

SUMMARY
Experience-driven plasticity of glutamatergic synapses on striatal spiny projection neurons (SPNs) is thought to be essential to goal-directed behavior and habit formation. One major form of striatal plasticity – long-term depression (LTD) – has long appeared to be expressed only pre-synaptically. Contrary to this view, nitric oxide (NO) generated by striatal interneurons was found to induce a post-synaptically expressed form of LTD at SPN glutamatergic synapses. This form of LTD was dependent on signaling through guanylyl cyclase and protein kinase G – both of which are abundantly expressed by SPNs. NO-LTD was unaffected by local synaptic activity or by antagonism of endocannabinoid (eCb) and dopamine receptors; all of which modulate canonical, pre-synaptic LTD. Moreover, NO signaling disrupted induction of this canonical LTD by inhibiting dendritic Ca2+ channels regulating eCb synthesis. These results establish an interneuron-dependent, heterosynaptic form of post-synaptic LTD that could act to promote stability of the striatal network during learning.
Keywords: striatum, synaptic plasticity, long-term depression, nitric oxide, post-synaptic, corticostriatal, thalamostriatal, guanylyl cyclase, protein kinase G, spiny projection neuron
Graphical Abstract
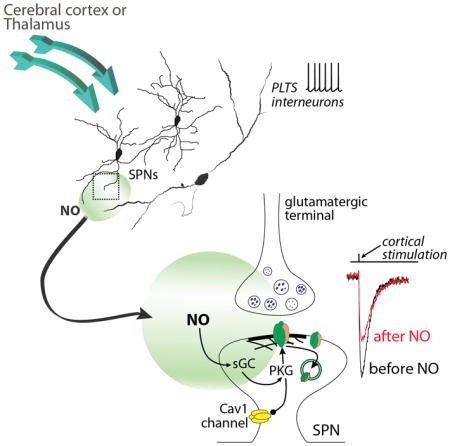
INTRODUCTION
The striatum has long been implicated in learning goal-directed behavior and habits (Balleine et al., 2007). This learning is thought to reflect changes in the strength of glutamatergic synapses that dictate the timing and pattern of activity of principal SPNs (Gerfen and Surmeier, 2011). In the dorsal striatum, axospinous glutamatergic synapses on SPNs are formed primarily by cortical pyramidal neurons. Long-term depression (LTD) of these synapses was initially described over 20 years ago and has been extensively studied (Calabresi et al., 1992; Centonze et al., 2001; Gerfen and Surmeier, 2011; Kreitzer and Malenka, 2008; Mathur and Lovinger, 2012). The best characterized form of LTD is induced by post-synaptic depolarization and activation of metabotropic glutamate receptors and L-type Ca2+ channels, which trigger the generation of endocannabinoids (eCbs) by SPNs; expression of LTD is mediated by eCb activation of pre-synaptic CB1 receptors, which result in sustained depression of glutamate release (Kreitzer, 2005; Lovinger et al., 1993). Although other neuromodulators can mimic the actions of eCbs at these synapses (Mathur et al., 2011), no post-synaptically expressed form of LTD has been described in SPNs.
One of the signaling molecules implicated in post-synaptically expressed LTD elsewhere in the brain is NO (Garthwaite, 2008). Striatal expression of NO signaling proteins – soluble guanylyl cyclase (sGC) and protein kinase G (PKG) – are among the highest of any brain region, making a role for NO signaling in striatal plasticity plausible (Ariano, 1983; Ding et al., 2004). Indeed, NO has been implicated in striatal synaptic plasticity, but its role is controversial (Calabresi et al., 1999; Sammut et al., 2007), with most of the available data suggesting it is a permissive modulator of eCb-dependent LTD (eCb-LTD) (Centonze et al., 2001; 1999).
