PubMed] [Google Scholar]
- 3. Corden JL. 1990. Tails of RNA polymerase II. Trends Biochem Sci 15: 383–7. [DOI] [PubMed] [Google Scholar]
- 4. McCracken S, Fong N, Yankulov K, Ballantyne S, et al. 1997. The C‐terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 385: 357–61. [DOI] [PubMed] [Google Scholar]
- 5. Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bentley DL. 2005. Rules of engagement: co‐transcriptional recruitment of pre‐mRNA processing factors. Curr Opin Cell Biol 17: 251–6. [DOI] [PubMed] [Google Scholar]
- 7. Phatnani HP, Greenleaf AL. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev 20: 2922–36. [DOI] [PubMed] [Google Scholar]
- 8. Buratowski S. 2003. The CTD code. Nat Struct Biol 10: 679–80. [DOI] [PubMed] [Google Scholar]
- 9. Saunders A, Core LJ, Lis JT. 2006. Breaking barriers to transcription elongation. Nat Rev Mol Cell Biol 7: 557–67. [DOI] [PubMed] [Google Scholar]
- 10. Egloff S, Murphy S. 2008. Cracking the RNA polymerase II CTD code. Trends Genet 24: 280–8. [DOI] [PubMed] [Google Scholar]
- 11. Egloff S, Dienstbier M, Murphy S. 2012. Updating the RNA polymerase CTD code: adding gene‐specific layers. Trends Genet 28: 333–41. [DOI] [PubMed] [Google Scholar]
- 12. Eick D, Geyer M. 2013. The RNA polymerase II carboxy‐terminal domain (CTD) code. Chem Rev 113: 8456–90. [DOI] [PubMed] [Google Scholar]
- 13. Yamaguchi Y, Takagi T, Wada T, Yano K, et al. 1999. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97: 41–51. [DOI] [PubMed] [Google Scholar]
- 14. Yamada T, Yamaguchi Y, Inukai N, Okamoto S, et al. 2006. P‐TEFb‐mediated phosphorylation of hSpt5 C‐terminal repeats is critical for processive transcription elongation. Mol Cell 21: 227–37. [DOI] [PubMed] [Google Scholar]
- 15. Marshall NF, Price DH. 1995. Purification of P‐TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 270: 12335–8. [DOI] [PubMed] [Google Scholar]
- 16. Marshall NF, Price DH. 1992. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol 12: 2078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Claudio PP, Cui J, Ghafouri M, Mariano C, et al. 2006. Cdk9 phosphorylates p53 on serine 392 independently of CKII. J Cell Physiol 208: 602–12. [DOI] [PubMed] [Google Scholar]
- 18. Radhakrishnan SK, Gartel AL. 2006. CDK9 phosphorylates p53 on serine residues 33, 315 and 392. Cell Cycle 5: 519–21. [DOI] [PubMed] [Google Scholar]
- 19. Grana X, De Luca A, Sang N, Fu Y, et al. 1994. PITALRE, a nuclear CDC2‐related protein kinase that phosphorylates the retinoblastoma protein in vitro . Proc Natl Acad Sci U S A 91: 3834–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garriga J, Segura E, Mayol X, Grubmeyer C, et al. 1996. Phosphorylation site specificity of the CDC2‐related kinase PITALRE. Biochem J 320: 983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagella L, MacLachlan TK, Buono RJ, Pisano MM, et al. 1998. Cloning of murine CDK9/PITALRE and its tissue‐specific expression in development. J Cell Physiol 177: 206–13. [DOI] [PubMed] [Google Scholar]
- 22. Shore SM, Byers SA, Maury W, Price DH. 2003. Identification of a novel isoform of Cdk9. Gene 307: 175–82. [DOI] [PubMed] [Google Scholar]
- 23. Liu H, Herrmann CH. 2005. Differential localization and expression of the Cdk9 42 k and 55 k isoforms. J Cell Physiol 203: 251–60. [DOI] [PubMed] [Google Scholar]
- 24. Fu TJ, Peng J, Lee G, Price DH, et al. 1999. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J Biol Chem 274: 34527–30. [DOI] [PubMed] [Google Scholar]
- 25. Zhou Q, Yik JH. 2006. The Yin and Yang of P‐TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol Mol Biol Rev 70: 646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Luca A, De Falco M, Baldi A, Paggi MG. 2003. Cyclin T: three forms for different roles in physiological and pathological functions. J Cell Physiol 194: 101–7. [DOI] [PubMed] [Google Scholar]
- 27. Bezstarosti K, Ghamari A, Grosveld FG, Demmers JA. 2010. Differential proteomics based on 18O labeling to determine the cyclin dependent kinase 9 interactome. J Proteome Res 9: 4464–75. [DOI] [PubMed] [Google Scholar]
