

SUMMARY
A balanced interaction between dopaminergic and cholinergic signaling in the striatum is critical to goal-directed behavior. But how this interaction modulates corticostriatal synaptic plasticity underlying learned actions remains unclear – particularly in direct-pathway spiny projection neurons (dSPNs). Our studies show that in dSPNs endogenous cholinergic signaling through M4 muscarinic receptors (M4Rs) promoted long-term depression of corticostriatal glutamatergic synapses, by suppressing regulator of G-protein signaling type 4 (RGS4) activity, and blocked D1 dopamine receptor dependent long-term potentiation (LTP). Furthermore, in a mouse model of L-3,4-dihydroxyphenylalanine (L-DOPA) induced dyskinesia (LID) in Parkinson’s disease (PD), boosting M4R signaling with positive allosteric modulator (PAM) blocked aberrant LTP in dSPNs, enabled LTP reversal, and attenuated dyskinetic behaviors. An M4R PAM also was effective in a primate LID model. Taken together, these studies identify an important signaling pathway controlling striatal synaptic plasticity and point to a novel pharmacological strategy for alleviating LID in PD patients.
Keywords: Spike-timing-dependent plasticity, long-term depression, long-term potentiation, depotentiation, spiny projection neuron, cholinergic interneuron, positive allosteric modulator, chemogenetics, RGS4
INTRODUCTION
The striatum is a key component of the basal ganglia circuitry controlling action selection and habit learning (Gerfen and Surmeier, 2011; Maia and Frank, 2011; Yin and Knowlton, 2006). It is widely assumed that activity-dependent alterations in the strength of corticostriatal glutamatergic synapses formed on principal spiny projection neurons (SPNs) underlie striatal learning (Lerner and Kreitzer, 2011; Wickens et al., 2003). Not only are these synapses important for normal learning, their dysregulation has been implicated in a number of psychomotor diseases, including Parkinson’s disease (PD) (Gerfen and Surmeier, 2011; Kreitzer and Malenka, 2007; Shen et al., 2008).
One of the most important modulators of corticostriatal synapses is dopamine (DA) (Calabresi et al., 2007; Gerfen and Surmeier, 2011; Kreitzer and Malenka, 2007; Lovinger, 2010; Shen et al., 2008). By virtue of their differential expression of G-protein linked DA receptors, striatal indirect-pathway SPNs (iSPNs) and direct-pathway SPNs (dSPNs) respond to DA in contrasting ways. In D2 DA receptor (D2R) expressing iSPNs, DA promotes the induction of Hebbian long-term depression (LTD) at corticostriatal synapses and opposes A2a adenosine receptor (A2aR) mediated induction of long-term potentiation (LTP) (Shen et al., 2008). This is accomplished by bidirectionally regulating adenylyl cyclase (AC) through Gi-coupled D2Rs and Golf-coupled A2aRs (Augustin et al., 2014; Higley and Sabatini, 2010; Lerner et al., 2010).
In dSPNs, Golf-coupled D1 DA receptors (D1Rs) are necessary for the induction of LTP. D1R signaling also disrupts the induction of Hebbian LTD (Fino et al., 2010; Pawlak and Kerr, 2008; Shen et al., 2008; Wu et al., 2015; Yagishita et al., 2014). But it is unclear whether there is a receptor that is homologous to the D2R in dSPNs that promotes LTD and opposes LTP induction. One candidate for this role is the Gi-coupled M4 muscarinic receptor (M4R). The M4R is the most abundant striatal muscarinic receptor and it is preferentially expressed in dSPNs where it is clustered near axospinous glutamatergic synapses (Bernard et al., 1992; Hersch et al., 1994). Giant cholinergic interneurons (ChIs) have dense terminal fields that overlap those of DA neurons, allowing M4R suppression of D1R signaling through AC (Jeon et al., 2010; Sánchez et al., 2009). Nevertheless, the role of M4Rs in regulating synaptic plasticity in dSPNs has not been determined.
Pinning down the role of M4Rs in modulating corticostriatal synaptic plasticity could have translational implications. One of the unmet clinical needs for PD patients is a strategy for reducing L-DOPA-induced dyskinesia (LID). L-DOPA treatment is a mainstay for early and mid-stage PD patients. Although it is initially effective in alleviating symptoms, as the disease progresses, L-DOPA becomes less effective and the dose required to achieve symptomatic benefit rises. In most patients, high doses of L-DOPA produce unwanted dyskinetic movements. Although it is unlikely that dysregulation of dSPN corticostriatal synapses is solely responsible for LIDs, many lines of evidence suggest that aberrant D1R-dependent synaptic plasticity is a major factor (Feyder et al., 2011; Jenner, 2008; Picconi et al., 2003). In particular, it is thought that repeated L-DOPA treatment, abnormally increases D1R signaling, leading to pathological LTP of corticostriatal synapses and inappropriately timed or scaled dSPN activity (Picconi et al., 2003). Antagonizing D1Rs is not a viable therapeutic strategy because it diminishes the symptomatic benefit of L-DOPA treatment. Hence, identifying an alternative means of normalizing D1R signaling could provide relief from LID.
The goal of the studies described here was to test two linked hypotheses. The first hypothesis was that dSPN M4Rs suppressed Hebbian LTP induction through Gi-coupled inhibition of AC and promoted LTD by diminishing regulator of G-protein signaling type 4 (RGS4) activity (Blazer et al., 2015; Lerner and Kreitzer, 2012). The second hypothesis was that boosting M4R signaling would diminish the deficits in dSPN synaptic plasticity and aberrant behavior in models of LID. The data presented confirm both hypotheses.
RESULTS
M4R signaling was necessary for LTD at dSPN glutamatergic synapses
To determine if M4Rs were modulating synaptic plasticity, a spike-timing-dependent plasticity (STDP) paradigm was used (Fino et al., 2010; Nazzaro et al., 2012; Pawlak and Kerr, 2008; Shen et al., 2008; Shindou et al., 2011; Wu et al., 2015; Yagishita et al., 2014). SPNs were interrogated in ex vivo corticostriatal parasagittal brain slices from Drd1a or Drd2 bacterial artificial chromosome (BAC) transgenic mice in which dSPNs and iSPNs (respectively) expressed tdTomato or enhanced green fluorescent protein (eGFP), allowing them to be reliably sampled. Once identified, neurons were monitored and controlled with perforated-patch recording, unless otherwise stated (Figure 1A). Synaptic plasticity was induced by pairing local stimulation of glutamatergic afferent fibers with postsynaptic spikes evoked by short bursts of intracellular current injection that were repeated at 5 Hz (Figures 1B, 5A). As showed in earlier work with dSPNs, pairing postsynaptic spiking with a trailing presynaptic volley failed to change excitatory postsynaptic potential (EPSP) amplitudes (Figure 1C). However, the same protocol led to LTD in dSPNs when D1Rs were antagonized by bath application of the D1R antagonist SCH23390 (3 μM; n = 7; p < 0.05, Wilcoxon signed rank test) (Figure 1D). The engagement of D1Rs in our experimental paradigm likely stems from the fact that local electrical stimulation activates not only glutamatergic fibers, but dopaminergic fibers as well (Threlfell et al., 2012). Regardless, these results confirm that D1R signaling disrupts the induction of LTD in dSPNs (Shen et al., 2008; Wu et al., 2015).
Figure 1. M4R signaling promotes induction of LTD at dSPN glutamatergic synapses.

(A) The experimental configuration. (B) The post-pre theta-burst pairing protocol for induction of LTD. (C) LTD was not induced by a post-pre timing pairing in dSPNs. Plots show EPSP amplitude (amp) and membrane input resistance (Ri) as a function of time. The dashed line represents the average EPSP amplitude before induction. STDP pairing is indicated by the vertical bar. Filled symbols specify the averages of 12 trials (± SEM). The averaged EPSP traces before and after induction are shown at the top. Scale bars, 2 mV x 80 ms. (D) In the presence of D1R antagonist SCH23390 (3 μM), a post-pre timing pairing revealed LTD; the LTD was disrupted by addition of the selective M4R antagonist MT3 (100 nM). Data are represented as mean ± SEM. Plot of the average EPSP amplitudes as a function of time. (E) With 20 repetitions of the pairing protocol, bath perfusion of VU10010 did not lead to LTD induction. However, when the number of repetitions was increased to 60 in the presence of the M4R PAM, LTD induction was robust. Plot of the average EPSP amplitudes as a function of time. Error bars represent ± SEM. (F) In D1-Cre mice, post-pre pairing led to LTD when VU10010 (5 μM) was bath applied; but the same protocol had no effect on the induction of LTD in D1-M4-KO mice. Data are shown as mean ± SEM. Plot of the average EPSP amplitudes as a function of time (see also Figures S1 and S2).