RESULTS
NO signaling produced LTD of glutamatergic synapses
To assess the potential role of NO signaling in striatal synaptic plasticity, excitatory post-synaptic currents (EPSCs) evoked in SPNs by electrical stimulation of corticostriatal axons were monitored before, during and after the application of the NO donor (S)-nitroso-N-acetyl-D,L-penicillamine (SNAP, 100 μM) to ex vivo parasagittal brain slices of mouse forebrain (Figure 1A). In this preparation, cortical axons can be stimulated electrically without directly activating striatal neurons, unlike the coronal brain slice (Kawaguchi et al., 1989). Transient application of SNAP led to a persistent depression of corticostriatal EPSCs (Figure 1B). To verify that our electrical stimulus was not directly exciting SPNs, cortical pyramidal neurons were optogenetically activated to evoke EPSCs; SNAP application also produced a persistent depression of optically evoked corticostriatal EPSCs (Figure S1A).
Figure 1.

NO induces LTD at corticostriatal synapses through activation of PKG. (A) Simplified diagram depicting the circuit components being examined. (B) Sample whole cell recording of an SPN before, during and after a 10-minute application of the NO donor SNAP (100 μM). Open circles represent single EPSC events and solid line represents average EPSC size over a one-minute interval. EPSC amplitudes are normalized to baseline responses. Access resistance plotted below. Example EPSCs from this SPN are shown to the right before (pre-i, min 1–5 averaged) and after (post-i, min 30–35 averaged) SNAP application. The stimulus artifact has been suppressed for clarity. (C) PKG inhibition (3 μM, Rp-8-Br-PET-cGMPS, Rp8Br; black trace) blocked SNAP induced LTD (red trace) (control: n = 16 cells; PKG inhibitor: n=6). *p<0.05, signed rank test. (D) Cartoon diagram illustrating the NO signaling cascade. (E) Bath application of 8-Br-cGMP (500 μM) induced LTD in dSPNs (n=7). *p<0.05, signed rank test. Example EPSCs are shown to the right. (F) 8-Br-cGMP induced LTD similarly in iSPNs and dSPNs (dSPNs, from 1E: n=7; iSPN: n=9). *p<0.05, rank sum test. (G) SNAP induced LTD was unaffected by the inclusion of mecamylamine (10 μM) and scopolamine (10 μM) in the bath (n=5). Scale bars (vertical, horizontal): (B) 10 pA, 20 ms; (E) 25 pA, 20 ms.
NO can have both direct and indirect effects on proteins involved in synaptic transmission (Garthwaite, 2008). Because its striatal expression is robust (Ariano, 1983), our working hypothesis was that the NO donor effects were mediated by soluble GC (sGC) activation. NO stimulates sGC, increasing cytoplasmic cyclic guanosine monophosphate (cGMP) production and the activation of PKG (Figure 1D). If NO was acting through this signaling cascade, its effects should be mimicked by analogs of cGMP and blocked by inhibitors of PKG. Indeed, antagonism of PKG by including Rp-8-Br-PET-cGMPS (3 μM) in the patch pipette blocked the effects of SNAP (Figure 1C) and brief bath application of the membrane permeable cGMP analog 8-bromo-cGMP (8Br-cGMP, 500 μM) produced a robust and persistent depression of corticostriatal EPSCs, mimicking SNAP (Figure 1E). Moreover, the ability of SNAP to decrease EPSC amplitudes was occluded in cells that had been pre-incubated in 8Br-cGMP (Figure S1B). The effects of cGMP analogs on corticostriatal EPSCs were similar in direct pathway SPNs (dSPNs) and indirect pathway SPNs (iSPNs) (Figure 1F), consistent with the broad striatal distribution of signaling molecules in the NO pathway (Bredt et al., 1990; Vincent, 1994).
Because activation of the NO/cGMP/PKG pathway has been shown to augment the activity of cholinergic interneurons (Centonze et al., 2001), the effect of SNAP was examined in the presence of antagonists of cholinergic signaling. Perfusion of the nicotinic receptor antagonist mecamylamine (10 μM) and the muscarinic receptor antagonist scopolamine (10 μM) prior to and throughout SNAP perfusion did not alter the depression of corticostriatal EPSCs (Figure 1G).