- 28. Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, et al. 2011. The cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 25: 2158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yik JH, Chen R, Nishimura R, Jennings JL, et al. 2003. Inhibition of P‐TEFb (CDK9/cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol Cell 12: 971–82. [DOI] [PubMed] [Google Scholar]
- 30. Michels AA, Fraldi A, Li Q, Adamson TE, et al. 2004. Binding of the 7SK snRNA turns the HEXIM1 protein into a P‐TEFb (CDK9/cyclin T) inhibitor. EMBO J 23: 2608–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Q, Price JP, Byers SA, Cheng D, et al. 2005. Analysis of the large inactive P‐TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P‐TEFb molecules containing Cdk9 phosphorylated at threonine 186. J Biol Chem 280: 28819–26. [DOI] [PubMed] [Google Scholar]
- 32. Markert A, Grimm M, Martinez J, Wiesner J, et al. 2008. The La‐related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes. EMBO Rep 9: 569–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nguyen VT, Kiss T, Michels AA, Bensaude O. 2001. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414: 322–5. [DOI] [PubMed] [Google Scholar]
- 34. Yang Z, Zhu Q, Luo K, Zhou Q. 2001. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414: 317–22. [DOI] [PubMed] [Google Scholar]
- 35. Barboric M, Lenasi T, Chen H, Johansen EB, et al. 2009. 7SK snRNP/P‐TEFb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc Natl Acad Sci U S A 106: 7798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhou Q, Li T, Price DH. 2012. RNA polymerase II elongation control. Annu Rev Biochem 81: 119–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jang MK, Mochizuki K, Zhou M, Jeong HS, et al. 2005. The bromodomain protein Brd4 is a positive regulatory component of P‐TEFb and stimulates RNA polymerase II‐dependent transcription. Mol Cell 19: 523–34. [DOI] [PubMed] [Google Scholar]
- 38. Yang Z, Yik JH, Chen R, He N, et al. 2005. Recruitment of P‐TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol Cell 19: 535–45. [DOI] [PubMed] [Google Scholar]
- 39. Field HJ, Vere Hodge RA. 2013. Recent developments in anti‐herpesvirus drugs. Br Med Bull 106: 213–49. [DOI] [PubMed] [Google Scholar]
- 40. Cherpes TL, Matthews DB, Maryak SA. 2012. Neonatal herpes simplex virus infection. Clin Obstet Gynecol 55: 938–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Herget GW, Riede UN, Schmitt‐Graff A, Lubbert M, et al. 2005. Generalized herpes simplex virus infection in an immunocompromised patient—report of a case and review of the literature. Pathol Res Pract 201: 123–9. [DOI] [PubMed] [Google Scholar]
- 42. Sears AE, Halliburton IW, Meignier B, Silver S, et al. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J Virol 55: 338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Poffenberger KL, Idowu AD, Fraser‐Smith EB, Raichlen PE, et al. 1994. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo . Arch Virol 139: 111–9. [DOI] [PubMed] [Google Scholar]
- 44. Weir JP. 2001. Regulation of herpes simplex virus gene expression. Gene 271: 117–30. [DOI] [PubMed] [Google Scholar]
- 45. Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guo L, Wu WJ, Liu LD, Wang LC, et al. 2012. Herpes simplex virus 1 ICP22 inhibits the transcription of viral gene promoters by binding to and blocking the recruitment of P‐TEFb. PLoS One 7: e45749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fraser KA, Rice SA. 2007. Herpes simplex virus immediate‐early protein ICP22 triggers loss of serine 2‐phosphorylated RNA polymerase II. J Virol 81: 5091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rice SA, Long MC, Lam V, Spencer CA. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J Virol 68: 988–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Long MC, Leong V, Schaffer PA, Spencer CA, et al. