

Abstract
Background
Declines in skeletal muscle structure and function are found in various clinical populations, but the intramuscular proteolytic pathways that govern declines in these individuals remain relatively poorly understood. The nematode Caenorhabditis elegans has been developed into a model for identifying and understanding these pathways. Recently, it was reported that UNC‐105/degenerin channel activation produced muscle protein degradation via an unknown mechanism.
Methods
Generation of transgenic and double mutant C. elegans, RNAi, and drug treatments were utilized to assess molecular events governing protein degradation. Western blots were used to measure protein content. Cationic dyes and adenosine triphosphate (ATP) production assays were utilized to measure mitochondrial function.
Results
unc‐105 gain‐of‐function mutants display aberrant muscle protein degradation and a movement defect; both are reduced in intragenic revertants and in let‐2 mutants that gate the hyperactive UNC‐105 channel. Degradation is not suppressed by interventions suppressing proteasome‐mediated, autophagy‐mediated, or calpain‐mediated degradation nor by suppressors of degenerin‐induced neurodegeneration. Protein degradation, but not the movement defect, is decreased by treatment with caspase inhibitors or RNAi against ced‐3 or ced‐4. Adult unc‐105 muscles display a time‐dependent fragmentation of the mitochondrial reticulum that is associated with impaired mitochondrial membrane potential and that correlates with decreased rates of maximal ATP production. Reduced levels of CED‐4, which is sufficient to activate CED‐3 in vitro, are observed in unc‐105 mitochondrial isolations.
Conclusions
Constitutive cationic influx into muscle appears to cause caspase degradation of cytosolic proteins as the result of mitochondrial dysfunction, which may be relevant to ageing and sarcopenia.
Keywords: Caspase, C. elegans, Degenerin, Mitochondria, Muscle
Background
Maintained muscle protein homeostasis (proteostasis) occurs through intricate regulation of balanced rates of protein synthesis and degradation. Proteostasis is required to maintain contractile ability and muscle as a metabolic reservoir.1 Declines in skeletal muscle mass, structure and function are associated with ageing (sarcopenia), cancer (cachexia), COPD, heart failure, and diabetes,2 but the proteolytic pathways that govern declines in each of these conditions remain relatively poorly understood.
Caenorhabditis elegans is an established laboratory animal in which genetics and genomics can be used to understand physiology and is the animal for which we know the most about genes controlling muscle protein degradation.3 In C. elegans muscle, increased traffic to the proteasomes is normally prevented by neuronal release of acetylcholine4 to presumably affect intramuscular calcium signalling5 and appears to occur in response to starvation,6 denervation,4 and neurodegeneration.7 Increased traffic to lysosomes via autophagy is controlled by a balance of constitutive fibroblast growth factor and insulin/insulin like growth factor8; roughly two dozen other protein kinases appear to oppose autophagic degradation.9 These signals converge, modulating activation of autophagy via mitogen‐activated protein kinase (MAPK),10 and appear relevant to a model of neurodegeneration11 and to ageing.12 Lastly, calpains are activated in response to muscle attachment complex disruption, which appears required to maintain muscle in response to use.13, 14
Recently, unc‐105 gain‐of‐function mutants were reported to display increased muscle protein degradation via an unknown mechanism.5 unc‐105 encodes a putative mechano‐sensitive ion channel of the ENaC/degenerin family.15 ENaC/Degenerin channels are implicated in the pathophysiology of some human neurodegenerative diseases,16 and hyperactivity of other ENaC/Degenerin channels causes necrotic‐like neuronal cell death in C. elegans. 17, 18, 19, 20 Dominant gain‐of‐function mutations in unc‐105 have previously been shown to cause worms to be small, hypercontracted, and paralyzed.21 These phenotypes are suppressed in unc‐105; let‐2 double mutants.15 LET‐2 is an alpha‐2 type IV basement membrane collagen22 that has been proposed to physically gate the mutationally activated UNC‐105 channel in the muscle plasma membrane and thereby relieve the anomalous ion influx and rescue the aforementioned phenotypes.15, 23
Here, we report that unc‐105 mutants but not unc‐105; let‐2 double mutants display caspase‐dependent muscle protein degradation and loss of normal muscle mitochondrial architecture, mitochondrial membrane potential, and maximal mitochondrial ATP production capacity. These changes are associated with decreased mitochondrial‐associated CED‐4. CED‐4, or cell death abnormal protein 4, is the C. elegans orthologue of mammalian apoptotic protease activating factor 1 (APAF1) and in vitro CED‐4 is sufficient to activate the caspase CED‐3. The discovery of CED‐3‐mediated and CED‐4‐mediated programmed cell death in C. elegans was the subject of the 2002 Nobel Prize in Physiology or Medicine as programmed cell death is now known to be crucial for regulating cell number. Indeed, lack of proper activation of cell death is associated with cancer, autoimmune disease, and neurodegenerative disease. Collectively, our data suggest that excessive cationic influx into muscle leads to pathological changes in mitochondrial architecture and function, and subsequent caspase activation. These data also complete our preliminary understanding of the regulation of activation and increased trafficking to the four major proteolytic systems in C. elegans muscle, and enable direct testing of the relevance of all four systems to various (patho)physiologic conditions.
Materials and methods
Strains and culture
Strains of C. elegans were handled, maintained, and age‐synchronized as described.6 RNAi was as described13 using sequence‐verified RNAi (Source BioScience LifeSciences Ltd.). Strains for protein degradation, maximal rates of ATP production, and mitochondrial membrane potential assays are as follows: PD55: ccls55 V, CC10: unc‐105(n490) II; ccls55 V, and CC7: unc‐105(n490) II; ccls55 V, lon‐2(e678) let‐2(n821) X. Strains for Mitotracker® and Caspase 3/7 substrate assays are as follows: CB5600: ccls4251 (myo‐3::Ngfp‐lacZ, myo‐3::Mtgfp) I; him‐8(e1489) IV, CC62: ccls4251 (myo‐3::Ngfp‐lacZ, myo‐3::Mtgfp) I; unc‐105(n490) II; him‐8(e1489) IV and CC63: ccls4251 (myo‐3::Ngfp‐lacZ, myo‐3::Mtgfp) I; unc‐105(n490) II; him‐8(e1489) IV; lon‐2(e678) let‐2(n821) X.
Mitochondrial function
MitoTracker® chloromethyl‐X‐rosamine (CMXRos) in vivo accumulation assay. Synchronized worms were grown to early adulthood. Twenty animals per trial were processed as described in 4.7 μM CMXRos.24
JC‐10 in vivo accumulation assay. Synchronized worms were grown from the L1 stage in the presence of 83 μM JC‐10 (Enzo Life Sciences, NY, USA).