To rule out any effects of cellular dialysis with the patch electrode, experiments using SNAP and 8Br-cGMP were also performed in the perforated patch recording configuration. The responses observed were similar to those seen in whole cell (Figure S1C). Because the NO-induced reduction in evoked EPSC amplitude persisted for as long as recordings could be maintained, it will be referred to as NO-LTD.
Optogenetic activation of PLTS interneurons induced NO-LTD
Although the effects of SNAP and cGMP analogs were consistent and robust, this does not prove that NO signaling is engaged by the striatal circuitry to control synaptic strength (Feelisch, 1998). In the striatum, neuronal nitric oxide synthase (nNOS) is expressed robustly only in interneurons that co-express somatostatin, neuropeptide Y and gamma amino butyric acid (GABA) (Tepper et al., 2010). Because of their distinctive physiological properties, these cells also are referred to as persistent and low threshold spiking interneurons (PLTSIs). If NO is a bona fide modulator of plasticity, the activation of PLTSIs and their generation of NO should induce NO-LTD. To test this hypothesis, we used optogenetic methods to selectively express channelrhodopsin2 (ChR2) in striatal PLTSIs (Holley et al., 2015; Witten et al., 2011; Zhang et al., 2006) (Figure 2A and S2A). In ex vivo brain slices from these mice, SPNs were sorted into two groups: those in which full-field optogenetic stimulation of PLTSIs evoked GABAergic responses (coupled) and those that showed no response (no coupling) (Figure S2B). In SPNs coupled to PLTSIs, optical stimulation for 5 minutes at 15 Hz, a firing frequency that spontaneously active PLTSIs do not normally achieve in slice, led to a robust LTD at corticostriatal synapses (Figures 2B and S2C). Exciting PLTSIs to firing frequencies lower than 15Hz, or stimulating for shorter durations, failed to elicit LTD (Figure S2D). As expected, when the nNOS inhibitor NG-Nitro-L-arginine methyl ester (L-NAME, 100 μM) or the PKG inhibitor Rp8Br (3 μM) were applied throughout the recording period, LTD induction was blocked (Figure 2C and 2D). Similarly, pre-incubation of slices with SNAP occluded PLTS induction of LTD (S2E). In contrast, in SPNs where optical stimulation failed to evoke GABAergic responses (possibly because optical stimulation was ineffective in activating PLTSIs), the same stimulation protocol had no effect on corticostriatal synaptic transmission (Fig. 2D and S2C–D). Taken together, these data argue that NO signaling originating from PLTSIs induces LTD at corticostriatal synapses.
Figure 2.

NO producing interneurons can induce NO-LTD. (A) Simplified diagram depicting the experimental design used to optogenetically probe PLTSI involvement in NO-LTD. (B) Full field LED activation of PLTSIs for 5 minutes at 15 Hz induced NO-LTD at corticostriatal synapses in SPNs synaptically coupled to PLTSIs. Example traces shown to the right. (C) LTD was blocked by continuous bath application of the nNOS inhibitor L-NAME (100 μM) or inclusion of Rp8Br (3 μM) in the patch pipette (control: n=5; L-NAME: n=6; Rp8Br: n=5). Example EPSCs are shown at right. (D) Summary data for NO-LTD induced by PLTSI activation. SPNs not coupled to PLTSIs showed no response to the PLTSI stimulation paradigm (n=4). **p<0.01 Mann-Whitney nonparametric test. Scale bars: (B), (C) 20 pA, 10 ms.
NO-LTD was independent of eCb signaling and was expressed post-synaptically
Phenomenologically, the LTD induced by NO signaling (NO-LTD) was very similar to that induced by eCb signaling (eCb-LTD). However, the two forms of LTD had very different determinants. First, NO-LTD induced by cGMP analogs in iSPNs and dSPNs was unaffected by antagonizing D2 dopamine receptors with sulpiride (1 μM) (Figure S3A). Second, NO-LTD induced by cGMP analogs in iSPNs and dSPNs was unaffected by antagonizing CB1 eCb receptors with AM251 (2 μM) (Figure S3A). Third, NO-LTD was unaffected by dialyzing neurons with the Ca2+ chelator BAPTA (20 mM) (Figure S3C). Each of these interventions is known to block eCb-LTD (Kreitzer and Malenka, 2008; Mathur and Lovinger, 2012). Another difference between these forms of plasticity was that NO-LTD was readily induced at thalamostriatal glutamatergic synapses, in contrast to eCb-LTD (Wu et al., 2015)(Figure S3C–D).