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus‐induced modification of the large subunit of RNA polymerase II. J Virol 73: 5593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zaborowska J, Baumli S, Laitem C, O'Reilly D, et al. 2014. Herpes Simplex Virus 1 (HSV‐1) ICP22 protein directly interacts with cyclin‐dependent kinase (CDK)9 to inhibit RNA polymerase II transcription elongation. PLoS One 9: e107654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rutkowski AJ, Erhard F, L'Hernault A, Bonfert T, et al. 2015. Widespread disruption of host transcription termination in HSV‐1 infection. Nat Commun 6: 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mostafa HH, Davido DJ. 2013. Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation. J Virol 87: 13510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maruzuru Y, Fujii H, Oyama M, Kozuka‐Hata H, et al. 2013. Roles of p53 in herpes simplex virus 1 replication. J Virol 87: 9323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang Y, Cesarman E, Pessin MS, Lee F, et al. 1994. Identification of herpesvirus‐like DNA sequences in AIDS‐associated Kaposi's sarcoma. Science 266: 1865–9. [DOI] [PubMed] [Google Scholar]
- 55. Soulier J, Grollet L, Oksenhendler E, Cacoub P, et al. 1995. Kaposi's sarcoma‐associated herpesvirus‐like DNA sequences in multicentric Castleman's disease. Blood 86: 1276–80. [PubMed] [Google Scholar]
- 56. Nador RG, Cesarman E, Chadburn A, Dawson DB, et al. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma‐associated herpes virus. Blood 88: 645–56. [PubMed] [Google Scholar]
- 57. Godden‐Kent D, Talbot SJ, Boshoff C, Chang Y, et al. 1997. The cyclin encoded by Kaposi's sarcoma‐associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol 71: 4193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ojala PM, Tiainen M, Salven P, Veikkola T, et al. 1999. Kaposi's sarcoma‐associated herpesvirus‐encoded v‐cyclin triggers apoptosis in cells with high levels of cyclin‐dependent kinase 6. Cancer Res 59: 4984–9. [PubMed] [Google Scholar]
- 59. Chang PC, Li M. 2008. Kaposi's sarcoma‐associated herpesvirus K‐cyclin interacts with Cdk9 and stimulates Cdk9‐mediated phosphorylation of p53 tumor suppressor. J Virol 82: 278–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tsai WH, Wang PW, Lin SY, Wu IL, et al. 2012. Ser‐634 and Ser‐636 of Kaposi's sarcoma‐associated herpesvirus RTA are involved in transactivation and are potential cdk9 phosphorylation Sites. Front Microbiol 3: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toth Z, Brulois KF, Wong LY, Lee HR, et al. 2012. Negative elongation factor‐mediated suppression of RNA polymerase II elongation of Kaposi's sarcoma‐associated herpesvirus lytic gene expression. J Virol 86: 9696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fortunato EA, McElroy AK, Sanchez I, Spector DH. 2000. Exploitation of cellular signaling and regulatory pathways by human cytomegalovirus. Trends Microbiol 8: 111–9. [DOI] [PubMed] [Google Scholar]
- 63. Zhou YF, Leon MB, Waclawiw MA, Popma JJ, et al. 1996. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med 335: 624–30. [DOI] [PubMed] [Google Scholar]
- 64. Ahn JH, Jang WJ, Hayward GS. 1999. The human cytomegalovirus IE2 and UL112‐113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein‐associated nuclear bodies (PODs or ND10). J Virol 73: 10458–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ishov AM, Stenberg RM, Maul GG. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J Cell Biol 138: 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kapasi AJ, Spector DH. 2008. Inhibition of the cyclin‐dependent kinases at the beginning of human cytomegalovirus infection specifically alters the levels and localization of the RNA polymerase II carboxyl‐terminal domain kinases CDK9 and CDK7 at the viral transcriptosome. J Virol 82: 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Feichtinger S, Stamminger T, Muller R, Graf L, et al. 2011. Recruitment of cyclin‐dependent kinase 9 to nuclear compartments during cytomegalovirus late replication: importance of an interaction between viral pUL69 and cyclin T1. J Gen Virol 92: 1519–31. [DOI] [PubMed] [Google Scholar]
