

Abstract
In hypoxic cells, dysfunctional mitochondria are selectively removed by a specialized autophagic process called mitophagy. The ER–mitochondrial contact site (MAM) is essential for fission of mitochondria prior to engulfment, and the outer mitochondrial membrane protein FUNDC1 interacts with LC3 to recruit autophagosomes, but the mechanisms integrating these processes are poorly understood. Here, we describe a new pathway mediating mitochondrial fission and subsequent mitophagy under hypoxic conditions. FUNDC1 accumulates at the MAM by associating with the ER membrane protein calnexin. As mitophagy proceeds, FUNDC1/calnexin association attenuates and the exposed cytosolic loop of FUNDC1 interacts with DRP1 instead. DRP1 is thereby recruited to the MAM, and mitochondrial fission then occurs. Knockdown of FUNDC1, DRP1, or calnexin prevents fission and mitophagy under hypoxic conditions. Thus, FUNDC1 integrates mitochondrial fission and mitophagy at the interface of the MAM by working in concert with DRP1 and calnexin under hypoxic conditions in mammalian cells.
Keywords: autophagy, ER–mitochondrial contact site, MAM, mitochondrial fission, mitophagy
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport, Metabolism
Introduction
The mitochondrion is at the core of cellular energy metabolism and controls a series of important metabolic pathways (Chan, 2006; Dimmer & Scorrano, 2006; Suen et al, 2008; Green et al, 2014). The vital role of mitochondria is mediated by mitochondrial fission and fusion, which consequently regulate embryonic development, metabolism, apoptosis, and autophagy (Green & Reed, 1998; Wang, 2001; Gomes et al, 2011; Nunnari & Suomalainen, 2012; Wasilewski et al, 2012; Escobar‐Henriques & Anton, 2013). Dysfunctional mitochondrial fission and fusion are implicated in neurodegeneration, diabetes, cardiovascular disorders, and cancer (Chan, 2006; Knott et al, 2008; Cho et al, 2010; Yoon et al, 2011; Feng et al, 2013; Chandel, 2014; Schneider & Cuervo, 2014).
Two dynamin‐related GTPases, MFN1 (Mitofusin 1) and MFN2 (Mitofusin 2), participate in mitochondrial outer membrane fusion by forming heterodimers with each other (Santel & Fuller, 2001; Chen et al, 2003; Koshiba et al, 2004). OPA1, another dynamin‐related GTPase, is involved in inner mitochondrial membrane fusion together with MFN1 (Cipolat et al, 2004). In addition, OPA1 also plays important roles in regulating crista morphology and apoptosis (Cipolat et al, 2006; Duvezin‐Caubet et al, 2006; Frezza et al, 2006). DRP1 (Dnm1 in yeast), a highly conserved dynamin‐related GTPase, is the crucial hub in the mitochondrial fission process (Smirnova et al, 2001; Taguchi et al, 2007). Ser637 of cytosolic DRP1 is dephosphorylated and DRP1 is subsequently recruited to mitochondria to drive mitochondrial constriction and membrane fragmentation (Smirnova et al, 2001; Roux et al, 2006; Cereghetti et al, 2008; Mears et al, 2011). DRP1 adaptors, which participate in mitochondrial fission together with DRP1, include MFF (mitochondrial fission factor), FIS1 (mitochondrial fission protein 1), and MiD49 and MiD51 (mitochondrial dynamics proteins of 49 and 51 kDa); however, the role of FIS1 and MID49/51 in mitochondrial fission and fusion is still controversial (Stojanovski et al, 2004; Gandre‐Babbe & van der Bliek, 2008; Otera et al, 2010; Palmer et al, 2011, 2013; Zhao et al, 2011; Loson et al, 2013).
The mitochondria‐associated membrane (MAM) connects the ER with mitochondria (Rowland & Voeltz, 2012; Kornmann, 2013; Klecker et al, 2014; Marchi et al, 2014). Recent data have shown that in yeast and mammalian cells, mitochondrial fission occurs at sites where the ER is tethered to the mitochondria. ER tubules wrap around mitochondria and mark the sites where Dnm1 (in yeast) or DRP1 (in mammals) is recruited by mitochondrial receptors. The assembled Dnm1 or DRP1 then forms a helix to constrict the mitochondria (Friedman et al, 2011; Murley et al, 2013). Another critical role of the MAM involves initiation of autophagosome formation in general autophagy in mammals or mitophagosome formation and mitophagy in yeast (Matsunaga et al, 2010; Hamasaki et al, 2013; Bockler & Westermann, 2014). Previously, we have shown that hypoxia could induce mitochondrial fragmentation and mitophagy (Liu et al, 2012; Wu et al, 2014b), but the mechanisms linking fission and mitophagy under hypoxic conditions remain poorly understood.
In this study, we found that the outer mitochondrial membrane protein FUNDC1 is a novel MAM protein, which accumulates at the ER–mitochondria contact sites by interacting with the ER membrane protein calnexin in hypoxic cells. Further, we found that FUNDC1 is specifically required for mitochondrial fragmentation: It recruits DRP1 to drive mitochondrial fission in response to hypoxic stress. In addition, siRNA silencing of FUNDC1, DRP1, or calnexin causes mitochondrial elongation and also reduces the association of the ER with LC3 and mitochondria, thus preventing mitophagy under hypoxic conditions. Taken together, these findings reveal that FUNDC1 is not only a novel MAM protein that acts as an adaptor for DRP1 during the mitochondrial fission process, but also involved in hypoxia‐induced mitophagy.
Results
FUNDC1 accumulates at ER–mitochondria tethering sites in response to hypoxia
The MAM plays an important role in autophagy or mitophagy (Hamasaki et al, 2013; Bockler & Westermann, 2014). In general, the mitochondrial ER tethering site with the average lengths of tethers between OMM and rough ER or between OMM and smooth ER are ranging from 9 to 30 nm (Csordas et al, 2006; Marchi et al, 2014; Naon & Scorrano, 2014). As shown in our previous studies, the outer mitochondrial membrane protein FUNDC1 mediates mitophagy in response to hypoxia (Liu et al, 2012; Li et al, 2014; Wu et al, 2014b). We thus speculated whether FUNDC1 is related to the MAM in response to hypoxia. We first confirmed the efficacy of our hypoxia induction protocol by measuring the expression of hypoxia‐inducible proteins (the transcription factor HIF‐1α and NIX) in cells in response to hypoxic stress (Appendix Fig S1). We then analyzed the subcellular distribution of FUNDC1 in normoxic and hypoxic HeLa cells by Percoll density‐gradient centrifugation. Different fractions were identified with the following organelle markers: calnexin (ER and MAM), calreticulin (ER), FACL4 (MAM), VDAC1 (MAM and mitochondria), MFN2 (ER, MAM, and mitochondria), TOM20, MID51/49, FIS1, MFF (mitochondria) (de Brito & Scorrano, 2008; Wieckowski et al, 2009; Hamasaki et al, 2013). We found that FUNDC1 substantially accumulates at the ER–mitochondria contact sites in response to hypoxia, although a small amount of FUNDC1 can be also detected in the MAM under normoxic conditions (Fig 1A–C). Surprisingly, the protein level of DRP1 in the MAM is also prominently elevated in response to hypoxia (Fig 1A–C). Immunogold labeling confirmed that FUNDC1 indeed accumulates at the ER–mitochondria interface since a well‐established MAM protein, FACL4, also concentrates at this site in response to hypoxia (de Brito & Scorrano, 2008; Wieckowski et al, 2009; Hamasaki et al, 2013) (Fig 1D–I). In addition, the number of MAM also increases under hypoxia condition (Fig 1H and I). As a negative control, TOM20, a membrane protein located in the outer mitochondrial membrane, does not accumulate in MAM under this condition (Fig 1E–G). We found that DRP1 is also enriched in the MAM fraction in hypoxic cells (Fig 1A and B) and localizes with FUNDC1 in the MAM as evidenced by double immunogold EM (Fig 1L). Triple‐color imaging further demonstrated that FUNDC1 and DRP1 puncta are closely associated with lacunae formed by ER lattices in hypoxic conditions (Appendix Fig S2).
Figure 1. FUNDC1 is a novel MAM protein and accumulates in the MAM under hypoxic conditions.
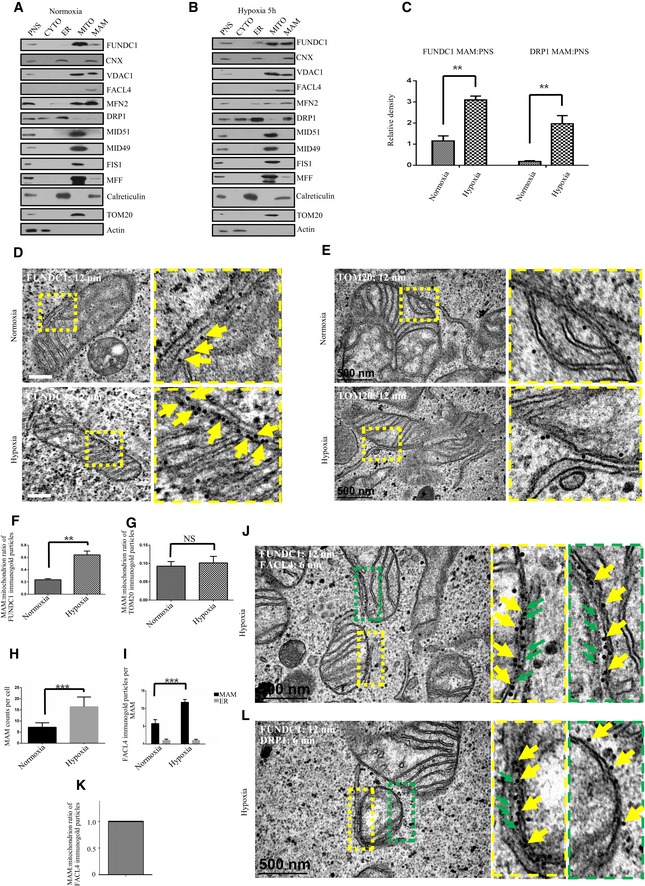
- Immunoblots of subcellular fractions from HeLa cells exposed to normoxia. P: post‐nuclear supernatant; C: cytosol; ER: endoplasmic reticulum; MT: mitochondria; MA: mitochondrial‐associated membranes.