Figure 5. M4R stimulation blunts LTP induction at dSPN synapses and enables depotentiation.

(A) The pre-post theta-burst pairing protocol for induction of LTP. (B) LTP was induced by a pre-post timing pairing in dSPNs. Plots show EPSP amplitude (amp) and membrane input resistance (Ri) as a function of time. The dashed line represents the average EPSP amplitude before induction. The vertical bar indicates STDP induction. Filled symbols specify the averages of 12 trials (± SEM). The averaged EPSP traces before and after induction are shown at the top. Scale bars, 4 mV x 80 ms. (C) LTP induction was disrupted by MEK inhibitor U0126 (10 μM). Plot of the average EPSP amplitudes as a function of time. (D) LTP induction was also blocked by the M4R PAM VU10010 (5 μM). Solid line (average) and gray shadow (± SEM) are control LTP from the panel (C) for reference. (E) Synaptic depotentiation was not induced by LFS (@2 Hz for 10 min) during the time indicated by the horizontal bar. However, pre-perfusion of a D1R antagonist SCH23390 (3 μM) facilitated depotentiation. (F) VU10010 (5 μM) also promoted the reversal of established LTP. Solid line (average) and gray shadow (± SEM) are control from the panel (E) for reference. Error bars represent SEM.
Although these experiments establish the ability of D1Rs to disrupt LTD induction in dSPNs, they do not make it clear whether activation of other G protein coupled receptors is necessary for induction. Previous work has shown that type 5 metabotropic glutamate receptors (mGluR5) are critical for STDP LTD (Fino et al., 2010; Nazzaro et al., 2012; Shen et al., 2008), but are there others? As outlined above, M4Rs might play a role in dSPNs that is analogous to that of D2Rs in iSPNs, which are necessary for LTD induction (Kreitzer and Malenka, 2007; Lerner and Kreitzer, 2012). To test this possibility, the selective M4R antagonist – muscarinic toxin 3 (MT3, 100 nM) – was bath applied (along with the D1R antagonist SCH23390) prior to stimulation. MT3 blocked induction of LTD (SCH23390 n = 7; SCH23390 + MT3 n = 6; p < 0.05, Mann-Whitney rank sum test) (Figure 1D), establishing the necessity of M4R stimulation for dSPN STDP LTD, even when D1Rs were blocked. In contrast, M4R antagonism with MT3 had no effect on STDP LTD in iSPNs (Figure S1).
If M4R signaling was robust, could it overcome concomitant D1R signaling? To amplify appropriately timed endogenous cholinergic signaling in the STDP protocol, an M4R positive allosteric modulator (PAM) was used; PAMs do not stimulate receptors directly but enhance their response to endogenous acetylcholine (ACh) (Brady et al., 2008; Shirey et al., 2008). In the absence of a D1R antagonist, 20 repetitions of the pairing protocol did not lead to LTD induction in the presence of the M4R PAM VU10010 (5 μM) (n = 7; p > 0.05, Wilcoxon test) (Figure 1E). This repetition number is adequate for LTD induction in iSPNs (Shen et al., 2008). However, when the number of repetitions was increased to 60 (matching the number of afferent volleys used in the LTP induction protocol, see Experimental Procedures) in the presence of the M4R PAM, LTD induction was robust in dSPNs (n = 6; p < 0.05, Wilcoxon test) (Figure 1E). Consistent with the work using conventional plasticity protocol (Calabresi et al., 2007; Lovinger, 2010), blocking N-methyl-D-aspartate receptors (NMDARs) with APV (50 μM) had no effect on the M4R-mediated LTD induction (n = 6; p < 0.05, Wilcoxon test) (Figure S2A). These results demonstrate that enhancing M4R signaling can lead to LTD induction even when D1Rs are activated.
Although the predominant expression site of striatal M4Rs is dSPNs, they are also positioned at other sites in the striatum where they are poised to exert concerted effects. For example, activation of M4Rs on corticostriatal terminals can diminish glutamate release (Higley et al., 2009; Pancani et al., 2014). To determine whether postsynaptic M4Rs mediate the timing-dependent induction of LTD in dSPNs, Cre/loxP based strategy was employed to generate mutant mice that lack M4Rs only in dSPNs [D1-M4-knockout (KO) mice] (Jeon et al., 2010). These mice, which lacked M4Rs in dSPNs, and D1-Cre mice were then injected with an adeno-associated virus (AAV) containing a double-floxed inverted tdTomato transgene, allowing dSPNs to be optically identified. Preceding synaptic stimulation (−10 ms) with a short burst of postsynaptic spikes resulted in STDP LTD in D1-Cre mice when VU10010 (5 μM) was bath applied, as described above. But the same protocol failed to alter EPSP amplitudes in D1-M4-KO mice (D1-Cre n = 6; D1-M4-KO n = 6; p < 0.05, Mann-Whitney test) (Figure 1F). These results demonstrate that postsynaptic, but not presynaptic, M4Rs promote the induction of LTD in dSPNs.
One of the potential limitations of these experiments is that in an ex vivo slice preparation at 30–31°C, spontaneous ChI activity is low. One way to compensate for lower than normal cholinergic signaling is to use designer receptors exclusively activated by designer drugs (DREADDs) to selectively increase ChI discharge rate (Rogan and Roth, 2011). To this end, bi-transgenic mice expressing tdTomato in dSPNs and Cre-recombinase under control of the choline acetyltransferase (ChAT) regulatory elements received striatal injections of an AAV containing an hM3D(q) expression construct with an upstream floxed STOP cassette; this STOP cassette is effectively excised by Cre-recombinase leading to DREADD expression (Figure 2A). When activated by its cognate ligand clozapine-N-oxide (CNO), the hM3D(q) receptor activates phospholipase C (PLC) signaling (Rogan and Roth, 2011). The PLC-dependent depletion of a membrane lipid phosphotidylinositol 4,5-bisphosphate (PIP2) decreases KCNQ (Kv7) and Kir2 K+ channel opening, enhancing action potential generation (Rogan and Roth, 2011). Five to six weeks after AAV injection, brain slices were made from these mice; in these slices, bath perfusion of CNO (10 μM) increased the discharge rate of ChIs into the physiological range [control median= 1.1 Hz (range: 0.7–2.2); CNO median = 6.3 Hz (range: 4.2–7.8); n = 6; p < 0.01, Mann-Whitney test] (Figure 2B, C). With elevated ChI activity, preceding synaptic stimulation with a short burst of postsynaptic spikes led to a robust LTD in adjacent dSPNs (n = 6; p < 0.05, Wilcoxon test) (Figure 2D). As expected, bath application of the M4R selective antagonist MT3 blunted the LTD induction (CNO n = 6; CNO + MT3 n = 5; p < 0.05, Mann-Whitney test) (Figure 2D). Moreover, this STDP LTD was dependent upon endocannabinoid (eCB) CB1 receptors (CB1Rs; AM251, 2 μM; CNO n = 6; CNO + AM251 n = 6; p < 0.05, Mann-Whitney test) (Figure 2E), suggesting a mechanistic parallel to D2R-dependent LTD induced in iSPNs (Kreitzer and Malenka, 2007; Lerner and Kreitzer, 2012; Lovinger, 2010; Shen et al., 2008; Wu et al., 2015).
Figure 2. Elevating endogenous ChI activity with DREADD hM3D(q) enhances dSPN LTD induction.