JC‐1 Fluorescence‐activated cell sorting (FACS). Animals were cultured and mitochondria isolated as described.14 Mitochondria were incubated in the dark for 15 min at 37°C in 20 μM glutamate, 2 mM malate, 6 μM JC‐1 (Invitrogen, UK). Mitochondria were centrifuged (10 000 g, 5 min), resuspended, and sorted in a Beckman Coulter FC500 flow cytometer (Ex 490 nm and Em 605 nm). Loss of JC‐1 in response to loss of membrane potential was confirmed by addition of 1 μM CCCP.
Maximal rates of ATP production (MRAP). Maximal rates of ATP production (MRAP) were assessed as described.14 Mixed‐stage populations were used to obtain sufficient quantity of mitochondria as unc‐105 mutants are very sick, taking at least 2–3 weeks to produce a population capable of exhausting food supplies on a plate.
Western blotting
β‐Galactosidase westerns were performed as described13 with quantification in Image J. CED‐4,25 cytochrome C,26 and ATP‐synthase27 antibodies were previously validated. Anti‐CED‐4 (Santa Cruz Biotechnology, USA), was used at a 1:100 dilution in 5% milk TBS‐T with secondary (Santa Cruz Biotechnology, USA) at a 1:10 000 dilution. Anti‐cytochrome C and anti‐ATP‐synthase (anti‐ATP5A) (Abcam®, UK) were used at a 1:1000 and 1:2500 dilution in 3% milk TBS, respectively, with secondary (Sigma‐Aldrich®, USA) at a 1:10 000 dilution.
Measurements of caspase activation
Synchronized adult worms were grown in the presence of 200 μM Z‐DEVD‐ProRed™ (AAT Bioquest, UK) at 20°C and analysed at t = 0, 12, 24, and 48 h with red fluorescence noted at all three later time points. For quantification of caspase activity, untreated animals were washed from single plates, homogenized in buffer containing 1% Triton, incubated with the caspase reagent buffer (Caspase‐Glo® 3/7 Assay Kit, Promega, UK) for 45 min at 37°C, and luminescence measured. Activity is expressed versus a standard curve of titrated recombinant human caspase 3 (Promokine, Germany).
Assessment of β‐galactosidase activity
Animals were stained for β‐galactosidase activity as described.6 Control animals (PD55) or unc‐105 (CC10 on standard Escherichia coli strain OP50 in the RNAi conditions) were utilized.
Whole genome sequencing
Genomic DNA was prepared as described28 and sequenced using Illumina Solexa GAII (BC Cancer Agency Genomic Sciences Centre). Sequences were aligned to C. elegans genome version WS200 using Burrows‐Wheeler Aligner (BWA)29 under default settings. Resulting BAM files were processed to identify single nucleotide variants (SNVs) and small insertions or deletions (indels) using VarScan.30 Variants were matched to WS200 annotation using CooVar31 and categorized as ‘nonsense’, ‘missense’, ‘synonymous’, ‘non‐coding’, ‘frame preserving indel’, or ‘frame shifting indel’. Non‐coding mutations were defined based upon intron and intergenic regions. Only homozygous mutations (a mutation with more than 90% read support for the change) were considered.
Movement assays
Movement was measured as described.10
Microscopy and statistics
Microscopy. All images were captured as described.13 Image analysis and figure preparation was conducted in GIMP and Photoshop.
Statistical analysis. All data are presented as means ± SEM from at least three replicates unless otherwise stated. Statistical differences were assessed using either one‐way ANOVA with Newman–Keuls corrections or two‐way ANOVA with Bonferroni corrections. Significance was determined as P < 0.05, and all statistics were completed using GraphPad Prism (USA).
Results
Activation of UNC‐105 causes muscle protein degradation
Recently, it was reported that UNC‐105 activation appears to cause muscle protein degradation that is not suppressed by treatment with proteasome inhibitors.5 The time dependent loss of a transgenic β‐galactosidase reporter protein's activity suggests that muscle protein degradation is occurring (Figure 1A). This transgenic reporter protein has been shown to be fully stable in muscle of well fed adult worms for at least 72–96 h post‐adulthood but is degraded upon activation of various proteolytic systems.6, 8, 13 To confirm that loss of enzymatic activity was due to degradation, we performed western blots (Figure 1B and C). RNAi against unc‐105 in wild‐type animals had no effect on proteostasis,5 while the dominant gain‐of‐function allele n490 caused protein degradation (Figure 1); these observations suggest that it is the activation of the UNC‐105 ion channel that induces muscle protein degradation. In support of this suggestion, we found that RNAi against unc‐105 attenuates the loss of β‐galactosidase activity in unc‐105(n490) mutants (Figure 1D).
Figure 1.
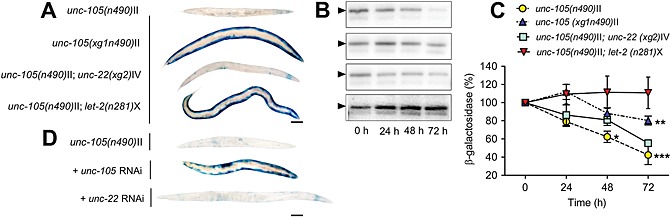
Activation of UNC‐105 causes protein degradation in muscle. Synchronized young adults were used (t = 0 h), and all experiments repeated at least three times. (A) Representative stains for β‐galactosidase at t = 72 h post‐adulthood; scale bar represents 100 µm. (B) Representative western blots of β‐galactosidase in 30‐worm lysates. Arrows show β‐galactosidase fusion protein at 146 kD. Wild‐type controls are not included as unc‐105; let‐2 results are not significantly different from published wild‐type values.4, 8, 10, 13 (C) Kinetics of β‐galactosidase degradation, quantified from three independent western blots. * denotes P < 0.05; ** denotes P < 0.01; *** denotes P < 0.001 between unc‐105 and unc‐105; let‐2, two‐way repeated measures ANOVA with Bonferroni correction. (D) Representative stains for β‐galactosidase at t = 72 h post‐adulthood; scale bar represents 100 µm.
