

Abstract
Background
Stricture formation occurs in ≈30% of patients with Crohn’s disease (CD) and is a significant cause of morbidity. Strictures are characterized by intestinal smooth muscle cell hyperplasia, smooth muscle cell hypertrophy, and fibrosis due to excess net extracellular matrix production, including collagen. Transforming growth factor-β1 (TGF-β1) has profibrotic effects in many tissues due to its ability to regulate collagen expression and extracellular matrix dynamics. We previously showed that both insulin-like growth factor (IGF) binding protein-3 (IGFBP-3) and TGF-β1 are expressed by normal human intestinal smooth muscle cells, bind to, and activate TGF-βRII/I receptors in these cells.
Methods
Smooth muscle cells isolated from the muscularis propria of patients were used to prepare RNA, protein lysates, or placed into primary culture. IGFBP-3, TGF-β1, and collagen IαI expression was measured with quantitative reverse-transcription polymerase chain reaction (RT-PCR) and protein levels by enzyme-linked immunosorbent assay (ELISA) or immunoblot.
Results
Expression and production of IGFBP-3, TGF-β1, and collagen IαI were significantly increased specifically in smooth muscle cells isolated from regions of strictured intestine in CD compared to nonstrictured histologically normal resection margin. IGFBP-3 and TGF-β1 regulated collagen IαI expression and production via a TGF-βRII/I-dependent and Smad2/3-dependent mechanism. Upregulated (excess) collagen IαI expression and production in smooth muscle cells of strictures and basal collagen IαI in smooth muscle cells of normal margin were inhibited by immunoneutralization of IGFBP-3 or TGF-β1.
Conclusions
The findings indicate that upregulated endogenous IGFBP-3 and TGF-β1 expression regulates excess collagen IαI production and contributes to fibrosis and stricture formation in CD.
Keywords: Crohn’s disease, stricture formation, fibrosis, IGFBP-3, TGF-β1
Crohn’s disease (CD) is characterized by three pathophysiologic changes within the smooth muscle of the muscularis propria of the intestine that contribute to stricture formation in the ≈30% of patients who experience this complication.1,2 Stricture formation is characterized by smooth muscle cell hyperplasia, smooth muscle cell hypertrophy, and excess net extracellular matrix formation by these cells.
Mounting evidence implicates members of the insulin-like growth factor (IGF) system as central to each of these events. Intestinal smooth muscle cells express endogenous IGF-I, which activates the cognate IGF-I receptor and regulates muscle cell hyperplasia by concomitantly stimulating proliferation and inhibiting apoptosis.3,4 Expression of IGF-I, a product of the IGF-IEa splice variant, is specifically up-regulated in smooth muscle cells in regions of stricturing CD compared to normal margins along with the expression of IGF binding protein (IGFBP)-5 that acts synergistically with IGF-I in these cells.5,6 Endogenous smooth muscle cell mechano-growth factor (MGF), a product of the IGF-IEc splice variant, is also upregulated in smooth muscle cells from regions of strictures and appears to regulate muscle cell hypertrophy.7 IGF-I and IGFBP-5 each independently stimulate collagen I expression and production in human intestinal smooth muscle and in rat intestinal smooth muscle cells.8 In the murine colon, as in humans, transforming growth factor-β1 (TGF-β1) regulates collagen synthesis.9,10 Collagen I, which accounts for 68%–70% of total collagen in the normal human intestine, and III, the major constituent isoforms in the normal human intestine, are overall increased in the intestine of stricturing CD.11
IGFBP-3, a 40–45-kDa glycoprotein, is produced both by the liver, which accounts for the majority of IGFBP present in serum, and by target tissues including human intestinal smooth muscle cells.12 IGFBP-3 expression and production in these cells is upregulated by endogenous IGF-I and TGF-β1.12,13 Recently we showed that IGFBP-3 has IGF-I-independent effects mediated by its ability to bind to the TGF-βRI/II receptor complex in human intestinal smooth muscle cells and activate receptor-based Smad signaling. The expression of both TGF-β1 and IGFBP-3 were increased specifically in the smooth muscle cells from stricturing CD compared to muscle cells from the normal margin of resected intestine. By contrast, while serum levels of IGFBP-3 are decreased, serumTGF-β1 levels are unchanged in these patients.14,15
The results show for the first time that endogenous IGFBP-3 and TGF-β1 activate the TGF-β1RI/II receptor complex coupled to Smad2/3 phosphorylation and regulate the excess expression and production of collagen IαI that occurs in smooth muscle cells of strictured intestine in CD. This pathophysiologic process leads to fibrosis and is one important factor in the development of strictures in CD.
MATERIALS AND METHODS
Isolation of Muscle Cells from Human Intestine in CD
Segments of intestine were obtained from patients undergoing ileal or ileocecal resection for stricturing CD according to a protocol approved by the VCU Institutional Review Board. Muscle cells were isolated from the circular muscle layer of the ileum using previously reported techniques from regions of stricturing CD and from the histologically normal proximal ileal resection margin.5,16–18 Demographic data on patients consenting to provide tissue for this study are presented in Table 1. Muscle cells isolated by enzymatic digestion were used to prepare RNA and whole cell lysates or placed into cell culture as reported previously.16,19 Epithelial cells, endothelial cells, neurons, and interstitial cells of Cajal do not contaminate cells isolated in this fashion.20 These cells possess a smooth muscle phenotype: immunostaining for smooth muscle markers but not fibroblast markers, expression of γ-enteric actin, and the physiologic characteristics of contractile intestinal smooth muscle; each of these properties are retained by the muscle cells in culture.20
TABLE 1.