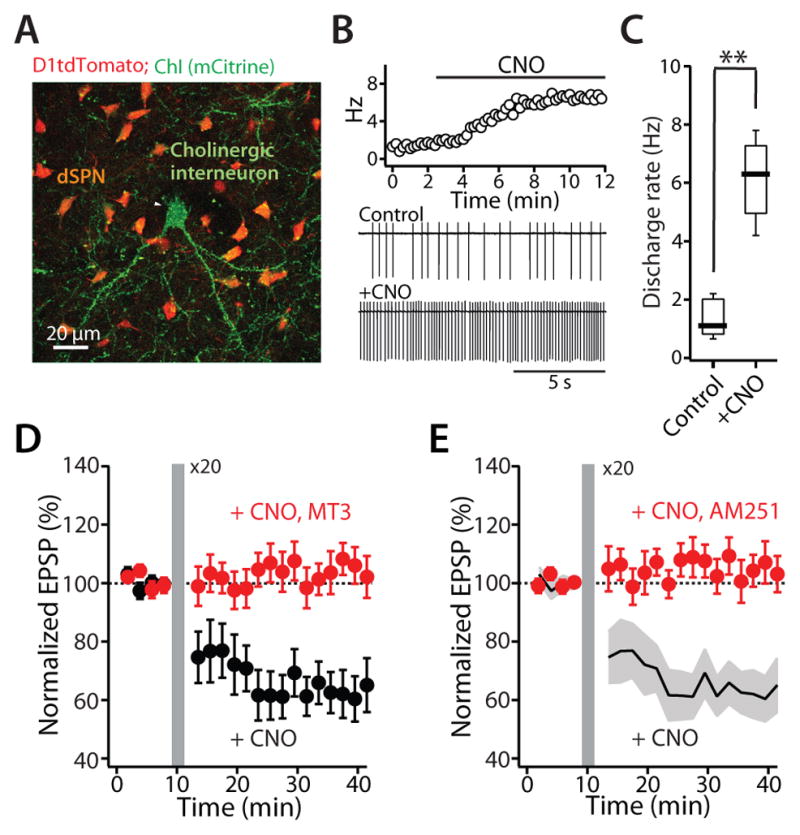
(A) ChAT Cre-dependent expression of DREADD hM3D(q) in ChIs (mCitrine reporter) in D1tdTomato BAC mouse. (B) DREADD cognate ligand CNO (10 μM) increased ChI spontaneous discharge rate recorded in cell-attached patches. (C) Box plot summary of the increase of discharge rate of ChIs. Box plot boxes indicate upper and lower quartiles; whiskers specify upper and lower 90%. **p < 0.01. (D) and (E) Enhancing local cholinergic signaling promoted the induction of LTD at neighboring dSPN glutamatergic synapses. Plot of the average EPSP amplitudes as a function of time. Error bars indicate SEM. The LTD was disrupted by (D) the M4R antagonist MT3 (100 nM) or (E) the CB1R antagonist AM251 (2 μM). Solid line (average) and gray shadow (± SEM) are control LTD from the panel (D) for reference.
Another way to counteract a weak cholinergic signaling is to lower ACh esterase activity in our ex vivo slice preparation with the specific inhibitor physostigmine (PHY, 10 μM) together with VU10010 (5 μM). This combination produced robust LTD induction with only 20 repetitions of the pairing protocol (n = 6; p < 0.05, Wilcoxon test) (Figure 3A). As anticipated, antagonizing M4Rs with MT3 (100 nM) disrupted the induction of LTD (VU + PHY n = 6; VU + PHY + MT3 n = 6; p < 0.05, Mann-Whitney test) (Figure 3A). However, blocking D2Rs with sulpiride (10 μM) had no effect on the induction of dSPN LTD (Figure S2B) (Shen et al., 2008; Wu et al., 2015).
Figure 3. M4R regulates dSPN LTD through RGS4 signaling.

(A) LTD was induced with fewer (20) repetitions of the post-pre pairing protocol by co-application of the ACh esterase inhibitor physostigmine (PHY; 10 μM) and VU10010 (VU; 5 μM). The induction was blunted in the presence of the M4R antagonist MT3 (100 nM). Plot of normalized EPSP amplitude as a function of time. The dashed line represents the average of EPSP amplitude before induction. STDP induction is indicated by the vertical bar. (B) Intracellular application of the RGS4 specific inhibitor CCG203769 (CCG; 10 μM) promoted the induction of LTD, even in the absence of the M4R PAM VU10010. The LTD was disrupted by the mGluR5 antagonist MPEP (10 μM). Moreover, (C) CCG enhanced LTD induction even in the presence of M4R antagonist. Solid line (average) and gray shadow (± SEM) are the LTD antagonism by MT3 from the panel (A) for reference. Plot of the average EPSP amplitudes as a function of time. Error bars indicate SEM. (D) Schematic showing the proposed signaling pathway through which M4Rs and D1Rs modulate the induction of dSPN LTD (see also Figure S2).
M4Rs regulate dSPN LTD induction through RGS4
The results thus far indicate that M4Rs are necessary participant in postsynaptic generation of eCBs that act at presynaptic CB1Rs to produce a sustained reduction in glutamate release, much like D2Rs in iSPNs (Kreitzer and Malenka, 2007). D2Rs diminish protein kinase A (PKA) phosphorylation of RGS4, which negatively modulates the ability of mGluR5 to activate Gαq and PLC isoforms coupled to the generation of eCBs (Lerner and Kreitzer, 2012). To test whether RGS4 is involved in regulation of the induction of dSPN LTD by M4Rs, we used a newly developed, selective RGS4 antagonist CCG203769 (Blazer et al., 2015). In this set of experiments, CCG203769 was applied through the patch pipette in whole-cell mode to restrict its action to the postsynaptic neuron. Intracellular application of CCG203769 (10 μM) enhanced induction of LTD during repeated pairing of postsynaptic spikes with presynaptic stimulation 10 ms later, even without boosting M4R stimulation with VU10010. This suggests that acute inhibition of RGS4 signaling dissociates LTD induction from M4Rs (CCG203769 n = 6; p < 0.05, Wilcoxon test) (Figure 3B). Indeed, when RGS4 signaling was blocked by CCG203769, antagonizing M4Rs had no effect on dSPN LTD (VU + PHY + MT3 n = 6; VU + PHY + MT3 + CCG203769 n = 7; p < 0.05, Mann-Whitney test) (Figure 3C, D). However, LTD induction still required mGluR5 stimulation, as blocking mGluR5 with MPEP (10 μM) prevented the LTD induction (CCG203769 n = 6; CCG203769 + MPEP n = 6; p < 0.05, Mann-Whitney test) (Figure 3B).
M4Rs inhibited NMDAR-mediated synaptic currents and Ca2+ transients
In iSPNs, D2R signaling is not only necessary for LTD induction, but also prevents the induction of LTP (Higley and Sabatini, 2010; Shen et al., 2008). In part, this modulation is likely mediated by suppression of NMDAR-mediated Ca2+ entry (Higley and Sabatini, 2010). M4Rs might also negatively modulate NMDARs. To test this hypothesis, dSPNs were filled with the Ca2+-insensitive red fluorophore Alexa Fluor 568 and the Ca2+-sensitive green fluorophore Fluo-5F to allow visualization of dendritic spines and simultaneous monitoring of NMDAR-mediated changes in intracellular Ca2+ concentration (Figure 4A). In these cells, glutamate was uncaged at spine heads in the presence of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR) antagonist NBQX (10 μM) and the muscarinic receptor agonist muscarine (3 μM), while monitoring the postsynaptic response with and without the M4R PAM VU10010 (5 μM) (Figure 4B). NMDAR-mediated Ca2+ transients were not altered by the M4R PAM in this situation (n = 10; p > 0.05, Wilcoxon test) (Figure S3). One interpretation of this negative result is that M4R signaling only affects NMDARs that have been positively modulated by D1R stimulation; D1R-mediated activation of PKA increases phosphorylation of the NMDAR NR2B subunit, enhancing receptor currents (Murphy et al., 2014). To test this possibility, the D1R agonist SKF81297 (3 μM) was applied and then muscarine and the M4R PAM co-applied. In this situation, M4R signaling decreased NMDAR-mediated Ca2+ transients by ~40% (n = 12; p < 0.05, Wilcoxon test) (Figure 4B, C), in agreement with previous studies (Higley and Sabatini, 2010).
Figure 4. M4R activation on dSPNs inhibits postsynaptic NMDAR-mediated Ca2+ transients and currents.

(A) Low (left) and high (right) magnification maximum-intensity projections of a dSPN filled with Alexa Fluor 568. Spine was stimulated with 1 ms uncaging pulse of 720–725 nm light. (B) In the presence of the D1R agonist SKF81297 (3 μM), average NMDAR Ca2+ transients in a distal dSPN spine induced by a single glutamate uncaging pulse were reduced by the M4R PAM VU10010 (5 μM). Solid lines are averages across multiple spines and the shaded areas represent the mean ± SEM. (C) Box plot summary of modulation of NMDAR-mediated Ca2+ transients. (D) Addition of VU 10010 (5 μM) suppressed NMDAR-mediated uEPSC currents triggered by uncaging pulses of 500 Hz to five distal spines. Solid lines are averages across multiple spines and the shaded areas represent the mean ± SEM. (E) NMDAR-mediated EPSCs recorded from dSPNs in the presence of muscarine (3 μM; upper traces) and muscarine + VU10010 (5 μM; lower traces). EPSCs are averages of 10 consecutive trials. (F) Left: time course of EPSC amplitude from the experiment shown in (E). Muscarine and VU10010 were applied during the time indicated by the horizontal bars. Right: box plot sum of M4R mediated modulation of NMDAR currents. All box plot boxes indicate upper and lower quartiles; whiskers specify upper and lower 90%. *p < 0.05 (see also Figure S3).