- Immunoblots of subcellular fractions from HeLa cells exposed to hypoxia (1% O2) for 5 h. P: post‐nuclear supernatant; C: cytosol; ER: endoplasmic reticulum; MT: mitochondria; MA: mitochondrial‐associated membranes.
- Quantification of the MAM:PNS ratio of FUNDC1 or DRP1 under normoxic or hypoxic conditions. All quantitative data are presented as mean ± s.e.m. from three independent experiments, **P < 0.01.
- Control HeLa cells or hypoxia‐treated HeLa cells (1% O2 for 5 h) were processed for immunogold electron microscopy with anti‐FUNDC1 antibody (12‐nm gold particles). Scale bar = 500 nm. The boxed areas in the left panels are magnified in the right panels. Arrows indicate gold particles.
- Control HeLa cells or hypoxia‐treated HeLa cells (1% O2 for 5 h) were processed for immunogold electron microscopy with anti‐TOM20 antibody (12‐nm gold particles). Scale bar = 500 nm. The boxed areas in the left panels are magnified in the right panels.
- Quantification of the MAM:mitochondrion ratio of FUNDC1 immunogold particles from (D) under normoxic and hypoxic (1% O2) conditions. Data (mean ± s.e.m.) are from three independent experiments. n = 60 MAMs (one MAM means one mitochondrial ER tethering site in the corresponding mitochondria), **P < 0.01.
- Quantification of the MAM:mitochondrion ratio of TOM20 immunogold particles from (E) under normoxic and hypoxic (1% O2) conditions. Data (mean ± s.e.m.) are from three independent experiments. n = 50 MAMs (one MAM means one mitochondrial ER tethering site in the corresponding mitochondria). NS means not significant.
- The quantification of MAM counts from 50 cells (mean ± s.e.m.), ***P < 0.001.
- The quantification of FACL4 immunogold particles from 50 cells (mean ± s.e.m.), ***P < 0.001.
- Hypoxia‐treated HeLa cells (1% O2 for 5 h) were processed for immunogold electron microscopy with anti‐FUNDC1 antibody (12‐nm gold particles) and anti‐FACL4 (6‐nm gold particles). Scale bar = 500 nm. The boxed areas in the left panels are magnified in the right panels. Yellow arrows indicate FUNDC‐gold particles, and green arrows indicate FACL4‐gold particles.
- The ratio of FACL4 immunogold particles on the MAM and mitochondrion. n = 80 MAMs (one MAM means one mitochondrial ER tethering site in the corresponding mitochondria).
- Hypoxia‐treated HeLa cells (1% O2 for 5 h) were processed for immunogold electron microscopy with anti‐FUNDC1 antibody (12‐nm gold particles) and anti‐DRP1 (6‐nm gold particles). Scale bar = 500 nm. The boxed areas in the left panels are magnified in the right panels. Yellow arrows indicate FUNDC‐gold particles, and green arrows indicate DRP1‐gold particles.
Source data are available online for this figure.
The ER membrane protein calnexin is required for the accumulation of FUNDC1 in the MAM
Why does FUNDC1 accumulate in the MAM in response to hypoxia? We hypothesized that the accumulation of FUNDC1 might be driven by certain MAM proteins under hypoxic conditions. We tested whether FUNDC1 could be immunoprecipitated by different MAM protein markers, including STX17, FACL4, IP3Rs, and calnexin and found that calnexin is a potential binding partner of FUNDC1 in the MAM. Using transient transfection, we found that endogenous calnexin, but not BAP31, FIS1, or MFF is co‐immunoprecipitated with FUNDC1‐MYC using anti‐MYC antibody (Fig 2B). Furthermore, we observed that FLAG‐FUNDC1 co‐immunoprecipitated with calnexin using an anti‐calnexin antibody, but not an IgG control (Fig 2C).
Figure 2. The ER membrane protein calnexin is required for the accumulation of FUNDC1 in the MAM .
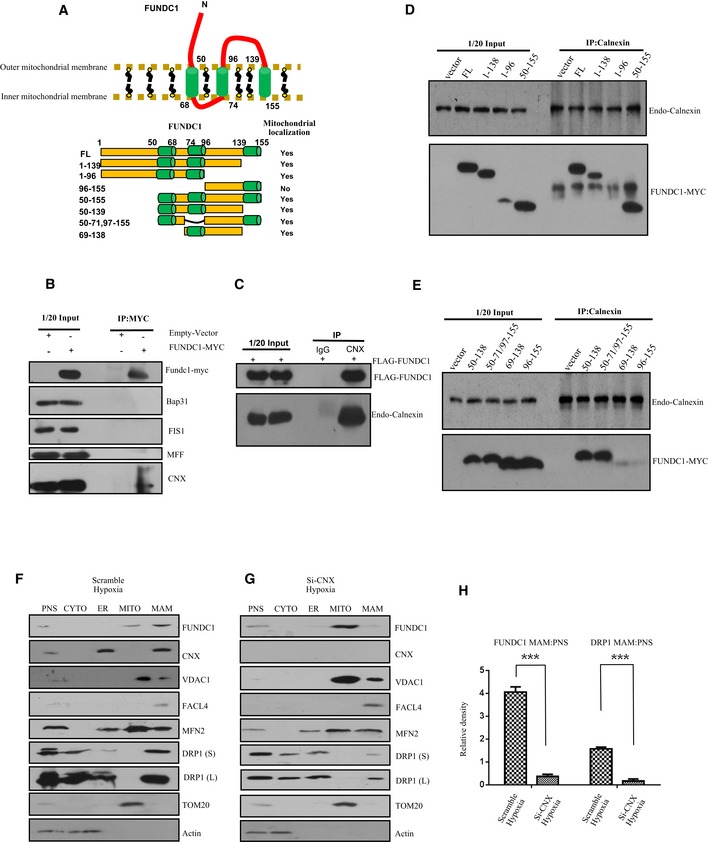
-
ATop: Cartoon representing the topology of the FUNDC1 protein in the mitochondrial membrane. The cytoplasmic domain that interacts with the ER protein calnexin is shown. Bottom: Illustration of the different FUNDC1 truncation variants.
-
BHeLa cells were transfected with FUNDC1‐MYC or empty vector for 24 h. Cell lysates were immunoprecipitated by anti‐MYC antibody and immunoblotted with the indicated antibodies.
-
CHeLa cells were transfected with FLAG‐FUNDC1. About 24 h post‐transfection, cell lysates were immunoprecipitated by anti‐calnexin and immunoblotted with anti‐FLAG and anti‐calnexin antibodies.
-
D, EHeLa cells were transfected with the indicated FUNDC1‐MYC constructs for 24 h. Cell lysates were immunoprecipitated using anti‐calnexin and immunoblotted using anti‐MYC and anti‐calnexin antibodies.
-
FImmunoblots of subcellular fractions from HeLa cells transfected with scramble siRNA and exposed to hypoxia (1% O2) for 5 h. PNS: post‐nuclear supernatant; CYTO: cytosol; ER: endoplasmic reticulum; MITO: mitochondria; MAM: mitochondrial‐associated membrane. DRP1 (S) and DRP1 (L) indicate short and long exposures, respectively.
-
GImmunoblots of subcellular fractions from HeLa cells transfected with calnexin siRNA (siCNX) and exposed to hypoxia (1% O2) for 5 h. PNS: post‐nuclear supernatant; CYTO: cytosol; ER: endoplasmic reticulum; MITO: mitochondria; MAM: mitochondrial‐associated membrane. DRP1 (S) and DRP1 (L) indicate short and long exposures, respectively.
-
HQuantification of the MAM:PNS ratio of FUNDC1 or DRP1 in HeLa cells treated with scramble siRNA or siCNX and exposed to hypoxia (1% O2) for 5 h. Data are presented as mean ± s.e.m. from three independent experiments, ***P < 0.001.
Source data are available online for this figure.
FUNDC1 includes three transmembrane domains, an N‐terminal region (AA 1–50) and a loop (AA 96–138) that is exposed to cytosol. Previously, we proved that AA 50–96, including the first two transmembrane domains, is crucial for localizing FUNDC1 to mitochondria (Fig 2A and Wu et al, 2014b). Here, to identify which domain of FUNDC1 is essential for the interaction with calnexin and whether localization of FUNDC1 to mitochondria is required for this interaction, we transfected various mutants of FUNDC1‐MYC into HeLa cells and then immunoprecipitated calnexin using anti‐calnexin antibody. We found that the cytosolic domain (AA 96–138) is essential for the interaction of FUNDC1 with calnexin (Fig 2D). To identify the minimal region for calnexin binding, we created smaller deletions within AA 96–138 of FUNDC1 and narrowed the key interaction region to AA 129–138 (Appendix Fig S3A). We then made a series of mutations in this region, but unfortunately, we found that they all associate with calnexin (Appendix Fig S3B). We also discovered that the first transmembrane domain (AA 50–68) is critical for the interaction of FUNDC1 with calnexin because the loss of this domain dramatically lessened the interaction (Fig 2E). Elimination of AA 50–96, which is required for the localization of FUNDC1 to mitochondria, severely interrupted the FUNDC1/calnexin association (Fig 2E). To identify the interaction region for FUNDC1 binding, we made C‐terminal (AA 504–586) and N‐terminal (AA 1–503) truncated forms of calnexin. We found that the N‐terminus of calnexin associates with FUNDC1 (Appendix Fig S4). Because the N‐terminus of calnexin is in the lumen of ER, it is unlikely that the hydrophilic domain of FUNDC1 (96–139) can interact directly with calnexin in the lumen, implying that their interaction is indirect.