- 68. Tamrakar S, Kapasi AJ, Spector DH. 2005. Human cytomegalovirus infection induces specific hyperphosphorylation of the carboxyl‐terminal domain of the large subunit of RNA polymerase II that is associated with changes in the abundance, activity, and localization of CDK9 and CDK7. J Virol 79: 15477–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kapasi AJ, Clark CL, Tran K, Spector DH. 2009. Recruitment of cdk9 to the immediate‐early viral transcriptosomes during human cytomegalovirus infection requires efficient binding to cyclin T1, a threshold level of IE2 86, and active transcription. J Virol 83: 5904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Graf L, Webel R, Wagner S, Hamilton ST, et al. 2013. The cyclin‐dependent kinase ortholog pUL97 of human cytomegalovirus interacts with cyclins. Viruses 5: 3213–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Richman DD, Whitley RJ, Hayden FG. 2009. Clinical virology. Washington, D.C.: ASM Press. [Google Scholar]
- 72. Young LS, Rickinson AB. 2004. Epstein–Barr virus: 40 years on. Nat Rev Cancer 4: 757–68. [DOI] [PubMed] [Google Scholar]
- 73. Henle W, Henle G. 1980. Epidemiologic aspects of Epstein–Barr virus (EBV)‐associated diseases. Ann N Y Acad Sci 354: 326–31. [DOI] [PubMed] [Google Scholar]
- 74. Damania B. 2007. DNA tumor viruses and human cancer. Trends Microbiol 15: 38–44. [DOI] [PubMed] [Google Scholar]
- 75. Palermo RD, Webb HM, Gunnell A, West MJ. 2008. Regulation of transcription by the Epstein–Barr virus nuclear antigen EBNA 2. Biochem Soc Trans 36: 625–8. [DOI] [PubMed] [Google Scholar]
- 76. Cohen JI, Kieff E. 1991. An Epstein–Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J Virol 65: 5880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cohen JI, Wang F, Kieff E. 1991. Epstein–Barr virus nuclear protein 2 mutations define essential domains for transformation and transactivation. J Virol 65: 2545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bark‐Jones SJ, Webb HM, West MJ. 2006. EBV EBNA 2 stimulates CDK9‐dependent transcription and RNA polymerase II phosphorylation on serine 5. Oncogene 25: 1775–85. [DOI] [PubMed] [Google Scholar]
- 79. Abbot SD, Rowe M, Cadwallader K, Ricksten A, et al. 1990. Epstein‐Barr virus nuclear antigen 2 induces expression of the virus‐encoded latent membrane protein. J Virol 64: 2126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang F, Tsang SF, Kurilla MG, Cohen JI, et al. 1990. Epstein–Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J Virol 64: 3407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Woisetschlaeger M, Jin XW, Yandava CN, Furmanski LA, et al. 1991. Role for the Epstein–Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc Natl Acad Sci U S A 88: 3942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Palermo RD, Webb HM, West MJ. 2011. RNA polymerase II stalling promotes nucleosome occlusion and pTEFb recruitment to drive immortalization by Epstein‐Barr virus. PLoS Pathog 7: e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Romano G, Massi D, Giordano A. 1998. The standpoint of AIDS research and therapy programs. Anticancer Res 18: 2763–78. [PubMed] [Google Scholar]
- 84. Venzke S, Keppler OT. 2006. Role of macrophages in HIV infection and persistence. Expert Rev Clin Immunol 2: 613–26. [DOI] [PubMed] [Google Scholar]
- 85. Chavez L, Calvanese V, Verdin E. 2015. HIV Latency is established directly and early in both resting and activated primary CD4 T cells. PLoS Pathog 11: e1004955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chomont N, El‐Far M, Ancuta P, Trautmann L, et al. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15: 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Altfeld M, Fadda L, Frleta D, Bhardwaj N. 2011. DCs and NK cells: critical effectors in the immune response to HIV‐1. Nat Rev Immunol 11: 176–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nelson PJ, D'Agati VD, Gries JM, Suarez JR, et al. 2003. Amelioration of nephropathy in mice expressing HIV‐1 genes by the cyclin‐dependent kinase inhibitor flavopiridol. J Antimicrob Chemother 51: 921–9. [DOI] [PubMed] [Google Scholar]