In some neurons, it has been found that Ca2+ permeability – but not total transmembrane current – is enhanced by phosphorylation of NMDARs (Higley and Sabatini, 2010). In other neurons, both Ca2+ permeability and total current are modulated in parallel (Murphy et al., 2014). To determine the situation in dSPNs, the effect of muscarine and VU10010 on NMDAR-mediated excitatory postsynaptic currents (EPSCs) evoked by uncaging glutamate on spine heads (uEPSCs) was examined in the presence of SKF81297. M4R signaling significantly reduced the amplitude of these postsynaptic NMDAR-mediated uEPSCs (n = 8; p < 0.05, Wilcoxon test) (Figure 4D). In addition, muscarine (3 μM) reduced the amplitudes of NMDAR-mediated EPSCs evoked by local electrical stimulation and this modulation was augmented by VU10010 (5 μM) (muscarine n = 10, muscarine + VU10010 n = 10; p < 0.05, Wilcoxon test) (Figure 4E, F). Taken together, these data show that dSPN M4R activation suppresses both total current and Ca2+ influx at spine NMDARs.
M4R signaling attenuated LTP induction in dSPNs
As shown previously (Pawlak and Kerr, 2008; Shen et al., 2008; Yagishita et al., 2014), pairing a presynaptic spike with a trailing postsynaptic spike induced a potent LTP in dSPNs (Figure 5A, B, C). This form of STDP LTP is dependent upon D1R signaling and Ca2+ entry through NMDARs (Pawlak and Kerr, 2008; Shen et al., 2008; Yagishita et al., 2014). One of the downstream targets of D1R and NMDAR signaling that has been implicated in LTP induction is extracellular signal-regulated kinase (ERK) (Pascoli et al., 2012; Plotkin et al., 2014). As expected from previous work, inhibition of ERK with the mitogen-activated protein kinase kinase or MEK inhibitor U0126 (10 μM) disrupted STDP LTP in dSPNs (control n = 8; U0126 n = 5; p < 0.05, Mann-Whitney test) (Figure 5C). Given its ability to antagonize D1R signaling and Ca2+ entry through NMDARs, M4R stimulation should impede the induction of LTP in dSPNs. As predicted, boosting M4R signaling with the M4R PAM disrupted STDP LTP induction (control n = 8; VU10010 n = 6; p < 0.05, Mann-Whitney test) (Figure 5D).
Shortly after LTP induction, synaptic strength is unstable. During this time, low-frequency afferent fiber stimulation (LFS; 2–5 Hz, 10 min) can reverse LTP; this reversal is called depotentiation (Otmakhova and Lisman, 1998). Depotentiation is triggered by Ca2+/calmodulin-mediated activation of protein phosphatase 2B (PP2B, or calcineurin); PP2B dephosphorylates DA and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), resulting in the dis-inhibition of protein phosphatase 1 (PP1) (Otmakhova and Lisman, 1998; Picconi et al., 2003); PP1 dephosphorylates GluA subunits recently trafficked into the synaptic membrane during LTP expression, leading to their removal (Lee et al., 2000). The serine residue on DARPP-32 targeted by PP2B is phosphorylated by PKA. So, in principle, D1R stimulation of PKA should prevent depotentiation (Otmakhova and Lisman, 1998). Consistent with this inference, 2 Hz intrastriatal stimulation, which also activates the axons of dopaminergic fibers, did not depotentiate dSPN glutamatergic synapses (n = 7; p > 0.05, Wilcoxon test) (Figure 5E). However, in the presence of the D1R antagonist SCH23390 (3 μM), the same protocol depotentiated glutamatergic EPSPs (control n = 7; SCH23390 n = 6; p < 0.05, Mann-Whitney test) (Figure 5E). More importantly within the context of the current study, depotentiation also was achieved by addition of the M4R PAM VU10010 (5 μM; control n = 7; VU10010 n = 6; p < 0.05, Mann-Whitney test) (Figure 5F), consistent with previous work suggesting that muscarinic receptor activation promotes depotentiation (Picconi et al., 2006).
Aberrant synaptic plasticity in dSPNs was corrected by an M4R PAM
Four to six weeks after a 6-hydroxydopamine (6-OHDA) lesion of the dopaminergic innervation to the striatum, conventional forms of striatal synaptic plasticity are lost (Picconi et al., 2003). Although present shortly after lesioning (Shen et al., 2008), STDP also was lost at longer survival times. In brain slices from D1-tdTomato and D2-eGFP BAC mice that had unilateral 6-OHDA lesions 3–4 weeks previously (Figure 6A), repeated pairing of synaptic stimulation with a postsynaptic spike 10 ms later did not change EPSP amplitude in dSPNs (n = 6; p > 0.05, Wilcoxon test) (Figure 6B) or iSPNs (n = 5; p > 0.05, Wilcoxon test) (Figure S4).
Figure 6. M4R PAM attenuates synaptic plasticity deficits in dSPNs from LID mice.

(A) Light microscopic image of a coronal section illustrating the loss of immunoreactivity for tyrosine hydroxylase (TH) after unilateral MFB 6-OHDA lesioning. CPu, caudate-putamen. (B) LTP induction was lost in prolonged 6-OHDA-lesioned animals. Shortly after the last injection of L-DOPA, LTP was recovered in dSPNs. Plot of average EPSP amplitude as a function of time. (C) The LTP was disrupted by U0126 (10 μM) or VU10010 (5 μM). Solid line (average) and gray shadow (± SEM) are LTP from the panel (B) for reference. (D) Low (left) and high (right) magnification maximum-intensity projections of a dSPN filled with Alexa Fluor 568. (E) NMDAR-mediated Ca2+ transients were decreased by addition of VU10010 (5 μM) in dSPNs from LID mice. Solid lines are averages across multiple spines and the shaded areas represent the mean ± SEM. *p < 0.05. (F) In dyskinetic animals, synaptic depotentiation was lost and restored by bath application of SCH23390 (3 μM) or VU10010 (5 μM). Horizontal bar indicates LFS (2 Hz, 10 min). Error bars represent SEM. Box plot boxes specify upper and lower quartiles; whiskers indicate upper and lower 90% (see also Figure S4).
Elevating striatal DA in lesioned rodents by systemic administration of the DA precursor, L-DOPA, restores some forms of striatal synaptic plasticity (Belujon et al., 2010; Picconi et al., 2003; Thiele et al., 2014). However, repeated L-DOPA treatment of parkinsonian rodents results in abnormal involuntary movements (AIMs), mimicking human LID (Cenci, 2007). In the rat LID model, high-frequency stimulation (HFS) of the striatum induces LTP at SPN glutamatergic synapses, but depotentiation of these synapses is disrupted (Picconi et al., 2003). However, it is unclear whether either of these effects generalizes to STDP and whether these effects are specific to dSPNs or iSPNs. To answer this question, a mouse model of LID was generated in which dSPNs and iSPNs were labeled (see Experimental Procedures). One hour after the last injection of L-DOPA, mice were sacrificed and ex vivo brain slices were prepared for patch clamping recordings, two-photon laser-scanning microscopy and glutamate uncaging. In dSPNs from L-DOPA-treated mice, the standard pre-post STDP induction protocol led to a robust LTP (n = 7; p < 0.05, Wilcoxon test) (Figure 6B). As in tissue from untreated mice, blocking ERK activity with U0126 prevented LTP induction – arguing that the plasticity was induced by the same mechanisms as in untreated mice (dyskinetic n = 7; dyskinetic + U0126 n = 5; p < 0.05, Mann-Whitney test) (Figure 6C). However, in iSPNs from LID mice, the same STDP protocol induced LTD (6-OHDA n = 5; L-DOPA n = 6; p < 0.05, Mann-Whitney test) (Figure S4). These results demonstrate that synaptic plasticity in SPNs following L-DOPA treatment is not grossly abnormal and continues to be governed by SPN subtype. Moreover, enhancing M4R signaling with VU10010 blocked the induction of STDP LTP in dSPNs from LID mice (dyskinetic n = 7; dyskinetic + VU10010 n = 6; p < 0.05, Mann-Whitney test) (Figure 6C), and reduced NMDAR-mediated Ca2+ transients (n = 10; p < 0.05, Wilcoxon test) (Figure 6D, E), just as in naïve mice.