Next, we examined whether calnexin plays a role in the accumulation of FUNDC1 at the MAM. When we knocked down calnexin and purified the MAM fraction, we found that the amount of FUNDC1 is significantly reduced compared to cells treated with scramble siRNA (Fig 2F–H). Double immunogold EM also suggests that FUNDC1 fails to accumulate in the MAM in calnexin‐depleted cells under hypoxia (Fig EV1). Interestingly, the amount of DRP1 in the MAM fraction also decreases in calnexin KD cells (Fig 2F–H).
Figure EV1. Depletion of calnexin prevents the accumulation of FUNDC1 in the MAM under hypoxia.
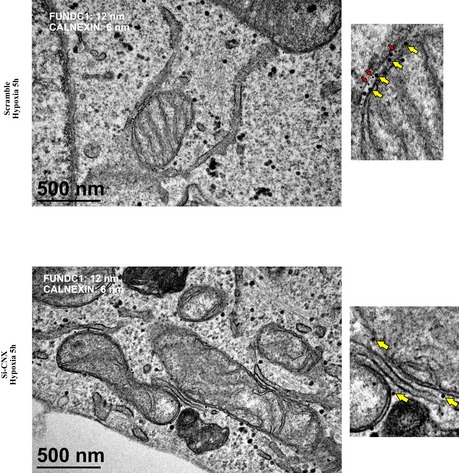
Immunogold electron microscopy of HeLa cells transfected with scramble siRNA or siCNX and exposed to hypoxia (1% O2) for 5 h. Endogenous FUNDC1 and endogenous CNX were detected by anti‐FUNDC1 antibody conjugated to 12‐nm gold particles (yellow arrows) and anti‐CNX antibody conjugated to 6‐nm gold particles (red arrows).
FUNDC1 mediates mitochondrial fission in response to hypoxia
Our previous study showed that FUNDC1 mediates hypoxia‐induced mitophagy. We examined the mitochondrial morphology in cells exposed to hypoxic conditions for different lengths of time (Fig EV2). The proportion of cells containing ruptured mitochondria increased dramatically between 5 and 12 h. At 24 h, over 90% cells had completely fragmented mitochondria (Fig EV2). It is generally believed that a mitochondrion should be fragmented before engulfment by the autophagosome to match the relatively small size of the latter (Tatsuta & Langer, 2008; Mao & Klionsky, 2013). Therefore, we wondered what drives mitochondrial fission before mitophagy in response to hypoxia. We silenced the main mitochondrial fission‐related proteins, MFF or FIS1, and discovered that the loss of MFF or FIS1 partially inhibit mitochondrial fission under hypoxic conditions. In hypoxic MFF and FIS1 KD cells, the percentage of mitochondria that were fragmented after 12 h was 65% and 80%, respectively, compared to 92% in hypoxic control cells (Fig 3A–C). Conversely, the loss of FUNDC1 dramatically inhibits mitochondrial fragmentation in response to hypoxia. Only 9% of mitochondria were fragmented, while 86% were elongated (Fig 3A–C). In addition, we tested whether FUNDC1 is involved in mitochondrial fission under conditions that are known to be affected by MFF, such as FCCP, apoptosis‐inducing compound (etoposide). Knocking‐down of FUNDC1 partially prevents mitochondrial fission induced by FCCP but not etoposide (Fig EV3). After silencing calnexin, mitochondrial fragmentation is also dramatically diminished compared to silencing of MFF and FIS1, but the effect is milder than that in FUNDC KD cells. The percentage of elongated mitochondria reached 60% in calnexin KD cells, compared to just 18% in MFF and 12% in FIS1 KD cells (Fig 3A–C) under hypoxic conditions.
Figure EV2. Mitochondrial morphology in cells exposed to hypoxia for different lengths of time.
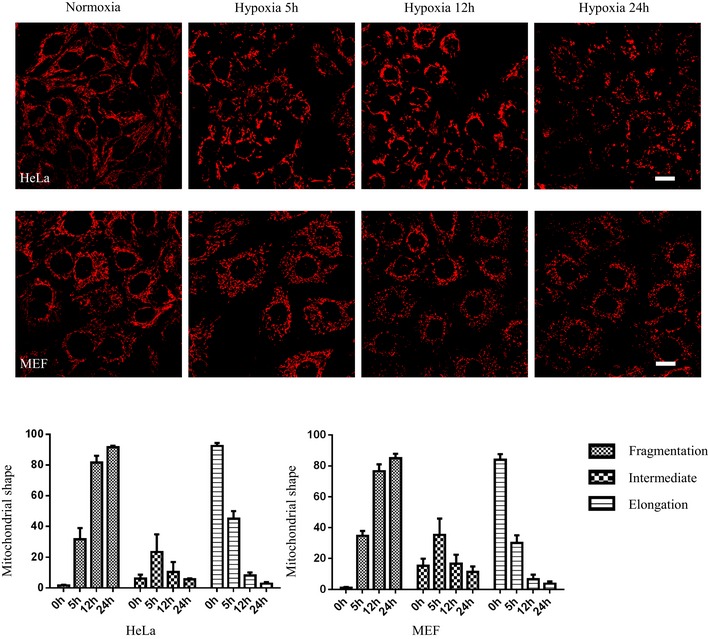
HeLa and MEF cells were exposed to hypoxia (1% O2) for the indicated times, then immunostained by anti‐TOM20. Scale bar = 20 µm. The bar charts show quantification of mitochondrial morphology (fragmented, intermediate, and elongated mitochondria) in 100 HeLa and MEF cells. Data represent mean ± s.e.m. from three independent experiments.
Figure 3. FUNDC1 mediates mitochondrial fission in response to hypoxia.
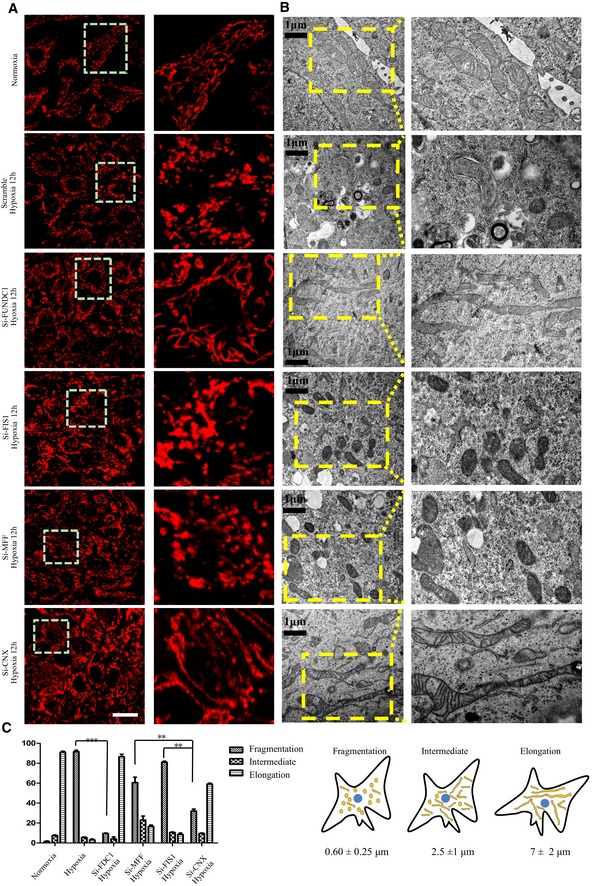
- HeLa cells were transfected with the indicated siRNAs. Twenty‐four hours post‐transfection, cells were exposed to hypoxia (1% O2) for 12 h and then immunostained by anti‐TOM20. Scale bar = 50 µm. Boxed regions in the left panels are enlarged in the right panels.
- Electron microscopy analysis of HeLa cells treated with the indicated siRNAs for 24 h and then exposed to hypoxia (1% O2) for 12 h. Scale bar = 1 μm. Boxed regions in the left panels are enlarged in the right panels.
- Quantification of mitochondrial morphology (fragmented, intermediate, and elongated mitochondria) in 100 cells from (A). Data represent mean ± s.e.m. (**P < 0.01, ***P < 0.001).
Figure EV3. The role of MFF or FUNDC1 in mitochondria collapse induced by CCCP, etoposide, or hypoxia, respectively.