Demographics of Patients with Stricturing Crohn’s Disease
| Demographics | Patient No. (% of Total) |
|---|---|
| Age, years | |
| <20 | 1 (6.7) |
| 20–29 | 1 (6.7) |
| 30–39 | 3 (20.0) |
| 40–49 | 4 (26.7) |
| 50–59 | 5 (33.3) |
| >60 | 1 (6.7) |
| Sex | |
| Male | 3 (20.0) |
| Female | 12 (80.0) |
| Race | |
| White | 9 (60.0) |
| African American | 5 (33.3) |
| Other/unknown | 1 (6.7) |
Preparation of RNA and Quantitative Real-time PCR
Quantitative real-time polymerase chain reaction (qRT-PCR) was used to measure RNA levels of: IGFBP-3 (NM_001013398), TGF-β1 (NM_000660), and collagen IαI (NM_000088). Briefly, total RNA was prepared from freshly isolated or cultured muscle cells using RNAqueous prep kits (Ambion, Austin, TX). First-strand cDNA synthesis was performed from two μg of total RNA using qScript cDNA prep kits (Quanta, Gaithersburg, MD). Quantitative RT-PCR reactions were performed in 25 μL total volume containing TAQman master mix, 2.5 pmol each of the forward and reverse primer (Applied Biosystems, Foster City, CA), and 2 μL of cDNA. MicroAmp 96-well optical plates are used for amplification in a GeneAmp 7300 Sequence Detection System (Applied Biosystems). Standard curves for each amplicon were generated from a dilution series of cDNA and results quantified and reported using the 2−ΔΔCt method based on β-actin amplification. β-Actin amplicon thresholds remained constant between control and strictured intestinal muscle.
Preparation of Whole Cell Lysates
Cell lysates were prepared as described previously in an immunoprecipitation buffer consisting of (in mM): 50 Tris-HCl (pH 7.5), 150 NaCl, 50 NaF, 1 Na orthovanadate, 1 dithiothreitol, 1 phenylmethylsulfonyl fluoride, and 0.5% NP-40 to which was added 1 μg/mL leupeptin, 1 μg/mL pepstatin A, and 1 μg/mL aprotinin.5,18,21 The resulting lysates were clarified by centrifugation at 14,000g for 10 minutes at 4°C prior to use for immunoprecipitation or immunoblot analysis.
Treatment of Cells for Preparation of RNA and Conditioned Medium
Quiescent cultured muscle cells were incubated in serum-free Dulbecco’s modified Eagle’s medium (DMEM) for 24 hours with added IGFBP-3 or TGF-β1 in the presence or absence of the selective TGF-βRI kinase inhibitor LY-364947 ([3-(pyridin-2-yl)-4-(4-quinonyl)]-1H-pyrazole inhibitor; Calbiochem, San Diego, CA)22 or the selective inhibitor of Smad3 phosphorylated SIS3 (6,7-dimethoxy-2-((2E)-3-(1-methyl-2-phenyl-1H-pyrrolo[2,3-b]pyridin-3-yl-prop-2-enoyl))-1,2,3,4-tetrahydroisoquinoline; Calbiochem).23 In experiments designed to identify the role of endogenous IGFBP-3 or TGF-β1, cells were incubated with neutralizing antibody to IGFBP-3 or TGF-β1 (R&D Systems, Minneapolis, MN) or preimmune isotype control as described previously.12,24 Media conditioned by cells treated for 24 hours was removed, clarified by centrifugation, and concentrated 10-fold in Sartorius Vivaspin 6 tubes (Sartorius, Bohemia, NY) at 4°C. Levels of collagen IαI protein were then determined by immunoblot analysis. Total RNA was also prepared from cells for qRT-PCR analysis.
Immunoblot Analysis
Collagen I, Smad 2 or 3, and phosphorylated Smad 2 or 3 levels were measured by immunoblot analysis using standard methods.3,5 Briefly, protein in cell lysate samples or concentrated conditioned medium containing equal amounts of total protein, determined using the BioRad DC protein assay kit (BioRad, Hercules, CA), were separated with sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions and electrotransferred to nitrocellulose. Membranes were incubated overnight with specific antibodies recognizing the protein of interest: collagen αI Type I (1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA), Smad2 and Smad 3, phospho-Smad2(Ser465/467), and phospho-Smad3(Ser423/425) or β-actin (1:1000–1:5000 dilution, Cell Signaling Technologies, Beverly, MA). Membranes were reblotted to measure total (phosphorylated plus nonphosphorylated) protein or β-actin. Placental human type I collagen or TGF-β activated HT1080 cells were used as a positive control for collagen IαI or Smad2/3, respectively. Bands were visualized with enhanced chemiluminescence using a FluoChem 8800 (Alpha Innotech, San Leandro, CA) and the digital images quantified using AlphaEaseFC v. 3.1.2 software. Densitometric values for protein bands are reported in arbitrary units above background values after normalization to total protein or β-actin.
Measurement of IGFBP-3 and TGF-β1 by Enzyme-linked Immunosorbent Assay (ELISA)
IGFBP-3 and T6F-β1 levels were measured in lysates prepared from muscle cells of stricturing CD or from normal margin as described previously.18 Briefly, IGFBP-3 and TGF-β1 were measured in samples containing equal protein using IGFBP-3 or TGF-β1-specific ELISAs (R&D Systems). These assays have no appreciable crossreactivity with IGF-I, IGF-II, insulin, or other IGF-binding proteins, or TGF-β2, TGF-β3, activins, inhibins, or BMPs. Results were calculated as ng/mg protein.
Statisical Analysis
Values represent means ± SE of n experiments, where n represents the number of experiments on cells derived from separate primary cultures. Statistical significance was tested by Student’s t-test for either paired or unpaired data as appropriate.