One of the most important features of eCb-LTD is that while it is induced post-synaptically in SPNs, it is expressed pre-synaptically as a reduction in glutamate release probability (Kreitzer and Malenka, 2008; Mathur and Lovinger, 2012). Four observations support the proposition that unlike eCb-LTD, NO-LTD was expressed post-synaptically. First, the expression of NO-LTD was not accompanied by a change in paired-pulse facilitation, a hallmark of pre-synaptic LTD (Figure S3E). Second, the induction of NO-LTD was independent of pre-synaptic activity; cessation of afferent fiber stimulation during the application of 8Br-cGMP had no effect on the magnitude of NO-LTD (Figure S3F), unlike eCb-LTD (Adermark et al., 2009). Third, intracellular dialysis of D15 – a polypeptide previously shown to disrupt the interaction between dynamin and amphiphysin, which is crucial for AMPAR endocytosis (Carroll et al., 1999; Morishita et al., 2005) – blocked NO-LTD expression while dialysis of a scrambled peptide had no effect on NO-LTD (Figures 3A–B). Moreover, D15 dialysis did not alter eCb-LTD induced by the metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine (DHPG, 100 μM) (Figure 3B). Fourth, transient application of 8Br-cGMP persistently decreased the amplitude of EPSCs evoked by uncaging of glutamate at visualized SPN spines (Figure 3C–3F). Taken together, these results provide strong evidence for a post-synaptic locus of NO-LTD expression, contrasting it with other forms of LTD described in the striatum.
Figure 3.

NO-LTD is post-synaptically expressed. (A) 8-Br-cGMP (500 μM) induced NO-LTD which was blocked by intracellular application of the endocytosis-disrupting peptide, D15 (1–2 mM; control: black trace, n=6; D15: red trace, n=6). (B) Quantification of the population data shown in (A). A scrambled peptide (sD15) had no effect on 8-Br-cGMP NO-LTD (1–2 mM; n=6). As expected, sD15 did not disrupt LTD induced by DHPG (100 μM; n=6). * p<0.05, signed rank test. (C) Simplified diagram illustrating 2PLUG experiments. (D) Representative 2PLSM image of iSPN dendritic spine at which 2PLUG was performed (blue circle). (E) 8-Br-cGMP induced a long-lasting decrease in uEPSC amplitude. Example uEPSCs before (pre-inc) and after transient bath application of 8-Br-cGMP (500 μM) are shown to the right. Time matched control traces (no 8-Br-cGMP application; post-control) are shown for comparison. * p<0.05, signed rank test. (F) Summary data for 8Br-cGMP effect in SPN dendritic spines (8-Br-cGMP: n=26 spines, 5 cells; control: n=29 spines, 3 cells). Scale bars: (D) 3 μm; (E) 3 pA (right panel, top) and 5 pA, 50 ms (right panel, bottom).