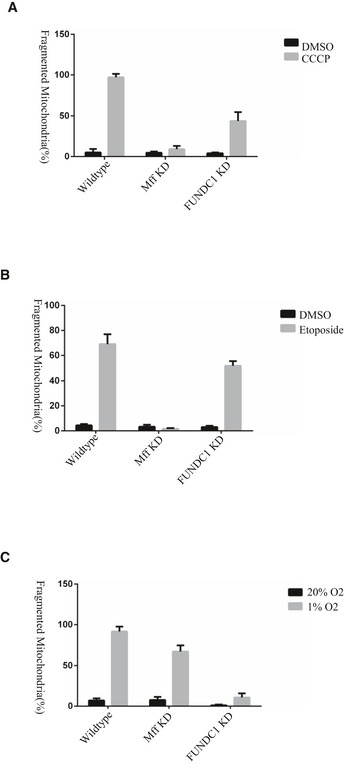
- Different HeLa cell lines (wild‐type, MFF KD, and FUNDC1 KD) were treated with 10 μM CCCP or DMSO for 1 h. The percentage of fragmented mitochondria is quantified from three independent experiments (averages ± s.e.m.), each with 100 cells. All statistical testing was performed with the Student's t‐test.
- Different HeLa cell lines (wild‐type, MFF KD, and FUNDC1 KD) were treated with 100 μM etoposide or DMSO for 5 h. The percentage of fragmented mitochondria is quantified from three independent experiments (averages ± s.e.m.), each with 100 cells. All statistical testing was performed with the Student's t‐test.
- Different HeLa cell lines (wild‐type, MFF KD, and FUNDC1 KD) were treated with 1% O2 or 20% O2 for 12 h. The percentage of fragmented mitochondria is quantified from three independent experiments (averages ± s.e.m.), each with 100 cells. All statistical testing was performed with the Student's t‐test.
FUNDC1 recruits DRP1 to facilitate mitochondrial fission during hypoxia
As DRP1 is at the core of regulating regular mitochondrial homeostasis, we wanted to test whether DRP1 is also involved in mitochondrial fragmentation together with FUNDC1 under hypoxic conditions. We therefore examined whether DRP1 translocates to mitochondria in hypoxic cells. Under normal conditions, DRP1 sporadically localizes with mitochondria to facilitate regular mitochondrial fission (Fig 4A). However, the level of mitochondrial DRP1 markedly increases after 12‐h hypoxic treatment (Fig 4A and B). Surprisingly, when FUNDC1 is silenced, the amount of DRP1 translocating to mitochondria in hypoxic cells sharply decreases and the mitochondria are observed to elongate (Fig 4A and B). Immunogold electron micrographs also show that in hypoxic siFUNDC1‐treated cells, less DRP1 translocates to mitochondria and less mitochondria are fragmented compared to control cells transfected with scramble siRNA (Fig 4C). However, knockdown of MFF, FIS1, or MID49/51 does not affect DRP translocation to mitochondria under hypoxic conditions (Fig 4A and B). Next, we evaluated hypoxia‐induced mitochondrial DRP1 translocation by subcellular fractionation and found that when FUNDC1 is silenced, less DRP1 co‐fractionates with crude mitochondria (mitochondria + MAM) than when MFF, FIS1, mid49/51 is silenced (Fig 4C–H). We further examined the role of APAP121 in hypoxia‐induced mitochondrial fission. APAP121 slightly decreases under hypoxia. However, mitochondrion still fragments in cells when much of AKAP121 is depleted (Fig EV4).
Figure 4. FUNDC1 recruits DRP1 to facilitate mitochondrial fission under hypoxic conditions.
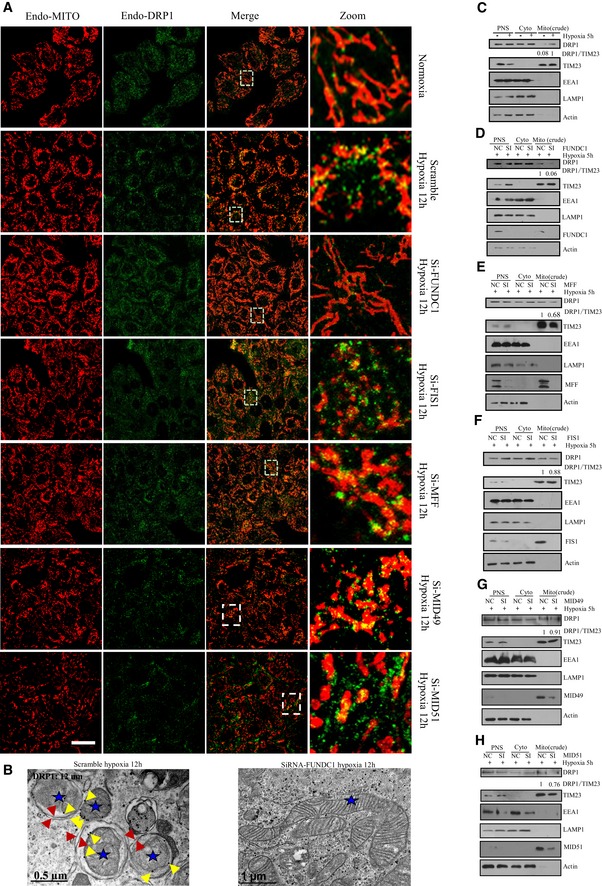
-
AHeLa cells were treated with the indicated siRNAs and exposed to hypoxia for 12 h. Cells were fixed and immunostained by anti‐TOM20 and anti‐DRP1 antibodies. Scale bar = 40 µm. Boxed regions are enlarged in the right panels.
-
BImmunogold electron micrographs of control HeLa cells (top) or HeLa cells transfected with siFUNDC1 (bottom) and then exposed to hypoxia (1% O2) for 12 h. DRP1 was detected with anti‐DRP1 (12‐nm gold particles). Blue stars indicate mitochondria. Yellow arrows indicate gold particles. Red arrows indicate double‐membraned autophagosomes.
-
C–HImmunoblots of subcellular fractions from control HeLa cells or HeLa cells treated with the indicated siRNAs and then exposed to hypoxia for 5 h. PNS: post‐nuclear supernatant; Cyto: cytosol; Mito (crude): crude mitochondrial fraction containing mitochondria and MAMs.
Source data are available online for this figure.
Figure EV4. The effect of AKAP121 on mitochondrial fragmentation under hypoxia.
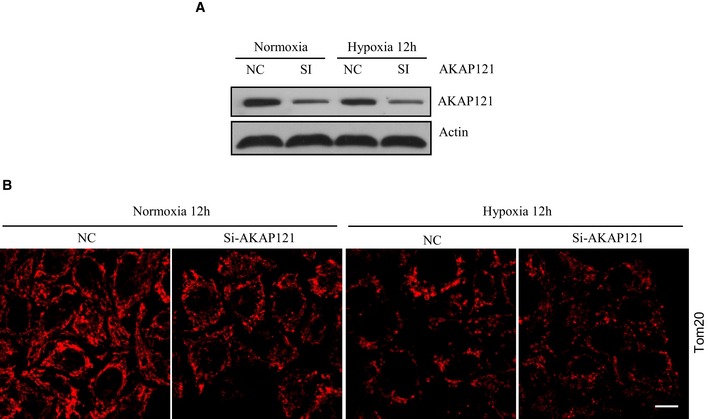
-
A, BHeLa cells were transfected using scramble or siAKAP121 RNAs. About 24 h post‐transfection, cells were exposed to hypoxia for 12 h and then cells were either subjected to immunoblot for knockdown efficacy (A) or immunostained by anti‐TOM20 antibody (B). Scale bar = 15 μm.
Next, we examined the localization of calnexin, DRP1, and FUNDC1 in hypoxic cells at different time points. Time‐course analysis of these proteins in different subcellular fractions, including ER, mitochondria, and MAMs, is shown in Appendix Fig S5. DRP1 and FUNDC1 have dynamic localization patterns. At 6 h, most DRP1 is found in the MAM fraction, while at 12 h, DRP1 mainly localizes to mitochondria, as compared MAM with mitochondria (Appendix Fig S5).
FUNDC1 binds directly to DRP1 and their role in mediating mitochondrial fission
Unlike FIS1, MID49/51, or MFF KD, FUNDC1 KD prevents DRP1 translocation and mitochondrial fission under hypoxic conditions, indicating that FUNDC1 may possibly be involved in the mitochondrial translocation of DRP1. To test this hypothesis, we cotransfected FLAG‐DRP1 and FUNDC1‐MYC into HeLa cells. Twenty‐four hours post‐transfection, we lysed the cells and found that FUNDC1‐MYC immunoprecipitated with FLAG‐DRP1 using anti‐FLAG antibody (Fig 5A). Meanwhile, we transiently cotransfected FLAG‐FUNDC1 and MYC‐DRP1 and found that MYC‐DRP1 was precipitated with FLAG‐FUNDC1 using anti‐FLAG antibody (Fig 5B). We further transfected FUNDC1‐MYC into MEFs and detected the colocalization of FUNDC1‐MYC and endo‐DRP1 by immunofluorescence. Interestingly, many endo‐DRP1 dots colocalizes with FUNDC1‐MYC‐positive puncta (Fig 5C). Next, we examined which domain of FUNDC1 is essential for its interaction with DRP1. We cotransfected FLAG‐DRP1 with different FUNDC1‐MYC constructs and found that the cytosolic loop (AA 96–138) of FUNDC1, which binds to calnexin, is also essential for the interaction with DRP1 (Fig 5D and E). In addition, deletion of AA 50–96 abolishes the interaction of FUNDC1 with DRP1 (Fig 5D and E). As DRP1 and calnexin interact with the same domain of FUNDC1, we were curious about how DRP1 and calnexin interact with FUNDC1 temporally. We exposed HeLa cells to hypoxic conditions for different lengths of time and found that no or very little DRP1 is precipitated by FUNDC1 antibody after 0 and 1 h (Fig 5F and Appendix Fig S6). When the cells were exposed to hypoxia for longer (3 or 6 h), the amount of DRP1 immunoprecipitated by anti‐FUNDC1 antibody increased considerably (Fig 5F and Appendix Fig S3A). In contrast, calnexin shows a different temporal pattern of association with FUNDC1. Calnexin is immunoprecipitated by anti‐FUNDC1 antibody at low but clearly detectable levels under normoxic conditions (0 h). After 1 h of hypoxia, the amount of calnexin immunoprecipitated by anti‐FUNDC1 antibody is significantly elevated, but the level of calnexin decreases after 3 h and is then sharply reduced after 6 h (Fig 5F and Appendix Fig S3A). We also observed an increased binding between LC3 and FUNDC1 at later stage of hypoxia (Fig 5F), and FUNDC1 (AA 1–50) is critical for its binding with LC3 (Appendix Fig S11). In vitro pull‐down assay verified the direct interaction between FUNDC1 and DRP1 because GST‐FUNDC1 co‐elutes with DRP1 but GST alone does not (Fig 5G).