- 89. Fujinaga K, Irwin D, Geyer M, Peterlin BM. 2002. Optimized chimeras between kinase‐inactive mutant CDK9 and truncated cyclin T1 proteins efficiently inhibit Tat transactivation and human immunodeficiency virus gene expression. J Virol 76: 10873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang Y, Dow EC, Liang YY, Ramakrishnan R, et al. 2008. Phosphatase PPM1A regulates phosphorylation of Thr‐186 in the Cdk9 T‐loop. J Biol Chem 283: 33578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ammosova T, Washington K, Debebe Z, Brady J, et al. 2005. Dephosphorylation of CDK9 by protein phosphatase 2A and protein phosphatase‐1 in Tat‐activated HIV‐1 transcription. Retrovirology 2: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nekhai S, Jerebtsova M, Jackson A, Southerland W. 2007. Regulation of HIV‐1 transcription by protein phosphatase 1. Curr HIV Res 5: 3–9. [DOI] [PubMed] [Google Scholar]
- 93. Wei P, Garber ME, Fang SM, Fischer WH, et al. 1998. A novel CDK9‐associated C‐type cyclin interacts directly with HIV‐1 Tat and mediates its high‐affinity, loop‐specific binding to TAR RNA. Cell 92: 451–62. [DOI] [PubMed] [Google Scholar]
- 94. Isel C, Karn J. 1999. Direct evidence that HIV‐1 Tat stimulates RNA polymerase II carboxyl‐terminal domain hyperphosphorylation during transcriptional elongation. J Mol Biol 290: 929–41. [DOI] [PubMed] [Google Scholar]
- 95. Peterlin BM, Price DH. 2006. Controlling the elongation phase of transcription with P‐TEFb. Mol Cell 23: 297–305. [DOI] [PubMed] [Google Scholar]
- 96. Barboric M, Yik JH, Czudnochowski N, Yang Z, et al. 2007. Tat competes with HEXIM1 to increase the active pool of P‐TEFb for HIV‐1 transcription. Nucleic Acids Res 35: 2003–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lin C, Smith ER, Takahashi H, Lai KC, et al. 2010. AFF4, a component of the ELL/P‐TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37: 429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. He N, Liu M, Hsu J, Xue Y, et al. 2010. HIV‐1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV‐1 transcription. Mol Cell 38: 428–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Chou S, Upton H, Bao K, Schulze‐Gahmen U, et al. 2013. HIV‐1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold. Proc Natl Acad Sci U S A 110: E123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lu H, Li Z, Xue Y, Zhou Q. 2013. Viral‐host interactions that control HIV‐1 transcriptional elongation. Chem Rev 113: 8567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lu H, Li Z, Zhang W, Schulze‐Gahmen U, et al. 2015. Gene target specificity of the super elongation complex (SEC) family: how HIV‐1 Tat employs selected SEC members to activate viral transcription. Nucleic Acids Res 43: 5868–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tahirov TH, Babayeva ND, Varzavand K, Cooper JJ, et al. 2010. Crystal structure of HIV‐1 Tat complexed with human P‐TEFb. Nature 465: 747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gu J, Babayeva ND, Suwa Y, Baranovskiy AG, et al. 2014. Crystal structure of HIV‐1 Tat complexed with human P‐TEFb and AFF4. Cell Cycle 13: 1788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kira J, Itoyama Y, Koyanagi Y, Tateishi J, et al. 1992. Presence of HTLV‐I proviral DNA in central nervous system of patients with HTLV‐I‐associated myelopathy. Ann Neurol 31: 39–45. [DOI] [PubMed] [Google Scholar]
- 105. Osame M, Usuku K, Izumo S, Ijichi N, et al. 1986. HTLV‐I associated myelopathy, a new clinical entity. Lancet 1: 1031–2. [DOI] [PubMed] [Google Scholar]
- 106. Yoshida M, Seiki M, Yamaguchi K, Takatsuki K. 1984. Monoclonal integration of human T‐cell leukemia provirus in all primary tumors of adult T‐cell leukemia suggests causative role of human T‐cell leukemia virus in the disease. Proc Natl Acad Sci U S A 81: 2534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Mochizuki M, Watanabe T, Yamaguchi K, Tajima K, et al. 1992. Uveitis associated with human T lymphotropic virus type I: seroepidemiologic, clinical, and virologic studies. J Infect Dis 166: 943–4. [DOI] [PubMed] [Google Scholar]