RESULTS
IGFBP-3, TGFβ-1, and Collagen IαI Expression Is Increased in Intestinal Smooth Muscle Cells of Stricturing CD
Quantitative RT-PCR showed that IGFBP-3 and TGF-β1 are expressed by human intestinal smooth muscle cells isolated from normal resection margins. The expression of IGFBP-3 was increased by 3.6 ± 2.2-fold in smooth muscle cells from strictures compared to normal margins (Fig. 1). The expression of TGF-β1 was increased by 6.1 ± 1.7-fold (Fig. 1A). Collagen Type IαI was expressed in smooth muscle cells isolated from normal resection margins. Collagen IαI expression was increased 6.3 ± 3.4-fold in muscle cells isolated from strictures compared to muscle cells from normal resection margin (Fig. 1A).
FIGURE 1.
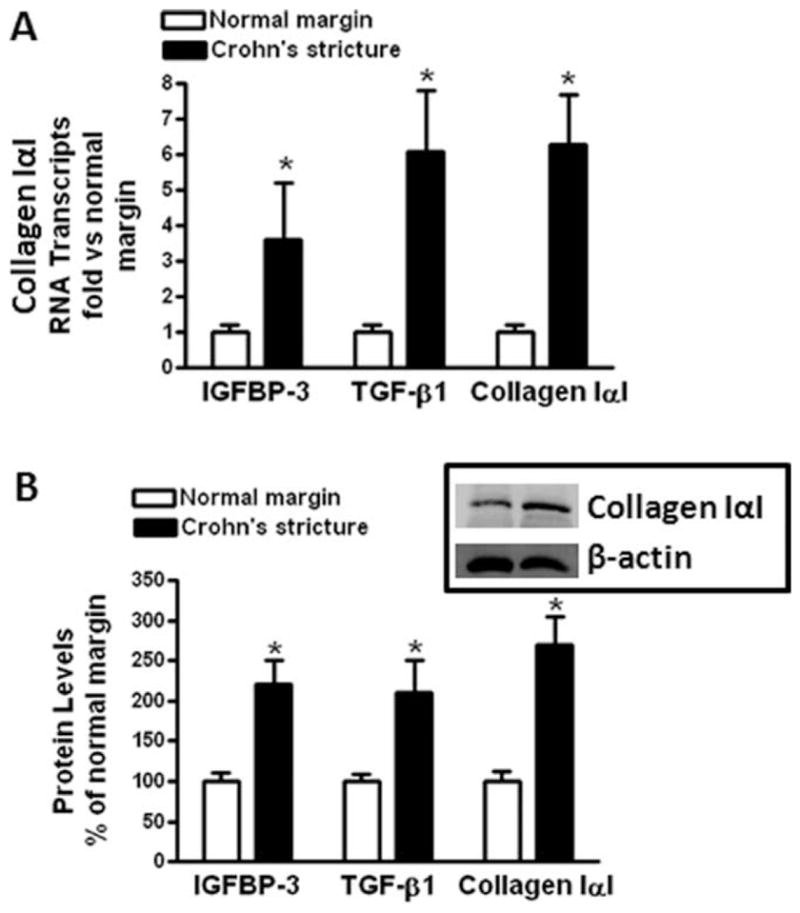
IGFBP-3, TGF-β1, and collagen IαI mRNA and protein levels are increased in strictured muscle cells of CD over normal intestine. (A) IGFBP-3, TGF-β1, and collagen IαI RNA transcript levels are increased in muscle cells isolated from stricturing CD compared to normal margins. qRT-PCR results are expressed as fold increase over normal muscle using the 2−ΔΔCt method with β-actin as control. (B) IGFBP-3, TGF-β1, and collagen IαI protein levels are increased in muscle cells isolated from stricturing CD compared to normal margins. IGFBP-3 and TGF-β1 levels were determined by ELISA and collagen IαI levels determined by immunoblot analysis with β-actin as control. Inset: representative immunoblot of collagen IαI and β-actin. Results are expressed as percent of normal margin muscle cells. Values represent the mean ± SEM of 4–5 separate experiments. *P < 0.05 versus normal margin.
IGFBP-3, TGF-β1, and collagen IαI protein levels were also increased in muscle cells isolated from stricturing CD. IGFBP-3 levels in muscle cells from normal margin, 210 ± 70 ng/mg protein, were increased 220 ± 30% in muscle cells from stricturing CD (Fig. 1B). TGF-β1 levels in muscle cells from normal margin 2.52 ± 0.44 ng/mg protein were increased 210 ± 40% in muscle cells from strictures and collagen IαI increased 270 ± 35% (Fig. 1B).
IGFBP-3 and TGF-β1 Stimulate Collagen IαI Expression and Production in Intestinal Smooth Muscle Cells
We have previously shown that IGFBP-3 binds to and activates TGF-βRI/II receptors. This effect is independent of TGF-β1.12,24,25 We hypothesized that IGFBP-3 and TGF-β1 might play a role in the regulation of collagen I production from human intestinal smooth muscle cells.
IGFBP-3 elicited a time-dependent increase in collagen IαI mRNA levels that was maximal after 24 hours (2.75 ± 0.25-fold above basal, Fig. 2A) and sustained for 48 hours. IGFBP-3 stimulated increase in collagen IαI expression corresponded to an increase in collagen I protein production at the 24-hour time period that was concentration-dependent (50 nM IGFBP-3: 183 ± 42% above basal, Fig. 2B). TGF-β1 elicited a concentration-dependent increase in collagen IαI expression (1 nM TGF-β1: 3.6 ± 0.3-fold above basal, Fig. 2A), that was accompanied by a concentration-dependent increase in collagen IαI protein production (1 nM TGF-β1: 226 ± 43% above basal) (Fig. 2C).