Figure 5. FUNDC1 binds directly to DRP1 and their role in mediating mitochondrial fission.
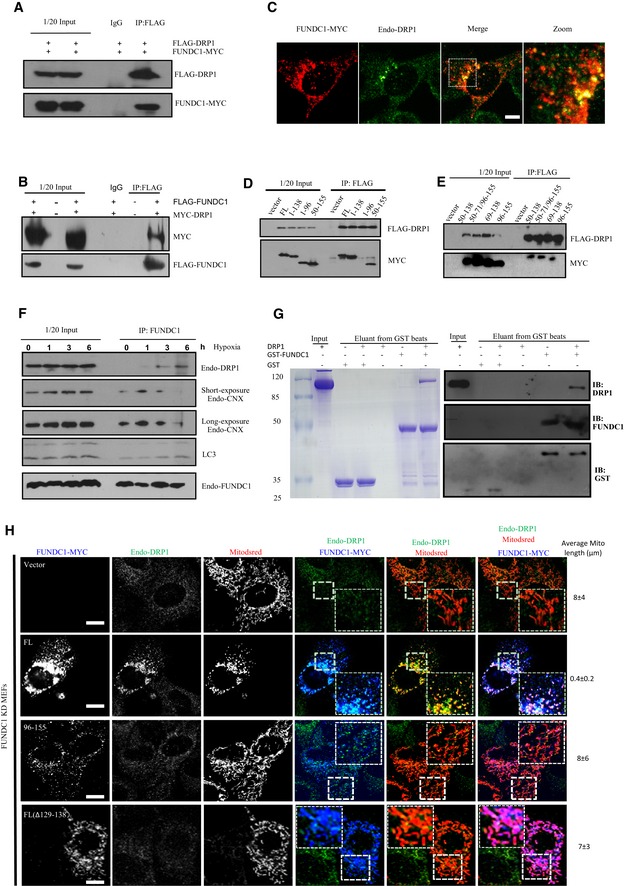
-
AHeLa cells were cotransfected with FLAG‐DRP1 and FUNDC1‐MYC for 24 h. Cells were lysed and immunoprecipitated using anti‐FLAG antibody. FLAG‐DRP1 and FUNDC1‐MYC were analyzed with the corresponding antibodies.
-
BHeLa cells were cotransfected with empty vector, MYC‐DRP1, and FLAG‐FUNDC1. Twenty‐four hours after transfection, cell lysates were immunoprecipitated using anti‐FLAG antibody and immunoblotted with anti‐MYC and anti‐FLAG antibodies.
-
CMEFs were transfected with FUNDC1‐MYC. Twenty‐four hours post‐transfection, cells were fixed and immunostained by anti‐MYC and anti‐DRP1. Scale bar = 10 µm.
-
D, EHeLa cells were cotransfected with the indicated FUNDC1‐MYC and FLAG‐DRP1 constructs for 24 h. Cell lysates were analyzed by immunoprecipitation using anti‐FLAG and immunoblotted with anti‐MYC and anti‐FLAG antibodies.
-
FHeLa cells were exposed to hypoxia (1% O2) for the indicated times, and cell lysates were immunoprecipitated using anti‐FUNDC1 antibody. Endogenous DRP1, calnexin, LC3, and FUNDC1 were detected by anti‐DRP1, anti‐calnexin, anti‐LC3, and anti‐FUNDC1 antibodies.
-
GAbout 10–20 μg GST‐FUNDC1 or GST was immobilized on glutathione sepharose resin (GE Healthcare). About 20–50 μg DPR1 was incubated with the GST‐FUNDC1‐ or GST‐bound resin overnight at 4°C. The resin was then extensively rinsed and eluted. Samples were subjected to SDS–PAGE and then visualized by Coomassie Blue staining and Western blot, respectively.
-
HFull‐length FUNDC1‐MYC, FUNDC1‐MYC (AA 96–155), FL (Δ129–138), or vector control was transfected with Mitodsred (red) into FUNDC1 KD MEFs. Twelve hours post‐transfection, cells were exposed to hypoxia for 12 h and then stained with anti‐MYC (blue) and anti‐DRP1 (green). Scale bar = 10 µm. Numbers at the right show the average length of mitochondria in Mitodsred‐positive cells.
Source data are available online for this figure.
We next compared the ability of full‐length FUNDC1 with truncated FUNDC1 (with a deletion of AA 1–95 or a deletion of AA 129–138) in inducing mitochondrial fragmentation. As shown in Fig 5H, full‐length FUNDC1 dramatically promotes mitochondrial DRP1 accumulation as well as mitochondrial fragmentation, in contrast to the two FUNDC1 deletion mutants. Similarly, hypoxia‐induced mitochondrial fission can be restored by expressing siRNA‐resistant calnexin in calnexin‐depleted cells (Appendix Fig S8). Notably, the distribution of FUNDC1, calreticulin, FACL4, or calnexin in MAM fraction is not affected in DRP1‐depleted cells (Appendix Fig S9).
Calnexin and DRP1 are involved in FUNDC1‐mediated mitophagy induced by hypoxia
Since calnexin and DRP1 interact with FUNDC1 and are required for mitochondrial fission, we next examined whether these two proteins also act in hypoxia‐induced mitophagy. We knocked down FUNDC1, DRP1, calnexin, MFF, or FIS1 in HeLa cells and found that silencing of FUNDC1, calnexin, or DRP1 causes mitochondrial elongation and reduces the colocalization of LC3 and mitochondria in response to hypoxia (Fig 6A). In contrast, mitochondrial fragmentation and mitophagy frequently occur in hypoxic MFF or FIS1 KD cells, or control cells treated with scramble siRNA. ER and mitochondrial morphology are not affected in FUNDC1 or calnexin KD cells under normoxic conditions (Appendix Figs S10 and S11). However, the colocalization of mitochondria and LC3 was impaired when we further knocked down FUNDC1 in MFN1/2 double KO cells, indicating that the mitochondrial fission is required but not sufficient for mitophagy (Appendix Fig S12). Biochemical results confirmed that silencing of FUNDC1, calnexin, or DRP1 dramatically impairs the clearance of fragmented mitochondria by autophagy, as BAF A1 is able to inhibit the degradation of inner and outer mitochondrial membrane proteins TIM23 and TOM20 as well as LC3 (Fig 6B–E). More specifically, when overexpressing the construct of FUNDC1 lacking either the DRP1/calnexin‐binding domain (1–96), or the construct lacking the mitochondrial localization sequence (96–155), it could prevent the mitophagy induced by FUNDC1 (Fig 6F). Taken together, our data show that FUNDC1, calnexin, and DRP1 cooperatively regulate mitochondrial dynamics and mitophagy in response to hypoxia.
Figure 6. Calnexin and DRP1 are involved in FUNDC1‐mediated mitophagy induced by hypoxia.
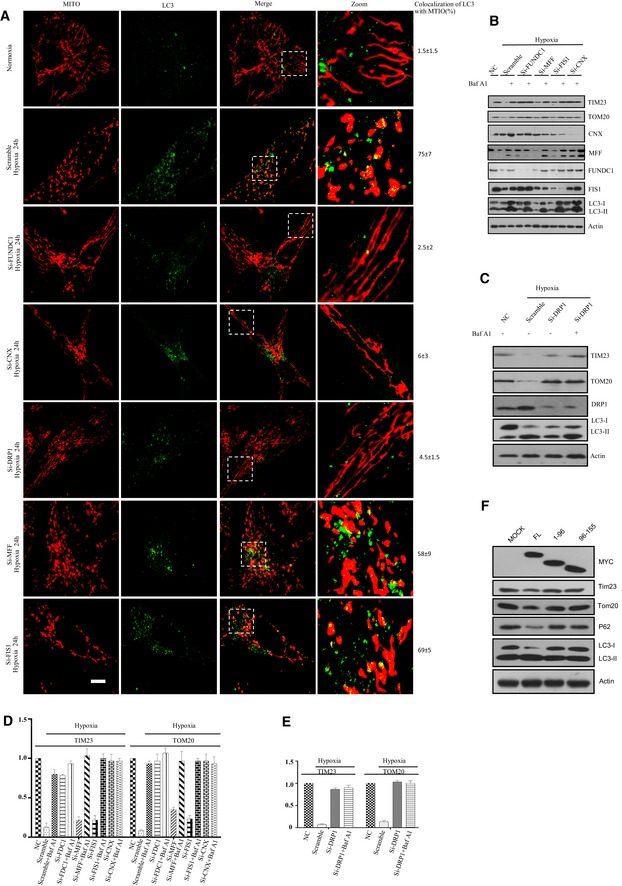
-
AHeLa cells were transfected with the indicated siRNAs for 24 h and then exposed to hypoxia for 24 h or not. Cells were fixed and immunostained by anti‐TOM20 and anti‐LC3 antibodies. The colocalization of LC3 and mitochondria was quantified and listed on the right side. Scale bar = 5 µm. Boxed regions are enlarged in the right panels.