- 108. Bex F, Gaynor RB. 1998. Regulation of gene expression by HTLV‐I Tax protein. Methods 16: 83–94. [DOI] [PubMed] [Google Scholar]
- 109. Franklin AA, Nyborg JK. 1995. Mechanisms of Tax regulation of human T cell leukemia virus type I gene expression. J Biomed Sci 2: 17–29. [DOI] [PubMed] [Google Scholar]
- 110. Zhou M, Lu H, Park H, Wilson‐Chiru J, et al. 2006. Tax interacts with P‐TEFb in a novel manner to stimulate human T‐lymphotropic virus type 1 transcription. J Virol 80: 4781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cho WK, Zhou M, Jang MK, Huang K, et al. 2007. Modulation of the Brd4/P‐TEFb interaction by the human T‐lymphotropic virus type 1 tax protein. J Virol 81: 11179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cho WK, Jang MK, Huang K, Pise‐Masison CA, et al. 2010. Human T‐lymphotropic virus type 1 Tax protein complexes with P‐TEFb and competes for Brd4 and 7SK snRNP/HEXIM1 binding. J Virol 84: 12801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fufa TD, Byun JS, Wakano C, Fernandez A, et al. 2015. The Tax oncogene enhances ELL incorporation into p300 and P‐TEFb containing protein complexes to activate transcription. Biochem Biophys Res Commun 465: 5–11. [DOI] [PubMed] [Google Scholar]
- 114. Dominguez O, Rojo P, de Las Heras S, Folgueira D, et al. 2005. Clinical presentation and characteristics of pharyngeal adenovirus infections. Pediatr Infect Dis J 24: 733–4. [DOI] [PubMed] [Google Scholar]
- 115. Boyer TG, Martin ME, Lees E, Ricciardi RP, et al. 1999. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 399: 276–9. [DOI] [PubMed] [Google Scholar]
- 116. Stevens JL, Cantin GT, Wang G, Shevchenko A, et al. 2002. Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 296: 755–8. [DOI] [PubMed] [Google Scholar]
- 117. Vijayalingam S, Chinnadurai G. 2013. Adenovirus l‐E1A activates transcription through mediator complex‐dependent recruitment of the super elongation complex. J Virol 87: 3425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Yamamoto M, Onogi H, Kii I, Yoshida S, et al. 2014. CDK9 inhibitor FIT‐039 prevents replication of multiple DNA viruses. J Clin Invest 124: 3479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA‐dependent RNA polymerase with cellular RNA polymerase II. J Virol 79: 5812–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chan AY, Vreede FT, Smith M, Engelhardt OG, et al. 2006. Influenza virus inhibits RNA polymerase II elongation. Virology 351: 210–7. [DOI] [PubMed] [Google Scholar]
- 121. Zhang J, Li G, Ye X. 2010. Cyclin T1/CDK9 interacts with influenza A virus polymerase and facilitates its association with cellular RNA polymerase II. J Virol 84: 12619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mackenzie JS, Gubler DJ, Petersen LR. 2004. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat Med 10: S98–109. [DOI] [PubMed] [Google Scholar]
- 123. Gubler DJ, Clark GG. 1995. Dengue/dengue hemorrhagic fever: the emergence of a global health problem. Emerg Infect Dis 1: 55–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Rigau‐Perez JG, Clark GG, Gubler DJ, Reiter P, et al. 1998. Dengue and dengue haemorrhagic fever. Lancet 352: 971–7. [DOI] [PubMed] [Google Scholar]
- 125. Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44: 649–88. [DOI] [PubMed] [Google Scholar]
- 126. Raghupathy R, Chaturvedi UC, Al‐Sayer H, Elbishbishi EA, et al. 1998. Elevated levels of IL‐8 in dengue hemorrhagic fever. J Med Virol 56: 280–5. [DOI] [PubMed] [Google Scholar]