NO signaling blocked eCb-LTD induction
Are NO-LTD and eCb-LTD completely independent? As shown above, NO-LTD does not depend upon signaling events (e.g., CB1R or D2R activation) known to be necessary for induction of eCb-LTD. But could NO signaling blunt eCb-LTD? This would help to explain why the two forms of LTD have not been distinguished to date. The core mechanisms controlling eCb-LTD are well described, making pursuit of this aim tractable (Figure 4A) (Mathur and Lovinger, 2012). As a first step toward characterizing their interaction, NO-LTD was induced by SNAP (100 μM) and a positive allosteric modulator of sGC (BAY-41-2272, 10 μM) (Stasch et al., 2001); BAY-41-2272 was used because it potentiated the ability of SNAP to induce LTD, was inhibited by Rp8Br, and had no effect on its own (Figure S4A–B) (Stasch et al., 2001). SPNs were then held at −50 mV, afferent fibers stimulated at low frequency and the mGluR agonist DHPG (100 μM) applied; normally, this is a very robust means of inducing eCb-LTD (Mathur and Lovinger, 2012; Surmeier et al., 2009) (Figure 4B). However, in this situation, the protocol failed to induce a lasting change in EPSC amplitude (Figure 4B), arguing that NO-LTD blocked eCb-LTD induction. Bath application of 8Br-cGMP had a very similar effect, disrupting eCb-LTD (Figure S4C). The ability of NO signaling to disrupt eCb-LTD was not pre-synaptic in origin as direct application of the CB1R agonist WIN55,212-2 mesylate (WIN) induced a robust LTD in the presence of SNAP and BAY-41-2272 (Figure 4C). The other possibility is that NO signaling blunted eCb generation. The production of eCbs by SPNs requires the opening of L-type Ca2+ channels and an elevation in intracellular Ca2+ concentration (Mathur and Lovinger, 2012). In endocrine cells, phosphorylation of L-type channels by PKG decreases their opening (Mahapatra et al., 2012), raising the possibility that NO signaling was suppressing dendritic L-type channels required for sustained eCb synthesis. Indeed, co-application of SNAP (100 μM) and BAY-41-2272 (10 μM) decreased SPN intraspine Ca2+ transients evoked by somatic depolarization (Figure 4D and 4E). Antagonism of L-type channels with isradipine eliminated the effects of SNAP/BAY on intraspine Ca2+ transients, confirming the L-type specificity of the modulation (Figure 4D). Thus, NO signaling disrupted the induction of eCb-LTD at least in part by attenuating activity-dependent, post-synaptic Ca2+ entry through L-type channels (Figure 4F).
Figure 4.

NO signaling occludes eCb-LTD through L-type current inhibition. (A) Simplified diagram outlining the mechanisms behind eCB-LTD. (B) Induction of eCb-LTD by DHPG (100 μM; black trace, n=6) was prevented by SNAP (100 μM) and BAY-41 (10 μM) perfused 10 min before and throughout recording (red trace: n=8). *p<0.05, rank sum test. Example EPSCs are shown to the right. (C) LTD induced by pharmacological activation of pre-synaptic CB1 receptors with WIN (2 μM) was not blocked by SNAP/BAY-41 (n=7). *p<0.05, signed rank test. (D) 2PLSM images of an iSPN (left) and dendritic spine from which a Ca2+ imaging line scan (middle, blue line) was performed. Bath application of SNAP/BAY-41 reduced peak calcium influx (n=26 spines, 3 cells). This reduction was occluded in the presence of 5 μM isradipine (n=22 spines, 3 cells). (E) Quantification of (D). * p<0.05, signed rank test. (F) Integrated diagram depicting the interaction between eCB and NO LTD. Scale bars represent: (B) 20 pA, 25 ms; (C) 40 pA, 20 ms; (D) 20 μm (left panel), 3 μm (middle panel), 4%ΔF/Fo and 200 ms (right panel).
DISCUSSION
The data presented make the case that NO release by PLTSIs induces a post-synaptically expressed form of LTD at SPN axospinous glutamatergic synapses. Although common in other regions of the brain (Garthwaite and Boulton, 1995; Holscher, 1997; Susswein et al., 2004), this is the first demonstration that striatal SPNs manifest this form of plasticity, complementing post-synaptically expressed LTP (Surmeier et al., 2009). The molecular mechanisms underlying NO-LTD remain to be fully elucidated, but our results point clearly to AMPA receptor endocytosis. Elsewhere, PKG, which was necessary for NO-LTD, is known to regulate the activity of DARPP-32 and protein phosphatase 1 – both of which have been implicated in phosphorylation of the GluA2 AMPAR subunit (Walaas et al., 2011).