-
B, CHeLa cells were transfected with the indicated siRNAs for 24 h and then exposed to hypoxia for 24 h. Cells were then treated with or without 50 nM BAF A1 (bafilomycin A1) for 6 h before harvesting. Cell lysates were immunoblotted with the indicated antibodies.
-
D, EQuantification of the TIM23:actin and TOM20:actin ratios from (B and C). All data are from three independent experiments (mean ± s.e.m.).
-
FHeLa cells were transfected by different truncated FUNDCY‐MYC. About 24 h post‐transfection, cell lysates were immunoblotted using the indicated antibodies.
Source data are available online for this figure.
Discussion
We previously showed that the mitochondrial protein FUNDC1 plays a key role in hypoxia‐induced mitophagy by binding LC3, an autophagy protein that is essential for autophagosome biogenesis. We have now shown how FUNDC1 mediates mitochondrial fragmentation under hypoxic conditions. We provide compelling evidences that FUNDC1 is not only a novel MAM‐related protein, but also a new mitochondrial receptor for DRP1 to facilitate mitochondrial fragmentation and mitophagy in response to hypoxia.
Many studies have demonstrated that contact between the mitochondria and the ER plays a very important role in mitochondrial fission (Friedman et al, 2011; Murley et al, 2013; Hatch et al, 2014; Naon & Scorrano, 2014). Several mitochondrial receptors, including MFF, FIS1, and MID49/51, have been reported to recruit DRP1 during mitochondrial fission under normal physiological conditions, but less is known about the mechanism of mitochondrial fission under hypoxic conditions (Kim et al, 2011; Loson et al, 2013). Until now, there was no evidence about how FUNDC1 mediates mitochondrial fission and further impacts on mitophagy under hypoxic conditions. Our data show that FUNDC1 specifically mediates DRP1‐dependent mitochondrial fission in hypoxic cells since knockdown of FUNDC1, but not MFF, MID49/51, or FIS1, prevents the translocation of DRP1 to mitochondria and causes mitochondrial elongation. This suggests that MFF, MID49/51, and FIS1 may play a lesser role in mitochondrial fission under hypoxic conditions and are mainly involved in normal mitochondrial dynamics or in other stressful conditions. As we know, PINK–PARKIN pathway is dominantly involved in FCCP‐induced mitophagy (Matsuda et al, 2010; Narendra et al, 2010; Iguchi et al, 2013; Kane et al, 2014; Koyano et al, 2014). However, they are not involved in hypoxia‐induced mitophagy since knockdown of Parkin does not prevent mitochondrial degradation and LC3‐I to II transition in response to hypoxia (Fig EV5). This indicates that FUNDC1‐mediated mitophagy is different from PINK–PARKIN mitophagy pathway.
Figure EV5. Mitophagy induced by hypoxia is independent of Parkin.
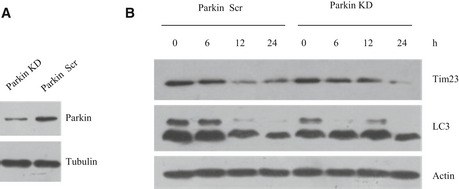
-
A, BHeLa cells were transfected using scramble siRNA or Parkin siRNA. About 24 h post‐transfection, cells were exposed to hypoxia for 0, 6, 12, or 24 h, respectively, and then samples were subjected to Western blot analysis with the indicated antibodies.
We also demonstrated a new signaling pathway mediating hypoxia‐induced mitochondrial fission. In this pathway, the mitophagy receptor FUNDC1 accumulates at ER–mitochondria contact sites by associating with the ER‐resident membrane protein calnexin. As mitophagy proceeds, FUNDC1/calnexin association attenuates and the cytosolic loop in FUNDC1 is released so that DRP1 can be recruited to the MAM where mitochondrial fission occurs (Fig 7). Of course, we cannot exclude the possibilities that there are other proteins in MAM contact may also facilitate the recruitment of Drp1 at ER contact sites to drive the fragmentation in other conditions.
Figure 7. Model of how FUNDC1, DRP1, and calnexin cooperate to mediate mitochondrial fission and mitophagy in response to hypoxia.
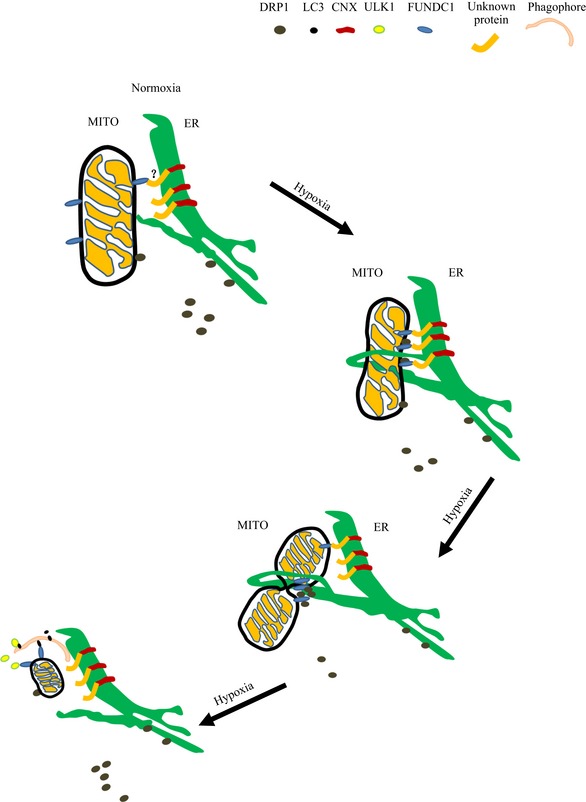
Under normoxic conditions, the ER and mitochondria remain in close contact. Small amounts of DRP1 are recruited to mitochondria to maintain the balance of mitochondrial fission and fusion. A small proportion of FUNDC1 associates with the ER membrane protein calnexin at the sites where ER and mitochondria are tethered. At the early stage of hypoxia, FUNDC1 increasingly accumulates at the MAM by interacting with calnexin mediated by an unknown intermediate protein. As mitophagy proceeds, FUNDC1 and calnexin dissociate, and DRP1 is recruited by FUNDC1 to the MAM to initiate mitochondrial constriction. Mitochondria are further constricted by self‐assembled DRP1 and fragment at the later stage of hypoxia. Meanwhile, the most upstream ATG complex (ULK1 complex) phosphorylates FUNDC1 to promote engulfment and clearance of damaged mitochondria by the autophagy pathway (Wu et al, 2014a).
As we know, the average diameter of an autophagosome is ~0.2–1 μm, but the average length of a mitochondrion in normal conditions is much greater, often ranging from several to tens of microns. Therefore, before a dysfunctional mitochondrion is engulfed by an autophagosome, it must be fragmented and then segregated from the mitochondrial network (Tatsuta & Langer, 2008; Mao & Klionsky, 2013). Thus, it is not surprising to see less mitochondria‐LC3 colocalization in FUNDC1, DRP1, or calnexin knockdown cells under hypoxic conditions because elongated mitochondria cannot be engulfed by autophagosomes in these cells. This phenomenon is in agreement with the finding that under starvation conditions, the mitochondria elongate to escape from autophagosome clearance (Gomes et al, 2011; Rambold et al, 2011). Our previous study has proved that in mammals, FUNDC1 is a mitophagy receptor, which mediates mitochondrial elimination by binding to LC3 under hypoxic conditions. Here, we further observed that FUNDC1‐mediated mitophagy in response to hypoxia is associated with the contacts between the ER and mitochondria. This agrees with recent studies showing that the MAM marks mitophagy initiation sites (Bockler & Westermann, 2014). It is likely that FUNDC1 and calnexin disrupt the constitution of the MAM and impact on mitophagy initiation. As our previous study showed, the most upstream Atg protein, ULK1, translocates to damaged mitochondria and directs mitophagosome formation per se by phosphorylating FUNDC1 (Wu et al, 2014a). Thus, it is possible that when cells are exposed to hypoxia, FUNDC1 accumulates at the MAM by interacting with calnexin and then binds to DRP1 to mediate mitochondrial fission. The ruptured mitochondrion subsequently recruits the ULK1 complex to initiate mitophagy (Wu et al, 2014a; Fig 7).
In conclusion, our findings reveal that under hypoxic conditions, FUNDC1 is the molecular link which integrates mitochondrial fission and mitophagy at the interface of the MAM by working in concert with DRP1 and calnexin in mammalian cells.