FIGURE 2.

IGFBP-3 and TGF-β1 cause time-dependent and concentration-dependent increases in collagen IαI mRNA and protein production. (A) IGFBP-3 (50 nM, closed circles) and TGF-β1 (1 nM, open circles) elicit time-dependent increases in collagen IαI mRNA transcript levels. Collagen IαI transcript levels were measured using qRT-PCR. Results are expressed as fold increase over normal using the 2−ΔΔCt method with β-actin as control. (B) IGFBP-3 elicits concentration-dependent increase in collagen IαI protein. Collagen IαI protein production was measured in media conditioned by IGFBP-3 using immunoblot analysis. Inset: representative immunoblot of collagen IαI and β-actin. (C) TGF-β1 elicits concentration-dependent increase in collagen IαI protein. Collagen IαI protein production was measured in media conditioned by TGF-β1 using immunoblot analysis. Inset: representative immunoblot of collagen IαI production and β-actin. Results in (B,C) are expressed as percent above untreated normal margin muscle cells. Values represent the mean ± SEM of four separate experiments. *P < 0.05 versus basal levels.
IGFBP-3 Stimulates Collagen I Expression and Production Via TGF-βRI/II and Smad 3-dependent Pathway
Our previous work has shown that IGFBP-3, like TGF-β1, elicited coordinate Smad2 and Smad3 phosphorylation following activation of TGF-βI/II.24 The signaling cascades activated by IGFBP-3 and TGF-β1 coupled to regulation of collagen IαI mRNA expression and protein production were therefore examined in the presence of the TGF-βRI kinase inhibitor, LY-364947,22 the immediate upstream kinase coupled to Smad 2 and Smad 3 phosphorylation, or in the presence of the Smad 3 inhibitor, SIS3.23 While Smad 2 and Smad 3 are activated coordinately in these cells by either IGFBP-3 or TGF-β1, the present study examined Smad 3 phosphorylation. A pharmacologic approach to Smad3 inhibition was used in these studies due to its potential therapeutic use rather than an RNAi approach, which we have used previously,24 with its inherent difficulties in translation to therapeutic use.
Treatment of normal human intestinal smooth muscle cells with 50 nM IGFBP-3 increased Smad 3 phosphorylation 148 ± 5% above basal within 15 minutes. The increase was inhibited 95 ± 3% by the TGF-βRI kinase inhibitor, LY-364947 (5 μM) and 81 ± 13% by the Smad 3 inhibitor, SIS3 (10 μM) (Fig. 3A). A similar pattern was observed in cells treated with 1 nM TGF-β1, which elicited a 125 ± 16% increase in Smad 3 phosphorylation that was inhibited 91 ± 4% by the TGF-βRI kinase inhibitor and 92 ± 4% by the Smad 3 inhibitor (Fig. 3B).
FIGURE 3.

IGFBP-3 and TGF-β1 activate Smad 3 via a TGF-βRI-dependent mechanism. (A) IGFBP-3 (50 nM) stimulated Smad 3 phosphorylation is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. Inset: representative immunoblot of Smad 3 phosphorylation and total Smad 3 levels. (B) TGF-β1 (1 nM) stimulated Smad 3 phosphorylation is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. Inset: representative immunoblot of Smad 3 phosphorylation and total Smad 3 levels. Quiescent normal muscle cells were stimulated for 15 minutes with 50 nM IGFBP-3 or 1 nM TGF-β1 and phosphorylated Smad 3 and total Smad 3 measured in whole cell lysates. Results are expressed a percent of basal in untreated normal margin muscle cells. Values represent the mean ± SEM of 4–5 separate experiments. *P < 0.05 versus untreated cells; **P < 0.05 versus IGFBP-3 or TGF-β1 stimulated cells.
The participation of this pathway in IGFBP-3-regulated and TGF-β1-regulated collagen IαI expression and production was therefore examined. In cells treated with 50 nM IGFBP-3 for 24 hours, collagen IαI mRNA levels increased by 2.4 ± 0.3-fold. In the presence of the TGF-βRI kinase inhibitor IGFBP-3-stimulated collagen IαI expression was abolished (100 ± 12% inhibition) and inhibited 51 ± 15% from basal levels in the presence of the Smad 3 inhibitor (Fig. 4A). Similarly, in cells treated with 1 nM TGF-β1 for 24 hours collagen IαI mRNA increased by 2.2 ± 0.3-fold. Collagen IαI mRNA levels declined to 80 ± 5% of basal in the presence of the TGF-βRI kinase inhibitor and declined to 57 ± 12% of basal in the presence of the Smad 3 inhibitor (Fig. 4B).
FIGURE 4.

IGFBP-3 and TGF-β1 regulate collagen IαI expression via a TGF-βRI-dependent and Smad 3-depedent mechanism. (A) The increase in collagen IαI expression elicited by 50 nM IGFBP-3 is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. (B) The increase in collagen IαI expression elicited by 1 nM TGF-β1 is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. Quiescent normal muscle cells were stimulated for 24 hours with 50 nM IGFBP-3 or 1 nM TGF-β1 and collagen IαI RNA transcript levels were measured by qRT-PCR. Results are expressed as fold increase over basal normal margin muscle cells using the 2−ΔΔCt method with β-actin as control. Values represent the mean ± SEM of 4–5 separate experiments. *P < 0.05 versus untreated cells; **P < 0.05 versus IGFBP-3 or TGF-β1 stimulated cells.