How does the existence of NO-LTD change our understanding of the striatal network? PLTSIs appear to be part of a corticostriatal feed-forward circuit that is activated in parallel with SPNs (Tepper et al., 2010). Thus, NO-LTD could counterpoise dopamine-dependent potentiation of glutamatergic synaptic strength in SPNs during goal-based learning, helping to maintain a stable level of regional responsiveness by diminishing the strength of inactive synapses neighboring those undergoing potentiation. For this mechanism to work, the signaling mechanisms governing LTP induction would have to oppose those of NO-LTD. As Ca2+ entry through NMDA receptors is necessary for LTP induction in SPNs (Shen et al., 2008), phosphodiesterase 1 – a striatally enriched, Ca2+/calmodulin dependent phosphodiesterase that preferentially degrades cGMP – might mediate this interaction (Bender, 2006). An additional possibility may be that NO released from PLTSIs also simultaneously tunes GABAergic inputs within the striatum, specifically from other interneurons onto SPNs, a possibility that has been described elsewhere (Nugent et al., 2007; Sagi et al., 2014).
In disease states, deficits in NO-LTD could contribute to pathological remodeling of the striatal circuitry. For example, in models of Parkinson’s disease (PD), striatal NO signaling falls (Sancesario et al., 2004). The loss of feed-forward NO-LTD could contribute to hyperactivity of iSPNs and hypokinetic symptoms of the disease (Albin et al., 1989). The sustained elevation in iSPN activity appears to drive profound homeostatic pruning of corticostriatal synapses (Day et al., 2006; Fieblinger et al., 2014), disrupting patterns of connectivity shaped by experience. An impairment in NO-LTD might also contribute to levodopa-induced strengthening of dSPN corticostriatal synapses and dyskinesia in PD (Picconi et al., 2011). How pathological changes in NO signaling preferentially effect dSPNs and iSPNs requires further study.
EXPERIMENTAL PROCEDURES
Tissue preparation
6–12 week old, male hemizygous C57Bl/6 mice expressing eGFP under control of either the Drd1a or Drd2 receptor regulatory elements; Thy1-ChR2 mice; SOM-IRES-Cre mice (Ssttm2.1(cre)Zjh/J), and KH288 mice developed by the GENSAT Project were used. Mice were killed by decapitation, the brain removed in artificial cerebral spinal fluid (ACSF), and para-sagittal brain slices containing striatum were cut at 275–300μM. Slices were incubated in 30–33°C ACSF until recording.
Electrophysiology
After incubation, slices were transferred to a recording chamber perfused with oxygenated ACSF at 30–33°C. Cells were filled with a Cs-based internal. SPNs were voltage-clamped at −70 mV and stimuli delivered at 0.1Hz through a concentric bipolar electrode placed in the cortex in the presence of 10 μM gabazine to block GABAA currents. Optical stimuli were delivered through the epifluorescent light path using an LED light source (CoolLED). Access resistance was monitored throughout recording and cells in which a change >20% baseline was observed were excluded.
Viral gene delivery
For optogenetic experiments a cre-dependent AAV-ChR2 vector was injected into the striatum or thalamus of SOM-Cre or KH288-Cre mice, respectively. Mice were sacrificed 2–3 weeks post infection.
2-photon laser scanning microscopy (2PLSM)
iSPNs were identified by somatic EGFP expression. Following patch rupture the internal solution was allowed to equilibrate for 10–15 minutes before imaging.
Ca2+ imaging
SPNs were loaded with 25 μM Alexa 568 and 600 μM Fluo 5F via the recording electrode, which contained a CsMeSO3 internal. Green fluorescent line scan signals were acquired from dendritic spines, 200 ms before a 300 ms voltage step from −70 to 0 mV. Changes in Fluo 5F fluorescence were measured as ΔF/Fo.
2-photon laser uncaging of glutamate (2PLUG)
Glutamate uncaging was achieved using a Verdi/Mira laser system. 5 mM MNI-glutamate was superfused over the slice at 0.4 ml/hr using a syringe pump and multi-barreled perfusion manifold (Cell MicroControls, Norfolk, VA). Glutamate was uncaged adjacent to individual spines using 1 ms pulses of 725 nm light. Photolysis power was tuned via a third Pockels cell modulator (Con Optics, Danbury, CT) to achieve uncaged-EPSCs (uEPSCs) ≤5 pA.uEPSC amplitudes were measured from averaged (5 repetitions) traces.