Materials and Methods
Reagents and antibodies
The following primary antibodies were used in this study: anti‐FIS1 polyclonal antibody (Proteintech, 10956‐1‐AP), anti‐DRP1 (D6C7) antibody (CST, #8570), anti‐DLP1 (BD Biosciences, 611112), anti‐EEA1 polyclonal antibody (Proteintech, 22266‐1AP), anti‐Mitofusin 2 monoclonal antibody (Epitomics, 3272‐1), anti‐MFF polyclonal antibody (Proteintech, 17090‐1‐AP), anti‐FUNDC1 polyclonal antibody (AVIVA, ARP53280_P050), anti‐FUNDC1 polyclonal antibody (for immunogold labeling) that was raised in rabbit and purified, anti‐FACL4 polyclonal antibody (Proteintech, 22401‐1‐AP), anti‐Lamp1 antibody (Abcam, ab24170), anti‐Bap31 antibody (Proteintech, 11200‐1‐AP), AKAP121 (Proteintech, 15618‐1‐AP), anti‐beta‐actin antibody (Santa Cruz, sc‐47778), anti‐beta‐tubulin monoclonal antibody (Earthox, E021040), anti‐LC3B polyclonal antibody (Sigma, L7543), anti‐LC3 polyclonal antibody (MBL, PM036), anti‐TIM23 (BD Biosciences, 611222), anti‐TOM20 (FL‐145) (Santa Cruz, sc‐11415), anti‐TOM20 (Santa Cruz (F‐10), sc‐17764), anti‐VDAC1 monoclonal antibody (Abcam, ab14734), anti‐FLAG M2 monoclonal antibody (Sigma, F1804), calreticulin (Proteintech, 10292‐1‐AP), MID49 (Proteintech, 16413‐1‐AP), MID51 (Proteintech, 20164‐1‐AP), anti‐FLAG (Sigma, F7425), anti‐c‐Myc (9E10) (Santa Cruz, sc‐40), anti‐c‐Myc monoclonal antibody (Sigma, C3956). Secondary antibodies used for Western blotting were the following: HRP affinipure goat anti‐mouse IgG (Earthox, E030110), HRP affinipure goat anti‐rabbit IgG (Earthox, E030120) and HRP‐conjugated goat anti‐rabbit IgG Fc (SouthernBiotech, 4041‐05). The following fluorescent secondary antibodies were used: Alexa fluor 555‐labeled donkey anti‐mouse IgG antibody (Life Technologies, A31570), Alexa fluor 555‐labeled donkey anti‐rabbit IgG antibody (Life Technologies, A31572), Alexa fluor 488‐labeled donkey anti‐mouse IgG antibody (Life Technologies, A21202), Alexa fluor 488‐labeled donkey anti‐rabbit IgG antibody (Life Technologies, A21206), Alexa fluor 647‐labeled donkey anti‐mouse IgG antibody (Life Technologies, A‐31571), Alexa fluor 647‐labeled donkey anti‐rabbit IgG antibody (Life Technologies, A‐31573). Bafilomycin A1 (B1793) and etoposide (E1383) were purchased from Sigma and used at a final concentration of 50 nM. Protein A/G plus agarose immunoprecipitation reagent (Santa Cruz, sc‐2003) and Lipofectamine 2000 (Invitrogen, 11668027) were also used according to the manufacturer's protocol.
Plasmids
DNM1L (MYC‐DDK‐tagged) human dynamin 1‐like vector (RC202046) was purchased from ORIGENE. Human calnexin (FL), CNX‐N (1–503, ▵cyto), and CNX‐C (504–586) were cloned into pCMV‐MYC plasmid using the following primers:
FL‐F: CGCGTCGACCATGGAAGGGAAGTGGTTG;
FL‐R: CGGGGTACCTCAGTTTCTTGGTGATCTGTTCAA;
CNX‐N‐F: CGCGTCGACCATGGAAGGGAAGTGGTTG;
CNX‐N‐R: CGGGGTACCTCAACAGCAGAAGAGGATAACCAG;
CNX‐C‐F: CGCGTCGACTTCTGGAAAGAAACAGACCAG;
CNX‐C‐R: CGGGGTACCTCAGTTTCTTGGTGATCTGTTCAA.
The FUNDC1‐MYC and FLAG‐FUNDC1 constructs and FUNDC1‐MYC truncations (1–139; 1–96; 96–155; ∆50; 50–139; 96–155; 50–71/96–155; 71–139) were described previously (Liu et al, 2012; Wu et al, 2014b). Deleted mutants and double‐point mutants were reconstructed from FUNDC1‐MYC, and the primers used were as follows:
Δ129–138 F: attgtgatatccagtggatttg
Δ129–138 R: actggatatcacaataattaaattgttgatttcaggtgctgct
Δ119–128 F: gaagaagcaacagaatttatca
Δ119–128 R: ttctgttgcttcttcgttcgctcgtttcttaatctgtctttttg
Δ109–118 F: aaagcagcacctgaaatcaaca
Δ109–118 R: ttcaggtgctgcttttttatttacatctttttcaactctcttc
Δ96–108 F: gcaaaaagacagattaagaaac
Δ96–108 R: aatctgtctttttgcctgcacatagccactatgactagcaatc
Mutation 137–138F: acagaatttatcaaggcggccattgtgatatccagt
Mutation 137–138 R: actggatatcacaatggccgccttgataaattctgt
Mutation 135–136F: gaagcaacagaatttgccgcgcagaacattgtgata
Mutation 135–136R: tatcacaatgttctgcgcggcaaattctgttgcttc
Mutation 133–134F: attgaagaagcaacagcagctatcaagcagaacatt
Mutation 133–134R: aatgttctgcttgatagctgctgttgcttcttcaat
Mutation 131–132F: aatttaattgaagaagcagcagaatttatcaagcag
Mutation 131–132R: ctgcttgataaattctgctgcttcttcaattaaatt
Mutation 129–130F: atcaacaatttaattgcagcagcaacagaatttatc
Mutation 129–130R: gataaattctgttgctgctgcaattaaattgttgat
Cell culture and transfection
MEFs and HeLa cells were cultured in high‐glucose DMEM (Hyclone) supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin/streptomycin at 37°C under 5% CO2. Hypoxic conditions were achieved with a hypoxic chamber (Billups‐Rothenberg) flushed with a pre‐analyzed gas mixture of 1% O2, 5% CO2, and 94% N2. The following target sequences for RNA interference were purchased from GenePharma: FUNDC1‐478 (sense 5′‐GCACCUGAAAUCAACAAUATT‐3′; antisense 5′‐UAUUGUUGAUUUCAGGUGCTT‐3′); CNX‐591 (sense 5′‐GUGGUGCCUAUGUGAAACUTT‐3′, antisense 5′‐AGUUUCACAUAGGCACCACTT‐3′); MFF‐519 (sense 5′‐GCUGGUCAGAAAUGAUUCUTT‐3′, antisense 5′‐AGAAUCAUUUCUGACCAGCTT‐3′); DRP1 (sense 5′‐AUUUGAGGCAGCUGGAUGAUGUCGG‐3′, antisense 5′‐CCGACAUCAUCCAGCUGCCUCAAAU‐3′); FIS1‐312 (sense 5′‐GGCUCAAGGAAUACGAGAATT‐3′, antisense 5′‐UUCUCGUAUUCCUUGAGCCTT‐3′). The scramble RNA interference sequence was as follows: sense 5′‐UUCUCCGAACGUGUCACGUTT‐3′; antisense 5′‐ACGUGACACGUUCGGAGAATT‐3′. RNA oligonucleotides were transfected into cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. The efficacy of different siRNA knocking‐downs is shown in Appendix Fig S13.
Protein expression and purification
GST‐FUNDC1 was transformed in Rosetta 2 (DE3) cells (Novagen). The target protein expression was induced by addition of IPTG (isopropyl‐beta‐D‐thiogalactopyranoside) into LB culture at 18°C when the OD600 approached 1.0. After 12 h of additional growth, cells were harvested and resuspended in buffer A (20 mM Tris–HCl pH 7.6, 500 mM NaCl) supplemented with 10 mM PMSF (Invitrogen). After cell lysis by sonication and centrifugation, soluble GST‐FUNDC1 fusion protein was purified with GST affinity beads (GE Healthcare), followed by size‐exclusion chromatography (SEC, Superdex‐200, GE Healthcare).
GST‐mediated pull‐down assay
About 10–20 μg GST‐FUNDC1 or GST was immobilized on glutathione sepharose resin (GE Healthcare). About 20–50 μg DPR1 was incubated with the GST‐FUNDC1‐ or GST‐bound resin overnight at 4°C. The resin was then extensively rinsed with buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.6, 0.5% Triton X‐100) to wash unbound and non‐specific bound proteins. The resin was resuspended with 50 μl of the above buffer containing 10 mM GSH in 15 min at room temperature and was centrifuged at 10,000 g for 5 min. Samples were subjected to SDS–PAGE and then visualized by Coomassie Blue staining and Western blot, respectively.
Immunofluorescence microscopy
Cells were grown to 60% confluence on a coverslip before treatment and then fixed with freshly prepared 4% paraformaldehyde at 37°C for 15 min, followed by permeabilization by treatment with 0.1% Triton X‐100 (Shanghai Sangon Biotech). After blocking with 1% BSA, cells were incubated with the indicated primary antibodies for 1 h at room temperature and, after washing with PBS, stained with corresponding secondary antibodies for further 50 min at room temperature. Cell images were captured with a TCS SPF5 II Leica confocal microscope.
Electron microscopy
Cells were grown and treated as described above. For immunoelectron microscopy, HeLa cells were fixed using 2% paraformaldehyde and 0.2% glutaraldehyde in Na cacodylate buffer (pH 7.4) at 37°C for 2 h and then dehydrated in a graded ethanol series and embedded in acrylic resin (LR White). After 70‐nm ultrathin sections were mounted on nickel grids, grids were incubated in 1% BSA in PBS containing anti‐DRP1, anti‐TOM20, anti‐FACL4, or anti‐FUNDC1 antibodies overnight at 4°C. After washing with 0.5% BSA in PBS, grids were probed using gold‐conjugated particles in 1% BSA in PBS for 2 h at room temperature. The samples were then stained and visualized using a 120‐kV Jeol electron microscope (JEM‐1400) at 80 kV. Images were captured using a Gatan‐832 digital camera. The number of Immunogold particles on the whole mitochondria or in the MAM was counted, and ratios were calculated as listed in figures.