Previous work has shown that excessive Na+ influx in mutants containing another dominant gain‐of‐function mutation in unc‐105, n506, causes sustained muscle depolarisation that is attenuated in unc‐105(n506) mutants containing a second mutation in let‐2. 23 LET‐2 is an alpha‐2 type IV basement membrane collagen22 and treatment of unc‐105; let‐2 double mutants with collagenase restores high levels of Na+ influx. These past results suggest that LET‐2 has a physical interaction with UNC‐105, which gates the channel.23 Therefore, to further test if UNC‐105 activation leads to increased muscle protein degradation, we tested if protein degradation was suppressed in unc‐105(n490); let‐2(n821) double mutants. As shown in Figure 1, protein degradation was suppressed in unc‐105; let‐2 double mutants, further suggesting that hyperactivity of the UNC‐105 channel causes muscle protein degradation.
While conducting this work, two spontaneous mutants that resulted in increased population growth rates in the unc‐105(n490) strain were isolated; designated as xg1 and xg2 ( Figure S1). xg1 mutants display reduced muscle protein degradation, while xg2 mutants do not (Figure 1). As unc‐105 mutants pick up suppressing mutations at a high rate,32 we presumed that xg1 and xg2 might be mutations in known suppressing genes. Previously identified suppressors of unc‐105 include mutations in unc‐15, unc‐22, unc‐54, unc‐96, crt‐1, let‐2, and unc‐105 itself15, 19, 32, 33, 34; these genes encode paramyosin, twitchin, myosin heavy chain A, a Lin11, Isl‐1 & Mec‐3 (LIM) domain containing protein that localizes to M‐lines, calreticulin, a collagen, and a degenerin channel, respectively. Because xg1 looked wild‐type and the most frequently isolated mutation that suppresses unc‐105 with a wild‐type appearance was unc‐105,32, 33 we presumed that xg1 might be an intragenic revertant. Similarly, as xg2 visibly twitched and unc‐22 is the only isolated suppressor that twitches,32 we presumed that xg2 might be an allele of unc‐22. Full genome sequencing ( Figure S2, Table 1, Dataset S1) confirmed a 4bp insertion unc‐105 at II: 8118943 in the strain containing xg1 and a nonsense mutation disrupted unc‐22 at IV: 11984751 in the strain containing xg2. RNAi against unc‐105 or unc‐22 produces the same effect on β‐galactosidase activity in unc‐105(n490) mutants (Figure 1D) as does allele xg1 or xg2 (Figure 1A). In the intragenic revertant, xg1, a premature stop codon is predicted, like RNAi against unc‐105, to reduce the abundance of UNC‐105. The decreased abundance of activated UNC‐105 presumably results in less Na+ influx, which suggests that hyperactivation of the channel causes muscle protein degradation.
Table 1.
Summary of single nucleotide variations and indels in strains CC24 (containing xg1) and CC50 (containing xg2)
| Strain | Synonymous | Silent | Missense | Nonsense | Frame‐preserving indel | Frame‐shifting indel |
|---|---|---|---|---|---|---|
| CC24 | 4 | 47 | 4 | 1 | 0 | 2 |
| CC50 | 1 | 20 | 2 | 1 | 0 | 0 |
Muscle protein degradation in response to UNC‐105 activation is distinct from previously described mechanisms
As unc‐105, other degenerin channels, and muscle protein degradation have all previously been studied in C. elegans, we were curious if other known suppressors of unc‐105 gain‐of‐function, mutations that reduce neuronal cell death in response to hyperactivity of other ENaC/Degenerin channels,17, 18, 19, 35 and/or suppression of previously identified proteolytic pathways could attenuate the protein degradation induced by UNC‐105 activation (Figure 2A). RNAi against unc‐15, unc‐96, and crt‐1 each also failed to suppress the protein degradation observed in unc‐105 gain‐of‐function mutants (Figure 2B). The failure to suppress degradation was not simply as the result of ineffective RNAi as RNAi against crt‐1 improved the movement defect in unc‐105 gain‐of‐function mutants (Figure 3), as expected.19 RNAi against itr‐1, asp‐3, asp‐4, asp‐6, cnx‐1, mec‐6, or unc‐68, also each failed to block protein degradation (Figure 2D); these genes encode the following: inositol trisphosphate receptor, three aspartyl proteases, calnexin, a degenerin channel subunit, and ryanodine receptor. Lastly, as shown in Figure 2C, protein degradation in response to UNC‐105 activation is not suppressed by treatment with proteasome inhibitor, calpain inhibitor or RNAi against calpains, or by RNAi against mpk‐1 that acts to control autophagic degradation.
Figure 2.
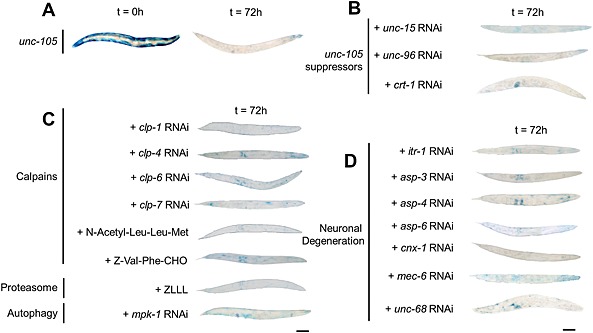
UNC‐105‐induced protein degradation is not suppressed by known suppressors of protein degradation or neurodegeneration in Caenorhabditis elegans. Synchronized young adults were used (t = 0 h), and all experiments repeated at least three times. Representative stains for β‐galactosidase at t = 72 h post‐adulthood; scale bar represents 100 µm. (A) Untreated unc‐105 mutants. (B) RNAi against known suppressors of unc‐105 growth or movement defect. (C) RNAi or drugs targeting known components of calpain,13 proteasome,4, 6 or autophagic8, 10 muscle protein degradation in C. elegans. (D) RNAi against known suppressors of MEC‐4 degenerin‐induced neuronal degeneration.
Figure 3.
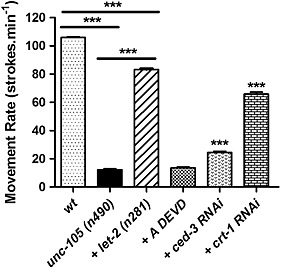
Caspase inhibition does not markedly restore the movement defect in unc‐105 mutants. Synchronized young adults were used (t = 0 h), and all experiments repeated at least three times. Movement of the head back and forth in liquid was determined for 10 adults each assessed 10 times. *** denotes P < 0.001, two‐way ANOVA with Bonferroni correction.