Was depotentiation altered in dSPNs from LID mice? As predicted by previous work (Picconi et al., 2003), intrastriatal LFS (2 Hz, 10 min) failed to depotentiate EPSPs in dSPNs from LID mice (n = 6; p > 0.05, Wilcoxon test) (Figure 6F). This could reflect a deficit in the intracellular machinery underlying depotentiation or it could simply be a consequence of sustained D1R stimulation following the last L-DOPA dose. To test the latter hypothesis, D1Rs were antagonized by bath application of SCH23390 and the LFS protocol repeated. In this situation, glutamatergic synapses depotentiated (dyskinetic n = 6; dyskinetic + SCH23390 n = 6; p < 0.05, Mann-Whitney test) (Figure 6F), arguing that the depotentiation machinery was intact. Boosting M4R signaling with the M4R PAM also restored depotentiation in the absence of a D1R antagonist (dyskinetic n = 6; dyskinetic + VU10010 n = 6; p < 0.05, Mann-Whitney test) (Figure 6F).
M4R PAM administration reduced dyskinetic behavior
Aberrant striatal LTP has been hypothesized to contribute to the development of dyskinetic behavior (Jenner, 2008; Picconi et al., 2003). The data presented to this point demonstrate that enhancing M4R signaling with a PAM can blunt D1R-mediated LTP in dSPNs of LID models, both by diminishing LTP induction and by enabling depotentiation. Thus, systemic administration of an M4R PAM should attenuate LID-like behavior. To test this hypothesis, two different animal models of LID were examined. First, mice were unilaterally lesioned with 6-OHDA and then given incremental doses of L-DOPA (1.5–6 mg kg−1, i.p.). These mice were randomly assigned to receive treatment with either L-DOPA and vehicle or L-DOPA and VU0467154 (10 mg kg−1, i.p.); VU0467154 is an M4R PAM with better pharmacokinetic properties than VU10010. As predicted, VU0467154 significantly attenuated peak AIM scores induced by each dose of L-DOPA (1.5 mg kg−1: L-DOPA n = 9; L-DOPA + VU0467154 n = 9; p < 0.05, Mann-Whitney test; 3 mg kg−1: L-DOPA n = 9; L-DOPA + VU0467154 n = 9; p < 0.01, Mann-Whitney test; 6 mg kg−1: L-DOPA n = 9; L-DOPA + VU0467154 n = 9; p < 0.01, Mann-Whitney test) (Figure 7A, B). The reduction in dyskinesia scores did not occur at the expense of the anti-parkinsonian effects of L-DOPA, as forelimb use asymmetry (Schallert et al., 2000) was improved by L-DOPA alone or when co-administered with VU0467154 (L-DOPA: n = 9, p < 0.01, Wilcoxon test; L-DOPA + VU0467154: n = 9; p < 0.01, Wilcoxon test).
Figure 7. M4R PAMs alleviate dyskinetic behaviors.

(A) Plot of sum of mouse AIM scores (n = 9, mean ± SEM) as a function of time. Systemic treatment with VU0467154 (10 mg kg−1) produced a significant overall reduction in AIM scores (time: p < 0.001; group: p < 0.01; interaction: p < 0.01; repeated measure 2-way ANOVA followed by Bonferroni’s post test). (B) Box plot summation shows that VU0467154 attenuates LID behaviors over a range of L-DOPA doses. Mice were treated with ascending doses of L-DOPA combined with either the M4R PAM or its vehicle (n = 9 per group). VU0467154 was effective on each of the three L-DOPA doses (1.5, 3, or 6 mg kg−1), indicated by horizontal bars. Box plot boxes indicate upper and lower quartiles; whiskers specify upper and lower 90%. *p < 0.05; **p < 0.01. (C) Plot of sum of primate dyskinesia scores (median) as a function of time (Fr = 11.64, n = 4; p < 0.01, Friedman’s test). (D) Box plot summary of antidyskinetic effects of VU0476406 (1, 3, or 10 mg kg−1) or amantadine (20 mg kg−1). Systemic administration of VU0476406 (10 mg kg−1) or amantadine (20 mg kg−1) ameliorates dyskinesia scores. *p < 0.05. (E) Proposed signaling model of L-DOPA-induced deficits in dSPN synaptic plasticity and dyskinesia (see also Figures S5, S6 and S7).
A separate group of L-DOPA-primed and dyskinetic mice were challenged with L-DOPA and another M4R PAM (VU0152100), or its vehicle, to examine the striatal induction of phosphorylated ERK1/2 (pERK1/2) and phosphorylated histone 3 (pH3) – neuronal markers that strongly correlated with LID severity (Santini et al., 2009). The M4R PAM significantly reduced the number of neurons immunoreactive for these markers in the lateral (motor) part of the striatum (pERK1/2: L-DOPA n = 9; L-DOPA + VU0152100 n = 9; p < 0.05; pH3: L-DOPA n = 9; L-DOPA + VU0152100 n = 9; p < 0.05, Mann-Whitney test) (Figure S5).
Next, four nonhuman primates were rendered parkinsonian with repeated intravenous (i.v.) injections of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) hydrochloride (Bezard et al., 2003) and then subjected to a chronic L-DOPA treatment regimen that reliably induces dyskinesia (Ko et al., 2014). Because VU0467154 has limited potency at primate M4Rs, VU0476406, a novel VU0467154 analog with improved potency (Conn et al., 2014), was used in these experiments. The NMDAR antagonist amantadine (20 mg kg−1, i.v.), which is used to treat dyskinesia in PD patients (Metman et al., 1999), was used as a positive control. Vehicle, VU0476406 (1, 3 and 10 mg kg−1) or amantadine was administered together with L-DOPA in a within-subject design (see Experimental Procedures). As anticipated, amantadine blunted dyskinetic behaviors (n = 4; p < 0.05, Friedman’s nonparametric repeated measure one-way ANOVA followed by Dunn’s post test) (Figure 7C, D) (Ko et al., 2014). VU0476406 at its highest concentration (10 mg kg−1), like amantadine, significantly reduced LID (n = 4; p < 0.05, Friedman’s test and Dunn’s test) (Figure 7C, D) and did not compromise the therapeutic effect of L-DOPA (n = 4; p > 0.05, Friedman’s test and Dunn’s test) (Figure S6).
DISCUSSION
There are three take-home points from the studies presented. First, endogenous cholinergic signaling through M4Rs promotes LTD and blunts LTP at dSPN glutamatergic synapses; in this respect, dSPN M4Rs serve a role that is analogous to that of iSPN D2Rs. Second, enhancing endogenous M4R signaling with an M4R PAM mitigates the synaptic plasticity deficits in dSPNs accompanying L-DOPA treatment. Third, systemic treatment with the M4R PAMs ameliorates LID in both mouse and primate models. These findings not only provide a new insight into the way in which DA and ACh interact to shape striatal plasticity, they point to a novel therapy for one of the major unmet medical needs for PD patients – a pharmacological strategy for alleviating LID.
M4R signaling promoted LTD induction in dSPNs
One of the fundamental unresolved questions about the striatal circuitry is how corticostriatal glutamatergic LTD is controlled in dSPNs. In iSPNs, the landscape is very well defined. D2Rs, mGluR5s and L-type calcium channels are necessary participants in the postsynaptic generation of eCBs that act at presynaptic glutamatergic terminals to bring about a sustained reduction in glutamate release. This is true in iSPNs, regardless of whether a STDP or HFS protocol is used. With HFS protocols utilizing macroelectrodes that directly stimulate the striatum, D2R signaling is also necessary for LTD induction in dSPNs. But, this dependence is indirect, reflecting interneuron engagement during induction (Tozzi et al., 2011; Wang et al., 2006).