The changes in collagen IαI mRNA expression were accompanied by similar changes in protein levels. The increase in collagen protein production induced by 50 nM IGFBP-3 (183 ± 42% above basal) declined to 81 ± 19% of basal in the presence of the TGF-βRI kinase inhibitor and declined to 57 ± 16% of basal in the presence of the Smad 3 inhibitor (Fig. 5A). A similar effect was observed with 1 nM TGF-β1-induced collagen IαI protein production (226 ± 43% above basal). TGF-β1-induced collagen IαI protein production declined to 79 ± 15% of basal in the presence of the TGF-βRI kinase inhibitor and declined to 74 ± 9% of basal in the presence of the Smad 3 inhibitor (Fig. 5B).
FIGURE 5.

IGFBP-3 and TGF-β1 regulate collagen IαI production via a TGF-βRI-dependent and Smad 3-depedent mechanism. (A) IGFBP-3 (50 nM) stimulated collagen IαI production is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. Inset: representative immunoblot of collagen IαI and β-actin. (B) TGF-β1 (1 nM) stimulated collagen IαI production is inhibited by the TGF-βRI kinase inhibitor, LY-364947, or the Smad 3 inhibitor, SIS3. Inset: representative immunoblot of collagen IαI and β-actin. Quiescent normal muscle cells were stimulated for 24 hours with 50 nM IGFBP-3 or 1 nM TGF-β1 in the presence and absence of test agents. Collagen IαI protein levels were measured in conditioned media by immunoblot analysis. Results are expressed as percent of untreated normal margin muscle cells. Values represent the mean ± SEM of 4–5 separate experiments. *P < 0.05 versus untreated cells; **P < 0.05 versus IGFBP-3 or TGF-β1 stimulated cells.
Endogenous IGFBP-3 Regulates Collagen I mRNA Expression in Normal Muscle Cells and in Stricturing CD
It is noteworthy that in the presence of the TGF-βRI kinase inhibitor or Smad 3 inhibitor, expression and production of collagen IαI declined to levels below basal. Since human intestinal smooth muscle cells produce both IGFBP-3 and TGF-β1, these findings imply that endogenous IGFBP-3 and TGF-β1 regulate collagen IαI expression and production.12 This notion was tested by immunoneutralization of endogenous IGFBP-3 and TGF-β1. In cultured muscle cells isolated from normal margin and incubated with a neutralizing antibody for IGFBP-3 (5 μg/mL) for 24 hours, collagen IαI mRNA expression was inhibited 47 ± 4% from control preimmune antibody-treated cells (Fig. 6A) and basal collagen IαI protein production decreased 53 ± 4% from control antibody-treated cells (Fig. 6B). In cells incubated with 1 μg/mL of a TGF-β1 neutralizing antibody for 24 hours, collagen IαI mRNA expression was inhibited by 67 ± 6% from control antibody-treated cells (Fig. 6A) and basal collagen IαI protein production decreased 66 ± 5% from control antibody-treated cells (Fig. 6B).
FIGURE 6.

Endogenous IGFBP-3 and TGF-β1 regulate excess collagen IαI expression and production in normal and stricturing muscle of CD. (A) Immunoneutralization of endogenous IGFBP-3 and TGF-β1 decreases collagen IαI expression in both normal margin muscle cells and in muscle cells isolated from strictures in CD. Collagen IαI RNA transcript levels were measured by qRT-PCR. Results are expressed as fold versus basal normal margin muscle cells using the 2−ΔΔCt method with β-actin as control. (B) Immunoneutralization of endogenous IGFBP-3 and TGF-β1 decreases collagen IαI protein production in both normal margin muscle cells and in muscle cells isolated from strictures of CD. Inset: representative immunoblots of collagen IαI and β-actin. Quiescent muscle cells were incubated for 24 hours with preimmune serum (control ab.), or neutralizing antibodies to IGFBP-3 or TGF-β1. Collagen IαI protein levels were measured in conditioned media by immunoblot analysis. Results are expressed as percent of control normal margin muscle cells. Values represent mean ± SEM of 4–5 separate experiments. *P < 0.05 versus normal margin; **P < 0.05 versus control antibody-treated cells.
The notion that upregulation of endogenous IGFBP-3 might contribute to excess collagen IαI production in muscle cells in stricturing CD and contribute to stricture formation was tested by immunoneutralization of endogenous IGFBP-3 and TGF-β1 in cultured muscle cells isolated from strictures of CD. Treatment of muscle cells for 24 hours with 5 μg/mL of an immunoneutralizing antibody to IGFBP-3 resulted in a 33 ± 7% decrease in collagen IαI mRNA levels (Fig. 6A) and a similar 41 ± 5% decrease in collagen IαI production (Fig. 6B). Treatment of muscle cells for 24 hours with 1 μg/mL of an immunoneutralizing antibody to TGF-β1 resulted in a 72 ± 8% decrease in collagen IαI mRNA levels (Fig. 6A) and a similar 67 ± 2% decrease in collagen IαI production (Fig. 6B).