Caspases are activated in response to UNC‐105 activation
Having determined that degradation did not appear to be proteasome, autophagy, or calpain based, we wished to determine if degradation was due to activation of caspases. While the activation of caspases has been shown in terminally differentiated mammalian cells,36 this has not been an area of much study in C. elegans and caspase activation in ageing C. elegans muscle was only recently demonstrated.37 RNAi against either of the executioner caspases, ced‐3 or csp‐1, attenuated protein degradation in unc‐105 mutants (Figure 4A). Similarly, unc‐105 mutants treated with caspase inhibitors (Quinolyl‐Valine‐Aspartic acid (QVD), Acetyl‐Aspartic acid‐Glutamic acid‐Valine‐Aspartic acid (ADEVD), and Acetyl‐Aspartic acid‐ Methionine ‐ Glutamine ‐Aspartic acid (ADMQD)) displayed reduced degradation (Figure 4A). Lastly, as CED‐3 exists as an inactive zymogen that is activated upon binding to CED‐4 in vitro,38 we confirmed that RNAi against ced‐4 also attenuated degradation (Figure 4A). Together, these results suggest that caspases are necessary for the increased protein degradation observed in response to UNC‐105 activation. We confirmed significantly greater caspase activity in unc‐105 mutants versus wild‐type and unc‐105; let‐2 mutants (Figure 4B). To confirm caspase activation specifically in muscles, we fed worms a caspase substrate that exhibits red fluorescence upon cleavage. No fluorescence was observed outside of the gut in wild‐type or unc‐105; let‐2 mutants, whereas red fluorescence was observed in tissue(s) outside of the gut in unc‐105 mutants (Figure 4C). Next, we used worms containing Green Fluorescent Protein (GFP) expressed only in body wall muscles and found unc‐105, but not unc‐105; let‐2 mutants display yellow/orange fluorescence, as the result of red and green fluorescence in the same tissue, in body wall muscles when fed the fluorescent caspase substrate (Figure 4D); note that yellow/orange fluorescence was not noted in all muscle cells at any individual time point, possibly reflective of inter‐muscle differences in caspase substrate uptake and/or degradation. Collectively, these results suggest that caspases are activated in body wall muscles in response to UNC‐105 activation and account for the increased muscle protein degradation.
Figure 4.
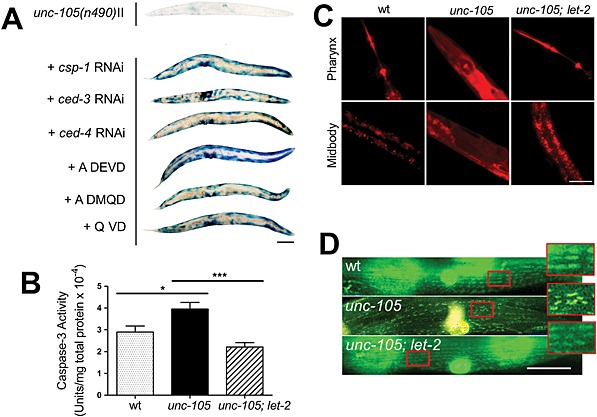
Caspases are activated in muscles of unc‐105 mutants. Synchronized young adults were used (t = 0 h), and all experiments repeated at least three times, for (B) seven independent experiments. (A) Representative stains for β‐galactosidase at t = 72 h post‐adulthood in unc‐105 mutants either untreated or treated with RNAi or drugs targeting caspases; scale bar represents 100 µm. (B) In vitro caspase 3 assay. * denotes P < 0.05 between unc‐105 and wild‐type; *** denotes P < 0.001 between unc‐105 and unc‐105; let‐2; one‐way ANOVA with Newman–Keuls correction. (C) Representative images of a fluorescent caspase 3 indicator substrate, Z‐DEVD‐ProRed™, from t = 24 h adult animals; scale bar represents 50 µm. (D) Representative images of a fluorescent caspase 3 indicator substrate, Z‐DEVD‐ProRed™, from t = 24 h adult animals expressing GFP localized to the mitochondria and nuclei of muscle; scale bar represents 25 µm, and the enlarged regions are an additional 2.5× magnification. Note that the GFP and red substrate combine to produce yellow/orange colour in muscle when viewed using a triple pass filter.
Blocking protein degradation does not substantially restore the movement defect in unc‐105 mutants
Since reducing caspase levels or activity attenuated the protein degradation observed in unc‐105 mutants, we tested if these treatments, like let‐2, also attenuated the movement defect. We confirmed that unc‐105 mutants have a significant reduction in movement in comparison with wild‐type animals at young adulthood21 and that this is substantially rescued in unc‐105; let‐2 double mutants15 (Figure 3). However, a caspase inhibitor (ADEVD) had no effect, and ced‐3 RNAi had only a small positive effect on movement in unc‐105 mutants (Figure 3). These results combined with the observations that crt‐1 RNAi rescues the movement defect but does not prevent protein degradation, suggest that caspase activation in unc‐105 mutants is occurring at least somewhat independently of the movement defect.
Activation of UNC‐105 causes fragmentation of the mitochondrial network in muscle
CED‐4 binds to and activates CED‐3 in vitro,38 and inactive CED‐4 has been suggested to be localized to mitochondria.39 Structural damage to mitochondria occurs in HEK cells overexpressing UNC‐10540 and excess Na+ influx causes functional damage to mitochondria in mammalian muscle.41 Thus, because ced‐4 RNAi blocked degradation (Figure 4A), we wanted to determine if mitochondria were damaged in response to UNC‐105 activation. As shown in Figure 5A, unc‐105 mutants display time‐dependent fragmentation of the mitochondrial network in adult muscle. This fragmentation is prevented by RNAi against unc‐105 and in unc‐105; let‐2 double mutants (Figure 5A), which suggests that it is UNC‐105 hyperactivation that is causing the fragmentation.
Figure 5.