- 127. Juffrie M, van Der Meer GM, Hack CE, Haasnoot K, et al. 2000. Inflammatory mediators in dengue virus infection in children: interleukin‐8 and its relationship to neutrophil degranulation. Infect Immun 68: 702–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Clyde K, Kyle JL, Harris E. 2006. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol 80: 11418–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li LL, Hu ST, Wang SH, Lee HH, et al. 2010. Positive transcription elongation factor b (P‐TEFb) contributes to dengue virus‐stimulated induction of interleukin‐8 (IL‐8). Cell Microbiol 12: 1589–603. [DOI] [PubMed] [Google Scholar]
- 130. Field HJ. 2001. Herpes simplex virus antiviral drug resistance—current trends and future prospects. J Clin Virol 21: 261–9. [DOI] [PubMed] [Google Scholar]
- 131. Malumbres M, Pevarello P, Barbacid M, Bischoff JR. 2008. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci 29: 16–21. [DOI] [PubMed] [Google Scholar]
- 132. Wang Y, Wang Q, Zhu Q, Zhou R, et al. 2011. Identification and characterization of acyclovir‐resistant clinical HSV‐1 isolates from children. J Clin Virol 52: 107–12. [DOI] [PubMed] [Google Scholar]
- 133. Chao SH, Fujinaga K, Marion JE, Taube R, et al. 2000. Flavopiridol inhibits P‐TEFb and blocks HIV‐1 replication. J Biol Chem 275: 28345–8. [DOI] [PubMed] [Google Scholar]
- 134. Schang LM, St Vincent MR, Lacasse JJ. 2006. Five years of progress on cyclin‐dependent kinases and other cellular proteins as potential targets for antiviral drugs. Antivir Chem Chemother 17: 293–320. [DOI] [PubMed] [Google Scholar]
- 135. Biglione S, Byers SA, Price JP, Nguyen VT, et al. 2007. Inhibition of HIV‐1 replication by P‐TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P‐TEFb from the large, inactive form of the complex. Retrovirology 4: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Salerno D, Hasham MG, Marshall R, Garriga J, et al. 2007. Direct inhibition of CDK9 blocks HIV‐1 replication without preventing T‐cell activation in primary human peripheral blood lymphocytes. Gene 405: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Diwan P, Lacasse JJ, Schang LM. 2004. Roscovitine inhibits activation of promoters in herpes simplex virus type 1 genomes independently of promoter‐specific factors. J Virol 78: 9352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bresnahan WA, Boldogh I, Chi P, Thompson EA, et al. 1997. Inhibition of cellular Cdk2 activity blocks human cytomegalovirus replication. Virology 231: 239–47. [DOI] [PubMed] [Google Scholar]
- 139. Pisell TL, Ho O, Lee G, Butera ST. 2001. Spectrum of cdk‐9 inhibitor activity against HIV‐1 replication among various models of chronic and latent infection. Antivir Chem Chemother 12: 33–41. [PubMed] [Google Scholar]
- 140. Hsieh WS, Soo R, Peh BK, Loh T, et al. 2009. Pharmacodynamic effects of seliciclib, an orally administered cell cycle modulator, in undifferentiated nasopharyngeal cancer. Clin Cancer Res 15: 1435–42. [DOI] [PubMed] [Google Scholar]
- 141. Thomas JP, Tutsch KD, Cleary JF, Bailey HH, et al. 2002. Phase I clinical and pharmacokinetic trial of the cyclin‐dependent kinase inhibitor flavopiridol. Cancer Chemother Pharmacol 50: 465–72. [DOI] [PubMed] [Google Scholar]
- 142. Durand LO, Roizman B. 2008. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J Virol 82: 10591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Rechter S, Scott GM, Eickhoff J, Zielke K, et al. 2009. Cyclin‐dependent kinases phosphorylate the cytomegalovirus RNA export protein pUL69 and modulate its nuclear localization and activity. J Biol Chem 284: 8605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Romano G, Kasten M, De Falco G, Micheli P, et al. 1999. Regulatory functions of Cdk9 and of cyclin T1 in HIV tat transactivation pathway gene expression. J Cell Biochem 75: 357–68. [PubMed] [Google Scholar]
- 145. Zhou M, Halanski MA, Radonovich MF, Kashanchi F, et al. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl‐terminal domain during human immunodeficiency virus type 1 transcription. Mol Cell Biol 20: 5077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Thomas D, Blakqori G, Wagner V, Banholzer M, et al. 2004. Inhibition of RNA polymerase II phosphorylation by a viral interferon antagonist. J Biol Chem 279: 31471–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item