DISCUSSION
This study shows for the first time that IGFBP-3 expression is upregulated specifically in the smooth muscle cells of the muscularis propria of stricturing CD compared to levels of constitutive expression in muscle cells from normal resection margins and that endogenous IGFBP-3 regulates excess collagen IαI production. TGF-β1 expression is also upregulated in smooth muscle cells of strictures with similar effect. Upregulated IGFBP-3 and TGF-β1 regulate excess collagen IαI production, a key constituent of extracellular matrix in the intestine and in the development of fibrosis and stricturing in CD, by binding to and activating the TGF-βRI/RII complex, and initiate intracellular signaling via Smad2/3 coupled to regulation of collagen IαI expression.24 IGFBP-3 exerts these effects independently of TGF-β1 in these cells,24 in contrast to other cell types where bioavailable TGF-β1 is required.25 The importance of this pathway was suggested by the resistance to the development of fibrosis in TNBS-induced colitis in Smad3 null mice.9 Expression of IGFBP-3 in human intestinal smooth muscle cells is further stimulated by endogenous TGF-β1, resulting in a positive feedback loop increasing not only IGFBP-3 expression but also possibly collagen production.12,24
The evidence supporting the conclusions that endogenous IGFBP-3 and TGF-β1 regulate collagen IαI expression and production in stricturing CD can be summarized as follows. 1) IGFBP-3 and TGF-β1 both regulate collagen IαI expression and production through TGF-βRI kinase-dependent and Smad 3-dependent mechanisms. 2) Collagen IαI is expressed and secreted by muscle cells isolated from normal margins; collagen IαI expression and production are significantly increased in muscle cells isolated from strictures. 3) Collagen IαI expression and production is decreased by immunoneutralization of endogenous IGFBP-3 or TGF-β1. 4) IGFBP-3 and TGF-β1 are constitutively expressed by intestinal smooth muscle cells with expression that is significantly upregulated in muscle cells of strictured intestine compared to muscle cells isolated from normal margins.
Regulation of extracellular matrix is central to the development of fibrosis in many organs, including in CD. In liver cirrhosis, pulmonary fibrosis, or systemic sclerosis the whole organ can be affected by the fibrosis. Fibrosis and stricture formation in CD are segmental and characteristically different than fibrosis in other organs. Despite intestinal inflammation, not all patients with CD develop fibrosis or strictures, implying that additional factors are involved. It is noteworthy that the upregulation of IGFBP-3 and TGF-β1 expression and the resulting excess collagen IαI production occurs specifically within the smooth muscle cells of strictured regions. In muscle cells from normal resection margins in these same patients, while IGFBP-3, TGF-β1, and collagen IαI are present, their levels are similar to that in normal muscle cells from non-CD controls (unpubl. obs.), and fibrosis and stricture formation do not occur in this adjacent intestine.
The components of extracellular matrix include structural proteins such as collagen and specialized proteins, such as fibronectin, and matricellular proteins, such as osteopontin and thrombospondin, which exert regulatory functions by interacting with cell surface receptors, including integrins.26–29 Regulation of collagen is a dynamic process involving its expression and production and its regulated turnover. While collagen is resistant to degradation by most proteases, specific matrix metalloproteinases (MMPs), collagenases regulate collagen degradation. MMPs are regulated in turn by tissue inhibitor of metalloproteinase (TIMP)-dependent inactivation. This system regulates not only collagen but also specialized extracellular matrix proteins such as fibronectin. Fibronectin and vitronectin secretion by human intestinal smooth muscle cells are significantly increased in the muscle cells of stricturing CD where they regulate, via αVβ3 integrin, excess smooth muscle growth.19
Three pathophysiologic events occurring within smooth muscle cells of the muscularis propria contribute to the stricture formation of CD: increased smooth muscle cell hyperplasia, increased smooth muscle cells hypertrophy, and excess net extracellular matrix proteins, including collagen. It is noteworthy that while serum levels of IGF-I and IGFBP-3 are decreased in patients with CD,30 and TGF-β1 remains unchanged, the expression and production of IGF-I, its binding proteins, IGFBP-3 and IGFBP-5, and TGF-β1 by smooth muscle cells of stricturing CD are all increased, where each plays a specific role in stricture formation. Upregulated IGF-I expression in smooth muscle cells from stricturing intestine regulates excess smooth muscle cell hyperplasia.6,19 Activation of αVβ3 integrin by specialized extracellular matrix proteins, vitronectin, or fibronectin maximizes the intensity and duration of IGF-I stimulated muscle cell growth and further stimulates excess smooth muscle cell hyperplasia in stricturing CD.5 IGFBP-5 has several regulatory functions. It directly stimulates muscle hyperplasia and collagen secretion, acts synergistically with IGF-I, and acts in concert with matricellular proteins, osteopontin and thrombospondin, to regulate IGF-I-stimulated smooth muscle cell growth.18,31,32
Increased expression of MMP-8 and MMP-9 has been demonstrated immunohistologically in mucosal ulcerations in CD where localized upregulation would increase degradation of extracellular matrix in the epithelial and subepithelial regions, contributing to ulcer formation.33 In subepithelial myofibroblasts TIMP-1 is increased in a TGF-β1/β2-dependent fashion and is accompanied by increased MMP-2; MMP-1 and MMP-3 were present only in inactive forms.34 Nienke et al35 have identified increased expression of MMP-1, MMP-3, and TIMP-1 in the muscle layer but the specific source was not identified. In contrast to the epithelial) procollagen I expression was increased in the muscularis propria of active CD and was accompanied by increased TIMP-1 and TIMP-2 expression, suggesting that upregulation of TIMPs which decrease extracellular matrix turnover, including collagen, could promote fibrosis.33
Gender and ethnic variation exists in the levels of IGFBP-3 and IGF-I in normal healthy adults and in the response of the intestine to inflammation.36 Serum levels of IGF-I are higher and IGFBP-3 levels lower in males than females. Levels of IGF-I and IGFBP-3 also tend to be higher in non-Hispanic white than in non-Hispanic black subjects.37 No differences in TGF-β1 levels have been observed.38 Inflammatory mediators and pharmacologic interventions in individual patients might also effect gene expression in smooth muscle cells. The available evidence, however, is conflicting. TNF-α, for example, has been shown to increase IGFBP-3 expression in vitro; however, immunoneutralization of TNF-α does not alter IGFBP-3 levels in vivo.36,39 In the present study these concerns were minimized by using muscle cells derived from normal resection margins and compared to responses in muscle cells from strictured intestine in the same patient.