In STDP protocols that use minimal local stimulation (MLS), D2R signaling is not necessary for LTD induction in dSPNs (Shen et al., 2008). In fact, a number of studies using MLS have failed to find LTD in dSPNs (Kreitzer and Malenka, 2007; Nazzaro et al., 2012; Shen et al., 2008). This failure is attributable to the inadvertent stimulation of dopaminergic fibers by MLS (Threlfell et al., 2012), as antagonism of D1Rs enables LTD induction in dSPNs with MLS (Shen et al., 2008). Postsynaptic M4R signaling enabled MLS-evoked LTD induction, even in the absence of a D1R antagonist. Enhancing M4R signaling in any of three ways – by use of an M4R PAM, by slowing ACh metabolism in the presence of the M4R PAM or by increasing the activity of ChIs with a DREADD – was sufficient to establish STDP LTD. The effects of M4R signaling appeared to be mediated by inhibition of AC (Jeon et al., 2010) and deactivation of RGS4, which attenuates mGluR5 signaling through Gαq (Saugstad et al., 1998); in this regard, the role of M4R signaling in LTD is very similar to that of D2Rs in iSPNs (Lerner and Kreitzer, 2012). Why there was an M4R signaling requirement for LTD induction in dSPNs in the absence of D1R signaling is not entirely clear. One possibility is that there are other signaling mechanisms driving AC (and PKA) activity in dSPNs (Yagishita et al., 2014) that maintain RGS4 activity.
Modulation of LTP in dSPN by M4Rs
In addition to promoting the induction of LTD, M4R signaling blocked D1R-mediated LTP induction in dSPNs. LTP induction in dSPNs requires co-activation of PKA and Ras-guanine nucleotide exchange factor 1 (Ras-GEF1), which converge upon ERK (Cerovic et al., 2015; Shen et al., 2008; Sweatt, 2004). Our studies suggest that M4R signaling blunts activation of both PKA and Ras-GEF1. First, M4Rs are well-known to inhibit AC isoforms by activating Gi proteins, limiting PKA activation (Gomeza et al., 1999; Jeon et al., 2010). Second, M4Rs reversed D1R-mediated enhancement of NMDAR Ca2+ currents, which is mediated by PKA phosphorylation of serine 1166 on GluN2B subunit (Murphy et al., 2014). This modulation should diminish synaptically driven activation of the Ca2+/calmodulin dependent Ras-GEF1 anchored to the GluN2B subunit (Krapivinsky et al., 2003). Thus, the strength of dSPN glutamatergic synapses is reciprocally modulated by M4Rs and D1Rs.
Of perhaps as much importance to global synaptic strength in the striatum is the ability of M4R signaling to promote LTP reversal or depotentiation in dSPNs. LTP at SPN glutamatergic synapses is postsynaptically expressed, resulting from AMPA trafficking into the synapse (Plotkin et al., 2014). The ability of M4Rs to depotentiate synapses is attributable to inhibition of AC and deactivation of DARPP-32, resulting in dis-inhibition of PP1 and removal of AMPARs that have been recently trafficked into the synapse (Lee et al., 2000; Otmakhova and Lisman, 1998; Picconi et al., 2003).
Although the basic determinants of depotentiation are widely agreed upon, poor experimental control of key experimental variables in the corticostriatal slice has led to apparent discrepancies. Perhaps of greatest importance, neither macroelectrode stimulation nor MLS allows for the selective activation of corticostriatal axons; both inevitably activate other axons, like dopaminergic and cholinergic axons coursing through the striatum, to varying degrees. The inability of minimal local LFS to induce depotentiation in the absence of a D1R antagonist undoubtedly reflects the stronger activation of dopaminergic fibers by this form of stimulation than white matter, macroelectrode LFS, where a D1R antagonist is not necessary (Picconi et al., 2003). However, macroelectrode stimulation of the white matter still activates dopaminergic fibers, as it is capable of inducing D1R LTP (Calabresi et al., 2007). This lack of control also complicates the interpretation of results from the disease models. For example, our results argue that the failure to see depotentiation with macroelectrode LFS in slices from LID models is attributable to an enhancement of D1R stimulation by drug treatment, not a fundamental alteration in the mechanisms governing depotentiation. This lack of experimental control and the cellular complexity of the striatum might also help explain the puzzling recent work suggesting that activation of the Ras-GEF1/ERK pathway is necessary for depotentiation (Cerovic et al., 2015). Optogenetic and chemogenetic approaches that allow precise control over the circuitry being engaged during the induction of synaptic plasticity should allow these apparent discrepancies to be resolved.
M4R PAM ameliorates L-DOPA-induced involuntary movements
A key unmet medical need of the PD community is a strategy for ameliorating LID. The hope is that a rationale therapy will come from understanding the mechanisms responsible for LID. Several lines of evidence suggest that aberrant D1R-dependent potentiation of dSPN glutamatergic synapses is a central feature of the LID pathophysiology. Our work is consistent with this conclusion. Specifically, in tissue taken from LID mice shortly after the last L-DOPA dose, STDP LTP was readily induced in dSPNs and did not depotentiate with LFS. However, this phenomenology is attributable simply to transiently elevated striatal DA levels after the last L-DOPA injection, rather than a fundamental change in plasticity signaling machinery. For example, depotentiation was immediately restored by D1R antagonism or M4R agonism, which is not basically different from the situation in tissue from naïve mice. Moreover, LTP induction was blocked by addition of the M4R PAM, much as in naïve tissue. What is different in L-DOPA treated mice is the spatio-temporal distribution of D1R stimulation. Rather than being briefly stimulated by burst spiking of DA neurons, D1Rs in the LID model are stimulated for long periods of time; this abnormally sustained stimulation is likely to underlie both the synaptic and biochemical signature of LID in dSPNs. The sustained elevation of extracellular DA following L-DOPA administration also prevents iSPNs from responding to patterned activity appropriately. In this state, STDP protocols that normally induce Hebbian LTP, induce LTD in iSPNs. This cell-specific biasing of synaptic plasticity – LTP for dSPNs and LTD for iSPNs – in the LID models is consistent with the hyperkinetic features of LID. But more importantly, the strength of SPN synapses should be unconstrained by action outcomes, leading to random alterations in strength. Randomizing synaptic strength or degrading information stored in synaptic strength could be a cause of involuntary movements characteristic of LID.
How might the aberrant plasticity in dSPNs be ameliorated? Clearly, counteracting D1Rs is not a feasible therapeutic strategy because it would diminish the symptomatic benefit of L-DOPA treatment. By amplifying endogenous cholinergic signaling, the M4R-PAMs disrupted STDP LTP in dSPNs and enabled previously potentiated synapses to be depotentiated. In addition, they diminished NMDAR-mediated Ca2+ signaling and phosphorylation of ERK in dyskinetic mice – a biomarker of the dyskinetic state (Santini et al., 2009). More importantly, systemic administration of M4R PAMs significantly diminished dyskinetic behaviors in both mouse and primate models following L-DOPA treatment. This was accomplished without compromising the symptomatic benefit of L-DOPA treatment. Although the anti-dyskinetic effects were modest, improvements in potency and pharmacokinetics could enhance the ability of an M4R PAM to alleviate LID. For example, matching the brain bioavailability of the M4R-PAM to that of dopamine, so that it effectively moderates D1R signaling during the on-state, could improve its behavioral effects.
At first glance, our results appear to be at odds with a recent report showing that ablating striatal ChIs after lesioning dopaminergic neurons, but prior to L-DOPA administration diminishes dyskinesia (Won et al., 2014). Although intriguing, this result is difficult to interpret. Beyond controlling synaptic plasticity in dSPNs, ChIs have a wide range of striatal effects through both muscarinic and nicotinic receptor signaling (Zhou et al., 2003). Undoubtedly, ablation of ChIs will induce striatal adaptations that could alter the effects of dopaminergic de-innervation and subsequent L-DOPA administration (Kaneko et al., 2000). For example, deletion of M1 muscarinic receptors significantly attenuates spine loss in iSPNs following dopamine depletion (Shen et al., 2007). In mice with a normal complement of ChIs, re-establishing iSPN axospinous connectivity following L-DOPA administration appears to be an important factor in the emergence of dyskinesia (Fieblinger et al., 2014). This re-wiring might be substantially altered by ChI ablation. So, while this study does not affect our conclusions, it does suggest that understanding the consequences of ChIs ablation on the striatal circuitry could identify new targets for anti-dyskinetic therapy.
In summary, M4Rs expressed by dSPNs play a central role in the regulation of synaptic plasticity of glutamatergic synapses in both healthy and parkinsonian states. These receptors promoted LTD and blocked LTP induction; they also enabled depotentiation. In parkinsonian mice rendered dyskinetic by L-DOPA treatment, an M4R-PAM blunted D1R-mediated LTP and enabled depotentiation. In both mouse and primate models, the dyskinetic behaviors were attenuated by systemic M4R PAM treatment as well. These results point to a novel therapy for one of the major unmet medical needs for PD patients – a pharmacological strategy for alleviating LID.