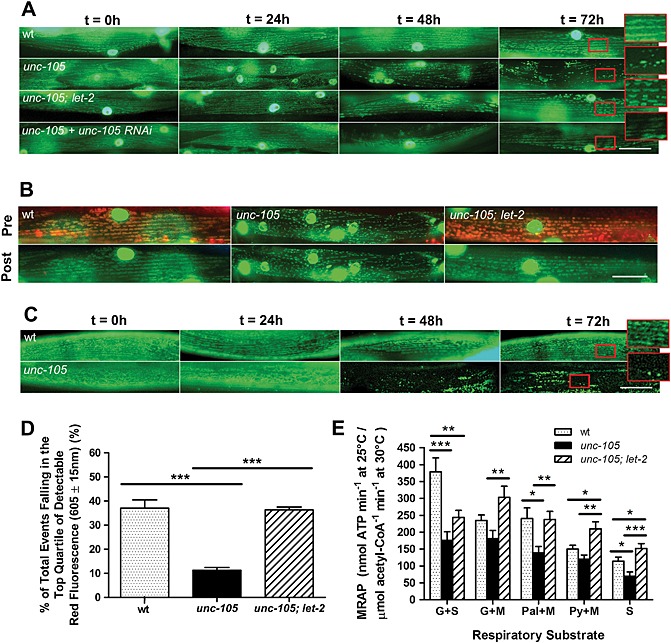
Mitochondria are dysfunctional in unc‐105 mutants. All experiments were repeated at least three times, for (D) seven and (E) 10 independent experiments. (A–C) Synchronized young adults were used (t = 0 h); scale bars represent 25 µm and enlarged regions an additional 2.5× magnification. (A) Strains containing GFP localized to mitochondria and nuclei in muscle were used to assess mitochondrial architecture. (B) Strains containing GFP localized to mitochondria and nuclei in muscle were used to assess mitochondrial membrane potential specifically in muscle. Accumulation of Mitotracker® CMXRos in mitochondria in muscle as indicated both by yellow/orange mitochondria and green mitochondria in muscle post‐photobleaching. Displayed images are for t = 0 h young adults. (C) Worms were grown in the presence of JC‐10 to assess in vivo loss of mitochondrial membrane potential. (D) Loss of membrane potential was confirmed in vitro in unc‐105 mutants when mitochondria were isolated from mixed populations of all three stains and were sorted using fluorescence‐activated cell sorting and JC‐1. Displayed are the percent of mitochondria showing the highest quartile of accumulation of JC‐1 as indicated by the extent of red fluorescence. (E) Measurement of maximal ATP production rates (MRAP). Displayed are data for mitochondria isolated from n = 250−300 mixed stage animals per sample. Substrate combinations were (G + S) glutamate and succinate; (G + M) glutamate and malate (Pal + M) palmitoyl‐l‐carnitine and malate; (Py + M) pyruvate and malate and (S) succinate. Data are expressed as a ratio to maximal citrate synthase (CS) activity; the standard marker of mitochondrial content. *P < 0.05; **P < 0.01; ***P < 0.001; one‐way ANOVA and Newman–Keuls correction.
UNC‐105 activation impairs maintenance of the mitochondrial membrane potential
We wanted to determine if the mitochondrial disturbance was purely structural or also functional. Mitotracker® Red CMXRos is a mitochondrial dye that accumulates within mitochondria dependent upon mitochondrial membrane potential. The accumulation of Mitotracker® Red CMXRos is reduced in unc‐105 but not in unc‐105; let‐2 double mutants (Figure 5B), suggesting that activation of UNC‐105 results in an inability to maintain mitochondrial membrane potential. However, because Mitotracker® requires a normal plasma membrane potential in order to accumulate in mitochondria and unc‐105 mutants have an altered membrane potential,23 it is possible that some or all of the reduced accumulation could be due to limited entry into muscle. Therefore, we used JC‐10, another dye that both accumulates and exits mitochondria dependent upon the mitochondrial membrane potential, thereby allowing assessment of the loss of mitochondrial membrane potential with time. When grown in the presence of JC‐10, unc‐105 mutants display accumulation of JC‐10 in muscle mitochondria at adulthood, with loss of JC‐10 over time (Figure 5C). These results suggest that UNC‐105 activation causes a time‐dependent fragmentation of the mitochondrial reticulum and concomitant failure to maintain the mitochondrial membrane potential. To confirm and quantify the extent of this impairment of the mitochondrial membrane potential and determine whether it was maintained ex vivo, we performed fluorescence activated cell sorting of isolated mitochondria from wild‐type, unc‐105, and unc‐105; let‐2 double mutants. In the top quartile of isolated mitochondria displaying JC‐1 accumulation in the presence of glutamate and malate, mitochondria from unc‐105 mutants display 25% less of the potential‐dependent dye JC‐1 than wild‐type or unc‐105; let‐2 animals (Figure 5D). These results further suggest impaired mitochondrial membrane potential in response to UNC‐105 activation.
Activation of UNC‐105 causes a depression in the maximal mitochondrial rate of ATP production
Inability to maintain mitochondrial membrane potential will lead to declines in mitochondrial ATP production as the result of a decrease in the proton gradient that drives H+ through ATP synthase. Thus, we measured the maximal rate of ATP production (MRAP) in isolated mitochondria. For all substrate combinations, the mean MRAP was reduced in mitochondria isolated from unc‐105 mutants versus wild‐type, and this reduction was significant for all substrates except glutamate + malate and pyruvate + malate (Figure 5E). Similarly, for all substrate combinations the reduction in mean MRAP in unc‐105 mutants was attenuated in mitochondria isolated from unc‐105; let‐2 double mutants, which was significant in all cases except glutamate + succinate (Figure 5E). Because MRAP is significantly depressed in unc‐105 mutants, we determined whether glycolytic ATP production increased as a compensatory adaptation to maintain cellular total ATP production. We find no increase in maximal Phosphofructokinase (PFK) activity in unc‐105 ( Figure S3), which suggests metabolic compensation via non‐mitochondrial ATP production is not occurring.
Specific mitochondrial proteins are decreased in mitochondria extracted from unc‐105 mutants
Having determined that mitochondria appear damaged in response to UNC‐105 activation, we tested if, as predicted by ced‐4‐dependent and ced‐3‐dependent cytosolic muscle protein degradation, less CED‐4 is associated with mitochondria following UNC‐105 activation. Mitochondria isolated from unc‐105 mutants contain less CED‐4 and cytochrome C compared with the amount present in mitochondria isolated from wild‐type or unc‐105; let‐2 double mutants (Figure 6). Given the reduced MRAP in unc‐105 mutants, we also examined the amount of ATP synthase. There is no comparable decline in ATP synthase in mitochondria isolated from unc‐105 mutants (Figure 6A), which confirms that reductions do not simply represent unequal loading of samples and suggests that decreased levels of CED‐4 and cytochrome C are not simply random loss of mitochondrial proteins in response to UNC‐105 activation. This observation appears to be consistent with the recent suggestion of a mitochondrial transition pore in C. elegans muscle.42 Additionally, our finding of less CED‐4 and cytochrome C associated with damaged mitochondria adds weight to the suggestion that CED‐4/CED‐3 dependent apoptosis, which is independent of a requirement of cytochrome C in C. elegans, reflects evolution of a lack of cytochrome C dependence in C. elegans versus evolutionary ancestors.43
Figure 6.