In summary, this study shows for the first time that collagen production, one of the characteristic features of stricture formation and fibrosis in CD, is regulated by endogenous IGFBP-3 and TGF-β1 expression that is specifically upregulated within smooth muscle cells. IGFBP-3 and TGF-β1 mediate excess collagen IαI production from these cells via activation of TGF-βRI/II and Smad 3 in regions of stricturing. The mechanism identified in this study also suggests potential targets for therapeutic interventions designed to impact fibrosis and stricture formation in CD.
Acknowledgments
Supported by grant DK49691 from the National Institutes of Diabetes and Digestive and Kidney Diseases (to J.F.K.).
References
- 1.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–1517. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 2.Ruthruff B. Clinical review of Crohn’s disease. J Am Acad Nurse Pract. 2007;19:392–397. doi: 10.1111/j.1745-7599.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 3.Kuemmerle JF. IGF-I elicits growth of human intestinal smooth muscle cells by activation of PI3K, PDK-1, and p70S6 kinase. Am J Physiol Gastrointest Liver Physiol. 2003;284:G411–422. doi: 10.1152/ajpgi.00310.2002. [DOI] [PubMed] [Google Scholar]
- 4.Kuemmerle JF. Endogenous IGF-I protects human intestinal smooth muscle cells from apoptosis by regulation of GSK-3 beta activity. Am J Physiol Gastrointest Liver Physiol. 2005;288:G101–110. doi: 10.1152/ajpgi.00032.2004. [DOI] [PubMed] [Google Scholar]
- 5.Kuemmerle JF. Occupation of αvβ3-integrin by endogenous ligands modulates IGF-I receptor activation and proliferation of human intestinal smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1194–1202. doi: 10.1152/ajpgi.00345.2005. [DOI] [PubMed] [Google Scholar]
- 6.Zimmermann EM, Li L, Hou YT, et al. Insulin-like growth factor I and insulin-like growth factor binding protein 5 in Crohn’s disease. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1022–1029. doi: 10.1152/ajpgi.2001.280.5.G1022. [DOI] [PubMed] [Google Scholar]
- 7.Berg KM, Bowers JG, Kuemmerle JF. The IGF-IEa (IGF-I) and IGF-IEc (MGF) splice variants of the IGF-I gene mediate hypertrophy and hyperplasia of human intestinal smooth muscle. Gastroenterology. 2007;132:A238. [Google Scholar]
- 8.Zimmermann EM, Li L, Hou YT, et al. IGF-I induces collagen and IGFBP-5 mRNA in rat intestinal smooth muscle. Am J Physiol. 1997;273:G875–882. doi: 10.1152/ajpgi.1997.273.4.G875. [DOI] [PubMed] [Google Scholar]
- 9.Latella G, Vetuschi A, Sferra R, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Investig. 2009;39:145–156. doi: 10.1111/j.1365-2362.2008.02076.x. [DOI] [PubMed] [Google Scholar]
- 10.Graham MF, Bryson GR, Diegelmann RF. Transforming growth factor beta 1 selectively augments collagen synthesis by human intestinal smooth muscle cells. Gastroenterology. 1990;99:447–453. doi: 10.1016/0016-5085(90)91028-5. [DOI] [PubMed] [Google Scholar]
- 11.Graham MF, Diegelmann RF, Elson CO, et al. Collagen content and types in the intestinal strictures of Crohn’s disease. Gastroenterology. 1988;94:257–265. doi: 10.1016/0016-5085(88)90411-8. [DOI] [PubMed] [Google Scholar]
- 12.Bushman TL, Kuemmerle JF. IGFBP-3 and IGFBP-5 production by human intestinal muscle: reciprocal regulation by endogenous TGF-beta1. Am J Physiol. 1998;275:G1282–1290. doi: 10.1152/ajpgi.1998.275.6.G1282. [DOI] [PubMed] [Google Scholar]
- 13.Kuemmerle JF. Endogenous IGF-I regulates IGF binding protein production in human intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2000;278:G710–717. doi: 10.1152/ajpgi.2000.278.5.G710. [DOI] [PubMed] [Google Scholar]
- 14.Eivindson M, Grønbaek H, Skogstrand K, et al. The insulin-like growth factor (IGF) system and its relation to infliximab treatment in adult patients with Crohn’s disease. Scand J Gastroenterol. 2007;42:464–470. doi: 10.1080/00365520601010115. [DOI] [PubMed] [Google Scholar]
- 15.Kanazawa S, Tsunoda T, Onuma E, et al. VEGF, basic-FGF, and TGF-β in Crohn’s disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. 2001;96:822–828. doi: 10.1111/j.1572-0241.2001.03527.x. [DOI] [PubMed] [Google Scholar]
- 16.Kuemmerle JF. Autocrine regulation of growth in cultured human intestinal muscle by growth factors. Gastroenterology. 1997;113:817–824. doi: 10.1016/s0016-5085(97)70176-8. [DOI] [PubMed] [Google Scholar]
- 17.Kuemmerle JF. Endogenous IGF-I promotes survival of human intestinal muscle cells by Akt-dependent inhibition of GSK-3b activity. Mol Biol Cell. 2002;13:165a. [Google Scholar]
- 18.Kuemmerle JF, Zhou H. Insulin-like growth factor-binding protein-5 (IGFBP-5) stimulates growth and IGF-I secretion in human intestinal smooth muscle by Ras-dependent activation of p38 MAP kinase and Erk1/2 pathways. J Biol Chem. 2002;277:20563–20571. doi: 10.1074/jbc.M200885200. [DOI] [PubMed] [Google Scholar]