EXPERIMENTAL PROCEDURES
Animal use procedures were approved by the Institutional Animal Care and Use Committees. See Supplemental Experimental Procedures for detailed methods.
Electrophysiology
Parasagittal slices were prepared from D1-tdTomato or D2-eGFP BAC transgenic mice (P80-P110). Whole-cell or perforated patch recordings from SPNs were obtained from visually identified SPNs in the dorsolateral striatum at a temperature of 30–31°C. EPSP/Cs were evoked by intrastriatal stimulation with a theta glass pipette placed 80–100 μm dorsolateral of the recorded neurons. Long-lasting synaptic plasticity was induced using protocols consisting of subthreshold synaptic stimulation paired with somatically induced action potentials (APs) at theta frequency (5 Hz). These protocols consisted of 20–60 trains of five bursts repeated at 0.1 Hz, with each burst composed of three APs preceded with three EPSPs at 50 Hz (pre-post timing pairing, +5ms) or three APs followed by one EPSP (post-pre timing pairing, −10 ms). To ensure induction of consistent synaptic plasticity, postsynaptic neurons were depolarized to −70 mV from their typical resting membrane potentials (−85 mV) during their induction. GABAA was blocked by the bath application of gabazine.
Mouse unilateral 6-OHDA model and LID
Mice were injected with 6-OHDA into the medial forebrain bundle at 8–10 weeks of age. Two weeks after surgery, the degree of damage to nigrostriatal DA neurons was assessed with a forelimb-use asymmetry test (Figure S7A) (Schallert et al., 2000). Striatal sections from a subset of mice were stained with tyrosine hydroxylase to verify successful lesion (Figure 6A). One day after the cylinder test, mice underwent behavioral testing for AIMs following L-DOPA treatment as previously described (Francardo et al., 2011). For ex vivo brain slice recording, behavioral testing occurred every other day for a total of five test sessions. Animals received 3 and 6 mg kg−1 L-DOPA for the first two and last three behavioral sessions, respectively. Benserazide was co-administered to inhibit peripheral conversion of L-DOPA to DA. AIMs (axial, limb and orolingual movements) were rated as previously described (Figure S7B) (Cenci and Lundblad, 2007). Physiological experiments were performed in mice sacrificed one hour after the last L-DOPA administration.
For behavioral pharmacology experiments as those shown in Figure 7, treatment with L-DOPA was given daily for 9 days using an incremental dose regimen (Francardo et al., 2011); at the doses of 1.5 mg kg−1 on days 1–3, 3 mg kg−1 on days 4–6, and 6 mg kg−1 on days 7–9. Animals were randomly allocated to receive treatment with L-DOPA and vehicle, or L-DOPA and VU0467154, which was given at the dose of 10 mg kg−1 for all treatment.
Primate MPTP model and LID
Primates received daily MPTP i.v. injections until PD motor symptoms were established (Bezard et al., 2003); after which twice daily L-DOPA treatment was given. L-DOPA was administered over 4–5 months for induction of stable dyskinesia (Bezard et al., 2003; Ko et al., 2014). Thereafter, L-DOPA was given twice weekly for maintaining a steady level of dyskinesia.
L-DOPA and amantadine were administered orally and the M4R PAM VU0476406 i.v. injected. Vehicle, VU0476406 or amantadine was administered with L-DOPA in a within-subject design. A two-day wash-out period was given between acute drug tests.
PD and dyskinesia were rated on the established behavioral rating scales (Fox et al., 2012) and calculated as previously described (Bezard et al., 2003; Ko et al., 2014).
Viral DREADD hM3D(q) expression and activation
Bi-transgenic mice expressing tdTomato in dSPNs and Cre-recombinase under control of the ChAT regulatory elements received striatal stereotaxic injections of an AAV (serotype 2/8) containing the hM3D(q) expression construct. Mice were allowed to recover for 5–6 weeks before recordings.
Ca2+ imaging and two-photon laser uncaging
dSPNs were loaded with 25 μM Alexa 568 and 300 μM Fluo 5F via the internal recording solution. The laser-scanned images were acquired with 810 nm light pulsed at 90 MHz (~250 fs pulse duration). Changes in Fluo 5F fluorescence were averaged from 3 trials and measured as ΔF/Fo.
Glutamate uncaging was achieved using a Verdi/Mira (Plotkin et al., 2014). 5 mM MNI-glutamate was superfused over the slice using a syringe pump and multi-barreled perfusion manifold. Glutamate was uncaged adjacent to individual spines using 1 ms pulses of 720–725 nm light typically 10–20 mW in power at the sample plane. uEPSC amplitudes were measured from averaged (3–5 repetitions) traces.
Data analysis and statistics methods
Data analysis was conducted with Igor Pro 6 and Clampfit 9. EPSP amplitude was calculated from 50 sweeps immediately before the start of induction and 20–30 min after the end of induction. In studies describing optical or uncaging responses measured from individual spines, the stated n indicates the number of spines. Compiled data were expressed as mean ± SEM. Nonparametric Mann-Whitney or Wilcoxon tests were used to assess the experiment results, using a probability threshold of 0.05.
Statistical analyses for behavioral data were carried out using Prism 6. For mice, data were analyzed using parametric repeated measure two-way ANOVA followed by post hoc Bonferroni’s test; for primates, data were analyzed by Friedman’s nonparametric one-way ANOVA followed by post hoc Dunn’s test.
Supplementary Material
Acknowledgments
This work was supported by the Michael J. Fox Foundation to W.S., D.J.S., the NIH NS034696 and MH074866 to D.J.S., the JPB Foundation to D.J.S., P.G., the USAMRAA grant W81XWH-09-1-0402 to P.G.; by the NIH Intramural Research Program to J.W.; by the grant to R.R.N. from NIH DA023252; by the grants to P.J.C. and C.W.L. from the NIH MH073676 and NS071669; by the grants from Association France Parkinson, the Foundation de France, and LABEX BRAIN ANR-10-LABX-43 to E.B.; and by the grants to M.A.C. from Swedish governmental funding of clinical research (grant ALFSKANE-450851), the Basal Ganglia Disorders Linnaeus Consortium (BAGADILICO), the Swedish Research Council, and the Olle Engkvist Foundation. We thank Shenyu Zhai and Savio Chan for their comments on the manuscript and Sasha Ulrich for technical assistance. P.J.C. and C.W.L. receive research support from AstraZeneca and are inventors on patents that protect multiple classes of M4R PAMs.