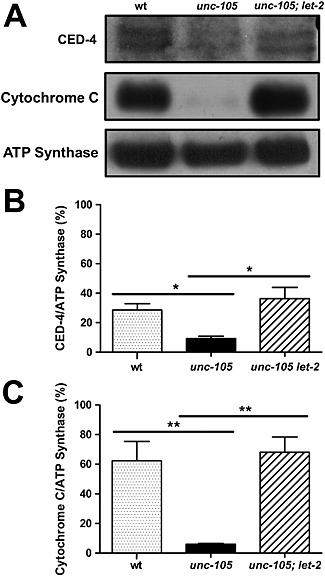
Less CED‐4 and cytochrome C are associated with mitochondria extracted from unc‐105 mutants. All experiments were conducted at least three times. Mitochondria extracted from wild‐type, unc‐105, and unc‐105; let‐2 double mutants were examined for mitochondrial protein content; n = 250−300 mixed stage animals per sample. (A) Representative western blots for CED‐4, cytochrome C, and ATP synthase. (B) Quantification of CED‐4 levels as a percentage of ATP synthase levels, n = 3. (C) Quantification of cytochrome C levels as a percentage of ATP synthase levels, n = 3. *P < 0.05; **P < 0.01, one‐way ANOVA with Newman–Keuls correction.
Discussion
Mitochondrial dysfunction and caspase activation in UNC‐105 mutants
We have shown that unc‐105 but not unc‐105; let‐2 double mutants display fragmentation of mitochondrial networks in muscle, decreased mitochondrial membrane potential, decreased maximal rates of mitochondrial ATP production, and reduced levels of cytochrome C and CED‐4 in extracted mitochondria. Because the second mutation in let‐2 relieves cationic influx in unc‐105 mutants,23 these results suggest that activation of UNC‐105 and consequent cationic influx into C. elegans muscle results in damage to mitochondrial structure and function in vivo. Additionally, because unc‐105 but not unc‐105; let‐2 double mutants display both caspase activation and pathological degradation of cytosolic muscle protein that is mediated by caspases, it appears that that activation of UNC‐105 and consequent cationic influx into C. elegans muscle results in activation of caspases in muscle cytosol in vivo. As CED‐4 is sufficient to activate the CED‐3 caspase in vitro,38 the findings of less CED‐4 in mitochondria extracted from unc‐105 but not unc‐105; let‐2 double mutants and RNAi against ced‐4 attenuating degradation in unc‐105 mutants, suggest that the pathological degradation in the muscle cytosol and the mitochondrial damage in unc‐105 mutants are causally linked by translocation of CED‐4 away from damaged mitochondria to cause activated CED‐3 to be present in the cytosol. Combined, our data suggest a model (Figure 7) whereby cationic influx through activated UNC‐105 results in mitochondrial damage, CED‐4 translocation away from damaged mitochondria, CED‐4 activation of CED‐3, and subsequent pathological degradation of proteins in the muscle cytosol.
Figure 7.
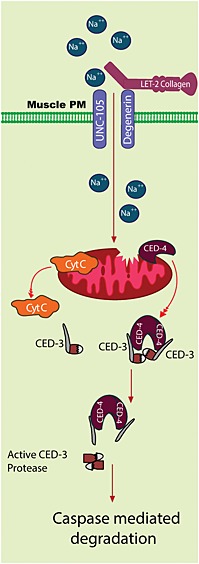
Graphical model of inferred consequences of UNC‐105 activation upon muscle mitochondria and cytosol. The UNC‐105 ion channel, when not gated by LET‐2, allows excess ion influx into muscle. In response to sustained ion influx, mitochondria are dysfunctional, and cytochrome C and CED‐4 translocate from the mitochondria. Translocated CED‐4 interacts with CED‐3 to cause CED‐3 activation. Activated CED‐3 causes cytosolic protein degradation.
Whole genome sequencing combined with subtractive analysis enables prospective studies of evolution in C. elegans
We used whole genome sequencing combined with subtractive analysis to identify two spontaneous mutants. We have confirmed the identity of the suppressing mutations both by known phenotypes of past suppressing mutations and by use of RNAi against each putative suppressor to phenocopy suppression of unc‐105 phenotypes. Given the ability to perform RNAi against every gene in the genome of C. elegans, whole genome sequencing combined with subtractive analysis and RNAi against a single or handful of genes identified provides a rapid way of identifying and confirming suppressing mutations of physiologic interest.
C. elegans as a model for studying genetic regulation of muscle protein degradation
Past studies have established C. elegans as a model in which genetics and genomics can be used to uncover the regulation of muscle protein degradation and revealed regulatory signals governing proteasomes, autophagy, and calpains.3 The present study now completes our preliminary picture of signals that regulate each of the four major proteolytic systems in C. elegans muscle. Combined, these studies enable future exploration of the relevance of these regulatory and proteolytic systems to different physiologic, pathophysiologic, or disease states.
Relevance of mitochondrial dysfunction and muscle protein degradation to sarcopenia
Mitochondrial dysfunction has been suggested to be a hallmark of ageing. The observations that unc‐105 mutants display disrupted sarcomeres, a movement defect, premature muscle protein degradation, and mitochondrial fragmentation and decline collectively suggest that unc‐105 mutants may be a model of accelerated ageing (progeria). This has previously been suggested for another mutant that displays mitochondrial dysfunction.44 Cellular degeneration of muscle appears to be similar to that of nerves in that both appear to be a disease of age, and there is fragmentation of mitochondrial networks in response to neuronal degeneration45, 46 and in ageing muscle.13, 46 These observations coupled with the fact that C. elegans is an accepted model for studying ageing47 and sarcopenia48 suggest that future study of the role of muscle mitochondrial dysfunction and muscle protein degradation in the onset and/or progression of sarcopenia can now be achieved using C. elegans. Accordingly, it is intriguing to note that caspase activation and consequent myosin degradation in ageing C. elegans muscle have recently been reported.37
Sodium channel, non‐voltage gated 1 alpha subunit (SCNN1A) is a human orthologue of UNC‐105 and is expressed in human skeletal muscle (http://www.proteinatlas.org/). Given that APAF1, caspases, and mitochondria are all also expressed in human skeletal muscle, there is significant scope for our finding of excessive sodium influx into C. elegans muscle leading to mitochondrial dysfunction and caspase activation (Figure 7) to be relevant to human muscle function and/or pathology. If SCNN1A is mechanosensitive in human skeletal muscle, then like mechanosensitive calcium channels, it may contribute to ionic imbalance in response to stretch and in individuals with muscular dystrophy.49 Given that cytoskeletal proteins are common targets of caspases, there is a reason to suspect that any ionic imbalance resulting from SCNN1A hyperactivation would lead to cytoskeletal remodelling and/or dystrophy; therefore, the role of SCNN1A in human skeletal muscle would seem to warrant further investigation. Similarly, if a past report of decreased chloride transport and mitochondrial function with age in human muscle is correct,50 then altered ionic balance may lead to both altered mitochondrial function and cytoskeletal alterations via loss of mitochondrial membrane potential and caspase activation with age, another area that would seem to warrant further investigation. As previously noted,50 altered mitochondrial function with age in human muscle, as opposed to C. elegans muscle, might be an adaptation rather than a simple path to eventual cellular demise.