SDS–PAGE and Western blotting
Cells were lysed in lysis buffer (50 mM Tris–Cl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP‐40, 10% glycerin) with 1 mM PMSF and protease inhibitors (Roche Applied Science). Thirty micrograms of equivalent protein was separated by SDS–PAGE and then transferred to PVDF membranes (Millipore). Target proteins were detected by incubating with the indicated primary antibodies, followed by the corresponding HRP‐conjugated secondary antibodies. Immunoreactive bands were detected with Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Immunoprecipitation
Cells were lysed with lysis buffer (1 ml) containing 1 mM PMSF and protease inhibitor cocktail (Roche Applied Science) for 30 min on ice. After 10,000 g centrifugation for 10 min, the lysates were immunoprecipitated with specific antibody for 2 h at room temperature and protein A/G plus agarose immunoprecipitation reagent (Santa Cruz) for 8 h at 4°C. Thereafter, the precipitants were washed five times with lysis buffer and boiled in sample buffer for 10 min at 100°C and then analyzed by SDS–PAGE.
Subcellular fractionation
The detailed procedure has been described previously (Jirasiritham & Tantivitayatan, 2004; Wieckowski et al, 2009). Briefly, 30 confluent plates (15‐cm diameter) of HeLa cells were collected and resuspended in ice‐cold IB cells‐1 buffer (225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA, and 30 mM Tris–HCl pH 7.4). After gentle homogenization with a Dounce homogenizer (about 200 times), cell extracts were centrifuged at 600 g for 5 min at 4°C. The supernatants were again centrifuged at 600 g for 5 min at 4°C (PNS). The supernatant was removed to a new 50‐ml polypropylene tube and centrifuged at 7,000 g for 10 min at 4°C to obtain the crude mitochondrial pellet. Further centrifugation of the obtained supernatant (100,000 g for 1 h) resulted in the isolation of ER (pellet) and cytosolic (supernatant) fractions. The crude mitochondrial pellet was resuspended gently in 2 ml of ice‐cold MRB buffer (250 mM mannitol, 5 mM HEPES pH 7.4, and 0.5 mM EGTA) and added on top of 8 ml of Percoll medium in an ultracentrifuge tube. MRB solution was then layered gently on top of the mitochondrial suspension to fill the centrifuge tube. Finally, centrifugation was carried out at 95,000 g for 60 min at 4°C to isolate the MAM and pure mitochondria.
Statistical analysis
For statistics, cells were randomly selected to calculate the number of MAM, gold particles, and colocalizations. Assays for characterizing cell phenotypes were analyzed by Student's t‐test, and correlations between groups were calculated using Pearson's test. P‐values < 0.01 were deemed statistically significant. All statistical data were calculated with GraphPad Prism software.
Author contributions
DF conceived and supervised the work. DF, LL and WW designed the experiments. DF, WW, KW, LJ, LL, JZ, XW, CL, WL, HZ, HC, SL, YY, YL, JW and RZ performed the experiments with the help of other co‐authors; DF, WW, LL, LZ, JZ, SS and BZ analyzed the data; WW, LL, JZ, BZ, NT and DF wrote the paper. All authors discussed the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Dr. Isabel Hanson for editing the manuscript. We also thank Prof. Hong Zhang (Institute of Biophysics, CAS), Prof. Li Yu (Tsinghua University), and Prof. Navdeep Chandel (Northwest University, USA) for critically reading the manuscript. This work was supported by grants from Guangdong Medical College (XB1342) and its affiliated hospital (701B01206), by the NSFC (No. 31401182, No. 81270196, No. 81171244, No. 81571203, No. 31370824, and No. 81470405), by the National Basic Research Program of China (2013CB910100), by the Natural Science Foundation of Guangdong Province, China (2014A030313533), by the Medical Scientific Research Foundation of Guangdong Province, China (No. A2014484), by the Foundation for Distinguished Young Talents in Higher Education of Guangdong, China (2013LYM_0035), by Yangfan Plan of Talents Recruitment Grant, Guangdong, China (Yue Ren Cai Ban [2014] 1, Yue Cai Jiao [2015] 216), by the Key Lab Funding from Guangdong Province (2012A061400019), and by the Shanghai Natural Science Foundation of China (No. 15ZR1437200).
The EMBO Journal (2016) 35: 1368–1384
See also: AM van der Bliek (July 2016)
References
- Bockler S, Westermann B (2014) Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 28: 450–458 [DOI] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA 105: 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC (2006) Mitochondria: dynamic organelles in disease, aging, and development. Cell 125: 1241–1252 [DOI] [PubMed] [Google Scholar]
- Chandel NS (2014) Mitochondria as signaling organelles. BMC Biol 12: 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Lipton SA (2010) Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci 67: 3435–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci USA 101: 15927–15932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio L, Derks C, Dejaegere T, Pellegrini L, D'Hooge R, Scorrano L, De Strooper B (2006) Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1‐dependent cristae remodeling. Cell 126: 163–175 [DOI] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmer KS, Scorrano L (2006) (De)constructing mitochondria: what for? Physiology 21: 233–241 [DOI] [PubMed] [Google Scholar]
- Duvezin‐Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS (2006) Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem 281: 37972–37979 [DOI] [PubMed] [Google Scholar]
- Escobar‐Henriques M, Anton F (2013) Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim Biophys Acta 1833: 162–175 [DOI] [PubMed] [Google Scholar]
- Feng D, Liu L, Zhu Y, Chen Q (2013) Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 319: 1697–1705 [DOI] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189 [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandre‐Babbe S, van der Bliek AM (2008) The novel tail‐anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309–1312 [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G (2014) Cell biology. Metabolic control of cell death. Science 345: 1250256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Hatch AL, Gurel PS, Higgs HN (2014) Novel roles for actin in mitochondrial fission. J Cell Sci 127(Pt 21): 4549–4560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N (2013) Parkin‐catalyzed ubiquitin‐ester transfer is triggered by PINK1‐dependent phosphorylation. J Biol Chem 288: 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirasiritham S, Tantivitayatan K (2004) A comparison of the efficacy of cisatracurium and atracurium in kidney transplantation operation. J Med Assoc Thai 87: 73–79 [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA (2011) Fine‐tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell 44: 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klecker T, Bockler S, Westermann B (2014) Making connections: interorganelle contacts orchestrate mitochondrial behavior. Trends Cell Biol 24: 537–545 [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy‐Wetzel E (2008) Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B (2013) The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol 25: 443–448 [DOI] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305: 858–862 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Li W, Zhang X, Zhuang H, Chen HG, Chen Y, Tian W, Wu W, Li Y, Wang S, Zhang L, Chen Y, Li L, Zhao B, Sui S, Hu Z, Feng D (2014) MicroRNA‐137 is a novel hypoxia‐responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem 289: 10691–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K, Klionsky DJ (2013) Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae . Autophagy 9: 1900–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S, Pinton P (2014) The endoplasmic reticulum‐mitochondria connection: one touch, multiple functions. Biochim Biophys Acta 1837: 461–469 [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T (2010) Autophagy requires endoplasmic reticulum targeting of the PI3‐kinase complex via Atg14L. J Cell Biol 190: 511–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears JA, Lackner LL, Fang S, Ingerman E, Nunnari J, Hinshaw JE (2011) Conformational changes in Dnm1 support a contractile mechanism for mitochondrial fission. Nat Struct Mol Biol 18: 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murley A, Lackner LL, Osman C, West M, Voeltz GK, Walter P, Nunnari J (2013) ER‐associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2: e00422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naon D, Scorrano L (2014) At the right distance: ER‐mitochondria juxtaposition in cell life and death. Biochim Biophys Acta 1843: 2184–2194 [DOI] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191: 1141–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep 12: 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288: 27584–27593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambold AS, Kostelecky B, Elia N, Lippincott‐Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux A, Uyhazi K, Frost A, De Camilli P (2006) GTP‐dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature 441: 528–531 [DOI] [PubMed] [Google Scholar]
- Rowland AA, Voeltz GK (2012) Endoplasmic reticulum‐mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol 13: 607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874 [DOI] [PubMed] [Google Scholar]
- Schneider JL, Cuervo AM (2014) Autophagy and human disease: emerging themes. Curr Opin Genet Dev 26C: 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin‐related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovski D, Koutsopoulos OS, Okamoto K, Ryan MT (2004) Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J Cell Sci 117: 1201–1210 [DOI] [PubMed] [Google Scholar]
- Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22: 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin‐related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282: 11521–11529 [DOI] [PubMed] [Google Scholar]
- Tatsuta T, Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27: 306–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922–2933 [PubMed] [Google Scholar]
- Wasilewski M, Semenzato M, Rafelski SM, Robbins J, Bakardjiev AI, Scorrano L (2012) Optic atrophy 1‐dependent mitochondrial remodeling controls steroidogenesis in trophoblasts. Curr Biol 22: 1228–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P (2009) Isolation of mitochondria‐associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: 1582–1590 [DOI] [PubMed] [Google Scholar]
- Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Li L, Zhang L, Sui S, Zhao B, Feng D (2014a) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D (2014b) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Galloway CA, Jhun BS, Yu T (2011) Mitochondrial dynamics in diabetes. Antioxid Redox Signal 14: 439–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Liu T, Jin S, Wang X, Qu M, Uhlen P, Tomilin N, Shupliakov O, Lendahl U, Nister M (2011) Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J 30: 2762–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6