- 19.Flynn RS, Murthy KS, Grider JR, et al. Endogenous IGF-I and αVβ3 integrin ligands regulate increased smooth muscle hyperplasia in stricturing Crohn’s disease. Gastroenterology. 2010;138:285–293. doi: 10.1053/j.gastro.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng B, Murthy KS, Kuemmerle JF, et al. Expression of endothelial nitric oxide synthase in human and rabbit gastrointestinal smooth muscle cells. Am J Physiol. 1998;275:G342–351. doi: 10.1152/ajpgi.1998.275.2.G342. [DOI] [PubMed] [Google Scholar]
- 21.Kuemmerle JF, Murthy KS. Coupling of the insulin-like growth factor-I receptor tyrosine kinase to Gi2 in human intestinal smooth muscle: Gbetagamma-dependent mitogen-activated protein kinase activation and growth. J Biol Chem. 2001;276:7187–7194. doi: 10.1074/jbc.M011145200. [DOI] [PubMed] [Google Scholar]
- 22.Singh J, Chuaqui CE, Boriack-Sjodin PA, et al. Successful shape-based virtual screening: the discovery of a potent inhibitor of the type I TGFβ receptor kinase (TβRI) Bioorgan Med Chem Lett. 2003;13:4355–4359. doi: 10.1016/j.bmcl.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 23.Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-β1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597–607. doi: 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
- 24.Kuemmerle JF, Murthy KS, Bowers JG. IGFBP-3 activates TGF-beta receptors and directly inhibits growth in human intestinal smooth muscle cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G795–802. doi: 10.1152/ajpgi.00009.2004. [DOI] [PubMed] [Google Scholar]
- 25.Fanayan S, Firth SM, Butt AJ, et al. Growth inhibition by insulin-like growth factor-binding protein-3 in T47D breast cancer cells requires transforming growth factor-beta (TGF-beta) and the type II TGF-beta receptor. J Biol Chem. 2000;275:39146–39151. doi: 10.1074/jbc.M006964200. [DOI] [PubMed] [Google Scholar]
- 26.Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Aspects Med. 2008;29:290–308. doi: 10.1016/j.mam.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pender SLF, MacDonald TT. Matrix metalloproteinases and the gut — new roles for old enzymes. Curr Opin Pharmacol. 2004;4:546–550. doi: 10.1016/j.coph.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 28.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 29.Singh M, Foster CR, Dalal S, et al. Osteopontin: role in extracellular matrix deposition and myocardial remodeling post-MI. J Mol Cell Cardiol. 2010;48:538–543. doi: 10.1016/j.yjmcc.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Katsanos KH, Tsatsoulis A, Christodoulou D, et al. Reduced serum insulin-like growth factor-1 (IGF-1) and IGF-binding protein-3 levels in adults with inflammatory bowel disease. Growth Horm IGF Res. 2001;11:364–367. doi: 10.1054/ghir.2001.0248. [DOI] [PubMed] [Google Scholar]
- 31.Flynn RS, Mahavadi S, Murthy KS, et al. Insulin-like growth factor-binding protein-5 stimulates growth of human intestinal muscle cells by activation of Gi3. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1232–1238. doi: 10.1152/ajpgi.00323.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nam T-J, Busby WH, Jr, Rees C, et al. Thrombospondin and osteopon-tin bind to insulin-like growth factor (IGF)-binding protein-5 leading to an alteration in IGF-I-stimulated cell growth. Endocrinology. 2000;141:1100–1106. doi: 10.1210/endo.141.3.7386. [DOI] [PubMed] [Google Scholar]
- 33.Arihiro S, Ohtani H, Hiwatashi N, et al. Vascular smooth muscle cells and pericytes express MMP-1, MMP-9, TIMP-1 and type I procollagen in inflammatory bowel disease. Histopathology. 2001;39:50–59. doi: 10.1046/j.1365-2559.2001.01142.x. [DOI] [PubMed] [Google Scholar]
- 34.McKaig BC, McWilliams D, Watson SA, et al. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162:1355–1360. doi: 10.1016/S0002-9440(10)63931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nienke W, Hofker HS, Mark HJM, et al. Matrix metalloproteinases as profibrotic factors in terminal ileum in Crohn’s disease. Inflamm Bowel Dis. 2006;12:863–869. doi: 10.1097/01.mib.0000231568.43065.ed. [DOI] [PubMed] [Google Scholar]
- 36.Elsasser TH, Caperna TJ, Ward PJ, et al. Modeling growth factor activity during proinflammatory stress: methodological considerations in assessing cytokine modulation of IGF binding proteins released by cultured bovine kidney epithelial cells. Domest Anim Endocrinol. 2007;33:390–399. doi: 10.1016/j.domaniend.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Berrigan D, Potischman N, Dodd KW, et al. Race/ethnic variation in serum levels of IGF-I and IGFBP-3 in US adults. Growth Hormone IGF Res. 2009;19:146–155. doi: 10.1016/j.ghir.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pam K, Elswick RK, Mitchell S. Ethnicity and cytokine production gauge response of patients with hepatitis C to interferon-alpha therapy. J Med Virol. 2001;65:510–516. [PubMed] [Google Scholar]
- 39.Sarzi-Puttini P, Atzeni F, Schölmerich J, et al. Anti-TNF antibody treatment improves glucocorticoid induced insulin-like growth factor 1 (IGF1) resistance without influencing myoglobin and IGF1 binding proteins 1 and 3. Ann Rheum Dis. 2006;65:301–305. doi: 10.1136/ard.2005.040816. [DOI] [PMC free article] [PubMed] [Google Scholar]