Acknowledgements
This work was supported by a grant from the US National Institutes of Health National Institute for Arthritis and Musculoskeletal and Skin Diseases (AR‐054342) to N. J. S., by a grant from the Canadian Institutes of Health Research to A. R., and by a grant from Natural Sciences and Engineering Research Council of Canada to D. L. B. C. G. was funded by a Doctoral Training Studentship provided by the University of Nottingham, and all metabolic measurement consumables were funded by the University of Nottingham. The unc‐105(n490) mutant allele utilized in this work was obtained from the Caenorhabditis Genetics Center, which is funded by the US NIH National Center for Research Resources. Bioinformatic work was conducted utilizing WormBase, which is funded by the US NIH National Human Genome Research Institute and the British Medical Research Council. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia, and Muscle 2010;1:7–8 (von Haehling S, Morley JE, Coats AJ and Anker SD).
Ethical Standards
The manuscript does not contain clinical studies or patient data. The use of invertebrate models of human disease is fully compliant with the replacement, reduction, and refinement of animal models and is therefore ethically preferred.
Conflict of interest
None declared.
Supporting information
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Gaffney, C. J. , Shephard, F. , Chu, J. , Baillie, D. L. , Rose, A. , Constantin‐Teodosiu, D. , Greenhaff, P. L. , and Szewczyk, N. J. (2016) Degenerin channel activation causes caspase‐mediated protein degradation and mitochondrial dysfunction in adult C. elegans muscle. Journal of Cachexia, Sarcopenia and Muscle, 7: 181–192. doi: 10.1007/jcsm.12040.
[Correction added after online publication: 16 June 2015: license statement altered to Creative Commons, CC‐BY wording].
References
- 1. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science 2008; 319: 916–919. [DOI] [PubMed] [Google Scholar]
- 2. Ruegg MA, Glass DJ Molecular mechanisms and treatment options for muscle wasting diseases. Annu Rev Pharmacol Toxicol 2011; 51: 373–395. [DOI] [PubMed] [Google Scholar]
- 3. Lehmann S, Shephard F, Jacobson LA, Szewczyk NJ. Integrated control of protein degradation in C. elegans muscle. Worm 2012; 1: 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Szewczyk NJ, Hartman JJ, Barmada SJ, Jacobson LA. Genetic defects in acetylcholine signalling promote protein degradation in muscle cells of Caenorhabditis elegans . J Cell Sci 2000; 113: 2003–2010. [DOI] [PubMed] [Google Scholar]
- 5. Shephard F, Adenle AA, Jacobson LA, Szewczyk NJ. Identification and functional clustering of genes regulating muscle protein degradation from amongst the known C. elegans muscle mutants. PLoS One 2011; 6: e24686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zdinak LA, Greenberg IB, Szewczyk NJ, Barmada SJ, Cardamone‐Rayner M, Hartman JJ, et al. Transgene‐coded chimeric proteins as reporters of intracellular proteolysis: starvation‐induced catabolism of a lacZ fusion protein in muscle cells of Caenorhabditis elegans . J Cell Biochem 1997; 67: 143–153. [PubMed] [Google Scholar]
- 7. Estevez AO, Mueller CL, Morgan KL, Szewczyk NJ, Teece L, Miranda‐Vizuete A, et al. Selenium induces cholinergic motor neuron degeneration in Caenorhabditis elegans . Neurotoxicology 2012; 33: 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Szewczyk NJ, Peterson BK, Barmada SJ, Parkinson LP, Jacobson LA. Opposed growth factor signals control protein degradation in muscles of Caenorhabditis elegans . EMBO J 2007; 26: 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lehmann S, Bass JJ, Szewczyk NJ. Knockdown of the C. elegans kinome identifies kinases required for normal protein homeostasis, mitochondrial network structure, and sarcomere structure in muscle. Cell Commun Signal 2013; 11: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szewczyk NJ, Peterson BK, Jacobson LA. Activation of Ras and the mitogen‐activated protein kinase pathway promotes protein degradation in muscle cells of Caenorhabditis elegans . Mol Cell Biol 2002; 22: 4181–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Florez‐McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin‐receptor signaling promotes the autophagic degradation of beta‐amyloid peptide in C. elegans . Autophagy 2007; 3: 569–580. [DOI] [PubMed] [Google Scholar]
- 12. Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life‐span extension in C. elegans . Science 2003; 301: 1387–1391. [DOI] [PubMed] [Google Scholar]
- 13. Etheridge T, Oczypok EA, Lehmann S, Fields BD, Shephard F, Jacobson LA, et al. Calpains mediate integrin attachment complex maintenance of adult muscle in Caenorhabditis elegans . PLoS Genet 2012; 8: e1002471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Etheridge T, Rahman M, Gaffney CJ, Shaw D, Shephard F, Magudia J, et al. The integrin‐adhesome is required to maintain muscle structure, mitochondrial ATP production, and movement forces in Caenorhabditis elegans . FASEB J 2014; doi: 10.1096/fj.14-259119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu J, Schrank B, Waterston RH. Interaction between a putative mechanosensory membrane channel and a collagen. Science 1996; 273: 361–364. [DOI] [PubMed] [Google Scholar]
- 16. Dworakowska B, Dolowy K. Ion channels‐related diseases. Acta Biochim Pol 2000; 47: 685–703. [PubMed] [Google Scholar]
- 17. Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans . Nature 2002; 419: 939–944. [DOI] [PubMed] [Google Scholar]
- 18. Driscoll M, Chalfie M. The mec‐4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 1991; 349: 588–593. [DOI] [PubMed] [Google Scholar]
- 19. Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 2001; 31: 957–971. [DOI] [PubMed] [Google Scholar]