Chemical and Reagents
Rp-8-Br-PET-cGMPS was obtained from Biolog Life Science Institute, L-NAME was obtained from Sigma Aldrich. All other reagents were obtained from Tocris – Custom peptides were synthesized by the Stanford peptide facility and used at a concentration of 1–2 mM.
Data Analysis and Statistics
The magnitude of LTD was calculated as a percentage of the baseline (minutes 0–5 of recording time). Unless otherwise noted, the average of minutes 30–35 of each recording was used to calculate effects of drug application and data are reported as Median, 1st and 3rd quartiles, minimum/maximum (whiskers), and outliers removed. dSPNs and iSPNs were combined in data sets unless specified (e.g. Figure 1E–F).
Supplementary Material
Acknowledgments
This work was supported by grants from NIH (NS34696), the JPB Foundation and the Cure Huntington’s Disease Initiative to D.J.S.; a grant from NSF (610-5108000-60032448) to I.V.R.; a fellowship from NIH (NS041234) to A.M.; and a fellowship from the Japan Society for the Promotion of Science Postdoctoral Fellow for Research Abroad to A.T. We thank Sasha Ulrich and David Wokosin for their technical assistance.
Footnotes
An extended description of the experimental procedures is included in the Supplemental Experimental Procedures.
AUTHOR CONTRIBUTIONS
The experiments were originally conceived and designed by I.R., A.M. and D.J.S. Experiments were performed and analyzed by I.R., A.M., J.P., A.T., and S.Z. The manuscript was written and figures constructed by I.R., A.M. and D.J.S. All authors participated in editing of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adermark L, Talani G, Lovinger DM. Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. European Journal of Neuroscience. 2009;29:32–41. doi: 10.1111/j.1460-9568.2008.06551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Ariano MA. Distribution of components of the guanosine 3′,5′-phosphate system in rat caudate-putamen. Neuroscience. 1983;10:707–723. doi: 10.1016/0306-4522(83)90212-9. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Delgado MR, Hikosaka O. The Role of the Dorsal Striatum in Reward and Decision-Making. Journal of Neuroscience. 2007;27:8161–8165. doi: 10.1523/JNEUROSCI.1554-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacological Reviews. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Centonze D, Sancesario G, Morello M, Giorgi M, Pisani A, Bernardi G. A critical role of the nitric oxide/cGMP pathway in corticostriatal long-term depression. J Neurosci. 1999;19:2489–2499. doi: 10.1523/JNEUROSCI.19-07-02489.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Maj R, Pisani A, Mercuri NB, Bernardi G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J Neurosci. 1992;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Lüscher C, Altschuler Y, Nicoll RA, Malenka RC, Von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centonze D, Gubellini P, Bernardi G, Calabresi P. Permissive role of interneurons in corticostriatal synaptic plasticity. Brain Res Brain Res Rev. 1999;31:1–5. doi: 10.1016/s0165-0173(99)00018-1. [DOI] [PubMed] [Google Scholar]
- Centonze D, Picconi B, Gubellini P, Bernardi G, Calabresi P. Dopaminergic control of synaptic plasticity in the dorsal striatum. Eur J Neurosci. 2001;13:1071–1077. doi: 10.1046/j.0953-816x.2001.01485.x. [DOI] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, et al. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Ding J-D, Burette A, Nedvetsky PI, Schmidt HHHW, Weinberg RJ. Distribution of soluble guanylyl cyclase in the rat brain. J Comp Neurol. 2004;472:437–448. doi: 10.1002/cne.20054. [DOI] [PubMed] [Google Scholar]