Footnotes
AUTHOR CONTRIBUTIONS:
W.S. and D.J.S. conceived the study and designed the experiments. M.A.C., V.F. and E.B. designed the behavioral pharmacology experiments. W.S. and J.L.P. carried out electrophysiological recordings, analyzed the data, and prepared the figures. V.F., W.K.D.K. and Q.L. performed behavioral study and histological processing, analyzed the data, and prepared the figures. Z.X. and T.F. conducted stereotaxic injections of viruses and 6-OHDA. J.W. generated D1-M4-KO mice. R.R.N. provided RGS4 selective antagonist. P.G. provided different transgenic animals and reagents. P.J.C and C.W.L. provided M4R PAMs. W.S. and D.J.S. wrote the paper. All authors discussed the results and implications and commented on the manuscript at all stages.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Augustin SM, Beeler JA, McGehee DS, Zhuang X. Cyclic AMP and afferent activity govern bidirectional synaptic plasticity in striatopallidal neurons. J Neurosci. 2014;34:6692–6699. doi: 10.1523/JNEUROSCI.3906-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belujon P, Lodge DJ, Grace AA. Aberrant striatal plasticity is specifically associated with dyskinesia following levodopa treatment. Mov Disord. 2010;25:1568–1576. doi: 10.1002/mds.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard V, Normand E, Bloch B. Phenotypical characterization of the rat striatal neurons expressing muscarinic receptor genes. J Neurosci. 1992;12:3591–3600. doi: 10.1523/JNEUROSCI.12-09-03591.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P. Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med. 2003;9:762–767. doi: 10.1038/nm875. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, Wang Q, Huang XP, Traynor JR, Husbands SM, et al. Selectivity and anti-Parkinson’s potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Neurosci. 2015;6:911–919. doi: 10.1021/acschemneuro.5b00063. [DOI] [PubMed] [Google Scholar]
- Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU, Breininger ML, Gentry PR, Yin H, Jadhav SB, et al. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Tozzi A, Di Filippo M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007;30:211–219. doi: 10.1016/j.tins.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Cenci MA. Dopamine dysregulation of movement control in L-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Cenci MA, Lundblad M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr Protoc Neurosci. 2007;Chapter 9(Unit9.25–9.25.23) doi: 10.1002/0471142301.ns0925s41. [DOI] [PubMed] [Google Scholar]
- Cerovic M, Bagetta V, Pendolino V, Ghiglieri V, Fasano S, Morella I, Hardingham N, Heuer A, Papale A, Marchisella F, et al. Derangement of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and extracellular signal-regulated kinase (ERK) dependent striatal plasticity in L-DOPA-induced dyskinesia. Biol Psychiatry. 2015;77:106–115. doi: 10.1016/j.biopsych.2014.04.002. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Lindsley CW, Wood MR, Hopkins CR, Salovich JM, Melancon BJ. Substituted pyrazolo[3′,4′,4,5]thieno[2,3-c]pyridazin-3-amine analogs as positive allosteric modulators of the muscarinic receptor M4. 0364409 WO. 2014
- Feyder M, Bonito-Oliva A, Fisone G. L-DOPA-Induced dyskinesia and abnormal signaling in striatal medium spiny neurons: focus on dopamine D1 receptor-mediated transmission. Front Behav Neurosci. 2011;5:71. doi: 10.3389/fnbeh.2011.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, et al. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun. 2014;5:5316. doi: 10.1038/ncomms6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Paille V, Cui Y, Morera-Herreras T, Deniau JM, Venance L. Distinct coincidence detectors govern the corticostriatal spike timing-dependent plasticity. J Physiol. 2010;588:3045–3062. doi: 10.1113/jphysiol.2010.188466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox SH, Johnston TH, Li Q, Brotchie J, Bezard E. A critique of available scales and presentation of the Non-Human Primate Dyskinesia Rating Scale. Mov Disord. 2012;27:1373–1378. doi: 10.1002/mds.25133. [DOI] [PubMed] [Google Scholar]
- Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, Cenci MA. Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol Dis. 2011;42:327–340. doi: 10.1016/j.nbd.2011.01.024. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M4 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci USA. 1999;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat Neurosci. 2010;13:958–966. doi: 10.1038/nn.2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Soler-Llavina GJ, Sabatini BL. Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci. 2009;12:1121–1128. doi: 10.1038/nn.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci. 2008;9:665–677. doi: 10.1038/nrn2471. [DOI] [PubMed] [Google Scholar]
- Jeon J, Dencker D, Wörtwein G, Woldbye DPD, Cui Y, Davis AA, Levey AI, Schütz G, Sager TN, Mørk A, et al. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J Neurosci. 2010;30:2396–2405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Hikida T, Watanabe D, Ichinose H, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Synaptic integration mediated by striatal cholinergic interneurons in basal ganglia function. Science. 2000;289:633–637. doi: 10.1126/science.289.5479.633. [DOI] [PubMed] [Google Scholar]
- Ko WKD, Pioli E, Li Q, McGuire S, Dufour A, Sherer TB, Bezard E, Facheris MF. Combined fenobam and amantadine treatment promotes robust antidyskinetic effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model of Parkinson’s disease. Mov Disord. 2014;29:772–779. doi: 10.1002/mds.25859. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. Neuromodulatory control of striatal plasticity and behavior. Curr Opin Neurobiol. 2011;21:322–327. doi: 10.1016/j.conb.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron. 2012;73:347–359. doi: 10.1016/j.neuron.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci. 2010;30:2160–2164. doi: 10.1523/JNEUROSCI.5844-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia TV, Frank MJ. From reinforcement learning models to psychiatric and neurological disorders. Nat Neurosci. 2011;14:154–162. doi: 10.1038/nn.2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol. 1999;56:1383–1386. doi: 10.1001/archneur.56.11.1383. [DOI] [PubMed] [Google Scholar]
- Murphy JA, Stein IS, Lau CG, Peixoto RT, Aman TK, Kaneko N, Aromolaran K, Saulnier JL, Popescu GK, Sabatini BL, et al. Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J Neurosci. 2014;34:869–879. doi: 10.1523/JNEUROSCI.4538-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazzaro C, Greco B, Cerovic M, Baxter P, Rubino T, Trusel M, Parolaro D, Tkatch T, Benfenati F, Pedarzani P, et al. SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nat Neurosci. 2012;15:284–293. doi: 10.1038/nn.3022. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancani T, Bolarinwa C, Smith Y, Lindsley CW, Conn PJ, Xiang Z. M4 mAChR-mediated modulation of glutamatergic transmission at corticostriatal synapses. ACS Chem Neurosci. 2014;5:318–324. doi: 10.1021/cn500003z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2012;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JND. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Centonze D, Håkansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat Neurosci. 2003;6:501–506. doi: 10.1038/nn1040. [DOI] [PubMed] [Google Scholar]
- Picconi B, Passino E, Sgobio C, Bonsi P, Barone I, Ghiglieri V, Pisani A, Bernardi G, Ammassari-Teule M, Calabresi P. Plastic and behavioral abnormalities in experimental Huntington’s disease: a crucial role for cholinergic interneurons. Neurobiol Dis. 2006;22:143–152. doi: 10.1016/j.nbd.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Plotkin JL, Day M, Peterson JD, Xie Z, Kress GJ, Rafalovich I, Kondapalli J, Gertler TS, Flajolet M, Greengard P, et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron. 2014;83:178–188. doi: 10.1016/j.neuron.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan SC, Roth BL. Remote control of neuronal signaling. Pharmacol Rev. 2011;63:291–315. doi: 10.1124/pr.110.003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Alcacer C, Cacciatore S, Heiman M, Hervé D, Greengard P, Girault JA, Valjent E, Fisone G. L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem. 2009;108:621–633. doi: 10.1111/j.1471-4159.2008.05831.x. [DOI] [PubMed] [Google Scholar]
- Saugstad JA, Marino MJ, Folk JA, Hepler JR, Conn PJ. RGS4 inhibits signaling by group I metabotropic glutamate receptors. J Neurosci. 1998;18:905–913. doi: 10.1523/JNEUROSCI.18-03-00905.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez G, Colettis N, Vázquez P, Cerveñansky C, Aguirre A, Quillfeldt JA, Jerusalinsky D, Kornisiuk E. Muscarinic inhibition of hippocampal and striatal adenylyl cyclase is mainly due to the M4 receptor. Neurochem Res. 2009;34:1363–1371. doi: 10.1007/s11064-009-9916-9. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively elevates dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Shindou T, Ochi-Shindou M, Wickens JR. A Ca2+ threshold for induction of spike-timing-dependent depression in the mouse striatum. J Neurosci. 2011;31:13015–13022. doi: 10.1523/JNEUROSCI.3206-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Thiele SL, Chen B, Lo C, Gertler TS, Warre R, Surmeier JD, Brotchie JM, Nash JE. Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and L-DOPA induced dyskinesia in mouse models. Neurobiol Dis. 2014;71C:334–344. doi: 10.1016/j.nbd.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threlfell S, Lalic T, Platt NJ, Jennings KA, Deisseroth K, Cragg SJ. Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron. 2012;75:58–64. doi: 10.1016/j.neuron.2012.04.038. [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F, Ghiglieri V, Giampà C, Fusco FR, Picconi B, et al. The distinct role of medium spiny neurons and cholinergic interneurons in the D2/A2A receptor interaction in the striatum: implications for Parkinson’s disease. J Neurosci. 2011;31:1850–1862. doi: 10.1523/JNEUROSCI.4082-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wickens JR, Reynolds JNJ, Hyland BI. Neural mechanisms of reward-related motor learning. Curr Opin Neurobiol. 2003;13:685–690. doi: 10.1016/j.conb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Won L, Ding Y, Singh P, Kang UJ. Striatal cholinergic cell ablation attenuates L-DOPA induced dyskinesia in Parkinsonian mice. J Neurosci. 2014;34:3090–3094. doi: 10.1523/JNEUROSCI.2888-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Kim JI, Tawfik VL, Lalchandani RR, Scherrer G, Ding JB. Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell Rep. 2015;10:75–87. doi: 10.1016/j.celrep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagishita S, Hayashi-Takagi A, Ellis-Davies GCR, Urakubo H, Ishii S, Kasai H. A critical time window for dopamine actions on the structural plasticity of dendritic spines. Science. 2014;345:1616–1620. doi: 10.1126/science.1255514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson C, Dani JA. Muscarinic and nicotinic cholinergic mechanisms in the mesostriatal dopamine systems. Neuroscientist. 2003;9:23–36. doi: 10.1177/1073858402239588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.