- 20. Hall DH, Gu G, Garcia‐Anoveros J, Gong L, Chalfie M, Driscoll M. Neuropathology of degenerative cell death in Caenorhabditis elegans . J Neurosci 1997; 17: 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Park EC, Horvitz HR. Mutations with dominant effects on the behavior and morphology of the nematode Caenorhabditis elegans . Genetics 1986; 113: 821–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sibley MH, Johnson JJ, Mello CC, Kramer JM. Genetic identification, sequence, and alternative splicing of the Caenorhabditis elegans alpha 2(IV) collagen gene. J Cell Biol 1993; 123: 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jospin M, Mariol M‐C, Segalat L, Allard B. Patch clamp study of the UNC‐105 degenerin and its interaction with the LET‐2 collagen in Caenorhabditis elegans muscle. J Physiol 2004; 557: 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaffney CJ, Bass JJ, Barratt TF, Szewczyk NJ. Methods to assess subcellular compartments of muscle in C. elegans . J Vis Exp 2014; 93: e52043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Contreras V, Richardson MA, Hao E, Keiper BD. Depletion of the cap‐associated isoform of translation factor eIF4G induces germline apoptosis in C. elegans . Cell Death Differ 2008; 15: 1232–1242. [DOI] [PubMed] [Google Scholar]
- 26. Suthammarak W, Morgan PG, Sedensky MM. Mutations in mitochondrial complex III uniquely affect complex I in Caenorhabditis elegans . J Biol Chem 2010; 285: 40724–40731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wirth M, Karaca S, Wenzel D, Ho L, Tishkoff D, Lombard DB, et al. Mitochondrial SIRT4‐type proteins in Caenorhabditis elegans and mammals interact with pyruvate carboxylase and other acetylated biotin‐dependent carboxylases. Mitochondrion 2013; 13: 705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chu JS, Johnsen RC, Chua SY, Tu D, Dennison M, Marra M, et al. Allelic ratios and the mutational landscape reveal biologically significant heterozygous SNVs. Genetics 2012; 190: 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009; 25: 2283–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vergara IA, Frech C, Chen N. CooVar: co‐occurring variant analyzer. BMC Res Notes 2012; 5: 1756–0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eide D, Anderson P. The gene structures of spontaneous mutations affecting a Caenorhabditis elegans myosin heavy chain gene. Genetics 1985; 109: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park EC, Horvitz HR. C. elegans unc‐105 mutations affect muscle and are suppressed by other mutations that affect muscle. Genetics 1986; 113: 853–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercer KB, Miller RK, Tinley TL, Sheth S, Qadota H, Benian GM. Caenorhabditis elegans UNC‐96 is a new component of M‐lines that interacts with UNC‐98 and paramyosin and is required in adult muscle for assembly and/or maintenance of thick filaments. Mol Biol Cell 2006; 17: 3832–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang M, Chalfie M. Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans . Nature 1994; 367: 467–470. [DOI] [PubMed] [Google Scholar]
- 36. Tiainen M, Spitkovsky D, Jansen‐Durr P, Sacchi A, Crescenzi M. Expression of E1A in terminally differentiated muscle cells reactivates the cell cycle and suppresses tissue‐specific genes by separable mechanisms. Mol Cell Biol 1996; 16: 5302–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fisher K, Gee F, Wang S, Xue F, Knapp S, Philpott M, et al. Maintenance of muscle myosin levels in adult C. elegans requires both the double bromodomain protein BET‐1 and sumoylation. Biol Open 2013; 2: 1354–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. del Peso L, Gonzalez VM, Nunez G. Caenorhabditis elegans EGL‐1 disrupts the interaction of CED‐9 with CED‐4 and promotes CED‐3 activation. J Biol Chem 1998; 273: 33495–33500. [DOI] [PubMed] [Google Scholar]
- 39. Chen F, Hersh BM, Conradt B, Zhou Z, Riemer D, Gruenbaum Y, et al. Translocation of C. elegans CED‐4 to nuclear membranes during programmed cell death. Science 2000; 287: 1485–1489. [DOI] [PubMed] [Google Scholar]
- 40. Garcia‐Anoveros J, Garcia JA, Liu JD, Corey DP. The nematode degenerin UNC‐105 forms ion channels that are activated by degeneration‐ or hypercontraction‐causing mutations. Neuron 1998; 20: 1231–1241. [DOI] [PubMed] [Google Scholar]
- 41. Iwai T, Tanonaka K, Inoue R, Kasahara S, Motegi K, Nagaya S, et al. Sodium accumulation during ischemia induces mitochondrial damage in perfused rat hearts. Cardiovasc Res 2002; 55: 141–149. [DOI] [PubMed] [Google Scholar]
- 42. Giacomotto J, Brouilly N, Walter L, Mariol MC, Berger J, Segalat L, et al. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome C release and IP3R activity in muscular dystrophy. Hum Mol Genet 2013; 25: 25. [DOI] [PubMed] [Google Scholar]
- 43. Bender CE, Fitzgerald P, Tait SW, Llambi F, McStay GP, Tupper DO, et al. Mitochondrial pathway of apoptosis is ancestral in metazoans. Proc Natl Acad Sci U S A 2012; 109: 4904–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ishii N, Fujii M, Hartman PS, Tsuda M, Yasuda K, Senoo‐Matsuda N, et al. A mutation in succinate dehydrogenase cytochrome B causes oxidative stress and ageing in nematodes. Nature 1998; 394: 694–697. [DOI] [PubMed] [Google Scholar]
- 45. Knott AB, Perkins G, Schwarzenbacher R, Bossy‐Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 2008; 9: 505–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kamp F, Exner N, Lutz AK, Wender N, Hegermann J, Brunner B, et al. Inhibition of mitochondrial fusion by alpha‐synuclein is rescued by PINK1, Parkin and DJ‐1. EMBO J 2010; 29: 3571–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guarente L, Kenyon C. Genetic pathways that regulate ageing in model organisms. Nature 2000; 408: 255–262. [DOI] [PubMed] [Google Scholar]
- 48. Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, et al. Stochastic and genetic factors influence tissue‐specific decline in ageing C. elegans . Nature 2002; 419: 808–814. [DOI] [PubMed] [Google Scholar]
- 49. Lansman JB, Franco‐Obregon A. Mechanosensitive ion channels in skeletal muscle: a link in the membrane pathology of muscular dystrophy. Clin Exp Pharmacol Physiol 2006; 33: 649–656. [DOI] [PubMed] [Google Scholar]
- 50. Zahn JM, Sonu R, Vogel H, Crane E, Mazan‐Mamczarz K, Rabkin R, et al. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet 2006; 2: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item