- Feelisch M. The use of nitric oxide donors in pharmacological studies. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:113–122. doi: 10.1007/pl00005231. [DOI] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nature Communications. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. European Journal of Neuroscience. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of Striatal Projection Systems by Dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holley SM, Joshi PR, Parievsky A, Galvan L, Chen JY, Fisher YE, Huynh MN, Cepeda C, Levine MS. Enhanced GABAergic Inputs Contribute to Functional Alterations of Cholinergic Interneurons in the R6/2 Mouse Model of Huntington’s Disease. eNeuro. 2015;2 doi: 10.1523/ENEURO.0008-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC. Dopamine Modulation of State-Dependent Endocannabinoid Release and Long-Term Depression in the Striatum. Journal of Neuroscience. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal Plasticity and Basal Ganglia Circuit Function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Tyler EC, Merritt A. Short- and long-term synaptic depression in rat neostriatum. J Neurophysiol. 1993;70:1937–1949. doi: 10.1152/jn.1993.70.5.1937. [DOI] [PubMed] [Google Scholar]
- Mahapatra S, Marcantoni A, Zuccotti A, Carabelli V, Carbone E. Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J Physiol. 2012;590:5053–5073. doi: 10.1113/jphysiol.2012.236729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur BN, Lovinger DM. Endocannabinoid-dopamine interactions in striatal synaptic plasticity. Frontiers in Pharmacology. 2012;3:66. doi: 10.3389/fphar.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–7411. doi: 10.1523/JNEUROSCI.6250-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Picconi B, Bagetta V, Ghiglieri V, Paille V, Di Filippo M, Pendolino V, Tozzi A, Giampa C, Fusco FR, Sgobio C, et al. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 2011;134:375–387. doi: 10.1093/brain/awq342. [DOI] [PubMed] [Google Scholar]
- Sagi Y, Heiman M, Peterson JD, Musatov S, Scarduzio M, Logan SM, Kaplitt MG, Surmeier DJ, Heintz N, Greengard P. Nitric oxide regulates synaptic transmission between spiny projection neurons. Proc Natl Acad Sci U S A. 2014;111:17636–17641. doi: 10.1073/pnas.1420162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammut S, Park DJ, West AR. Frontal cortical afferents facilitate striatal nitric oxide transmission in vivo via a NMDA receptor and neuronal NOS-dependent mechanism. J Neurochem. 2007;103:1145–1156. doi: 10.1111/j.1471-4159.2007.04811.x. [DOI] [PubMed] [Google Scholar]
- Sancesario G, Giorgi M, D’Angelo V, Modica A, Martorana A, Morello M, Bengtson CP, Bernardi G. Down-regulation of nitrergic transmission in the rat striatum after chronic nigrostriatal deafferentation. European Journal of Neuroscience. 2004;20:989–1000. doi: 10.1111/j.1460-9568.2004.03566.x. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasch JP, Becker EM, Alonso-Alija C, Apeler H, Dembowsky K, Feurer A, Gerzer R, Minuth T, Perzborn E, Pleiss U, et al. NO-independent regulatory site on soluble guanylate cyclase. Nature. 2001;410:212–215. doi: 10.1038/35065611. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Current Opinion in Neurobiology. 2009;19:621–628. doi: 10.1016/j.conb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susswein AJ, Katzoff A, Miller N, Hurwitz I. Nitric oxide and memory. Neuroscientist. 2004;10:153–162. doi: 10.1177/1073858403261226. [DOI] [PubMed] [Google Scholar]
- Tepper JM, Tecuapetla F, Koós T, Ibañez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. doi: 10.3389/fnana.2010.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent SR. Nitric oxide: a radical neurotransmitter in the central nervous system. Prog Neurobiol. 1994;42:129–160. doi: 10.1016/0301-0082(94)90023-x. [DOI] [PubMed] [Google Scholar]
- Walaas SI, Hemmings HCJ, Greengard P, Nairn AC. Beyond the dopamine receptor: regulation and roles of serine/threonine protein phosphatases. Front Neuroanat. 2011;5:50. doi: 10.3389/fnana.2011.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IB, Steinberg EE, Lee SY, Davidson TJ, Zalocusky KA, Brodsky M, Yizhar O, Cho SL, Gong S, Ramakrishnan C, et al. Recombinase-Driver Rat Lines: Tools, Techniques, and Optogenetic Application to Dopamine-Mediated Reinforcement. Neuron. 2011;72:721–733. doi: 10.1016/j.neuron.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB. Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 2015;10:75–87. doi: 10.1016/j.celrep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Meth. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.