

Abstract
Proteomic profiling on subcellular fractions provides invaluable information regarding both protein abundance and subcellular localization. When integrated with other data sets, it can greatly enhance our ability to predict gene function genome-wide. In this study, we performed a comprehensive proteomic analysis on the light-sensing compartment of photoreceptors called the outer segment (OS). By comparing with the protein profile obtained from the retina tissue depleted of OS, an enrichment score for each protein is calculated to quantify protein subcellular localization, and 84% accuracy is achieved compared with experimental data. By integrating the protein OS enrichment score, the protein abundance, and the retina transcriptome, the probability of a gene playing an essential function in photoreceptor cells is derived with high specificity and sensitivity. As a result, a list of genes that will likely result in human retinal disease when mutated was identified and validated by previous literature and/or animal model studies. Therefore, this new methodology demonstrates the synergy of combining subcellular fractionation proteomics with other omics data sets and is generally applicable to other tissues and diseases.
Owing to the rapid development of high-throughput technologies, the pace of omics sciences has been greatly accelerated. Direct proteomic characterization of final gene products is considered one of the most informative and invaluable tools that confirms and complements other omics data (Aebersold and Mann 2003). In addition to the entire cell, proteomic profiling has been conducted for specific cell organelles and compartments providing important information of protein subcellular localization (Andersen et al. 2003). Compared to the traditional immunohistochemistry methods in which a limited number of targets are examined, mass spectrometry (MS)-based proteomics is high throughput and less biased (Sadowski et al. 2006).
Vertebrate photoreceptor cells of the retina are specialized sensory neurons that consist of four primary structural compartments: the outer segment (OS), the inner segment (IS), cell body, and synaptic terminal (Mustafi et al. 2009). The OS is filled with stacks of photosensitive membrane discs that are essential for capturing and sensing light. The IS contains mitochondria, endoplasmic reticulum, and Golgi, and is the compartment where proteins are synthesized and sorted. The OS is joined to the IS by a connecting cilium (CC) that allows protein transportation between the two compartments (Fig. 1A).
Figure 1.
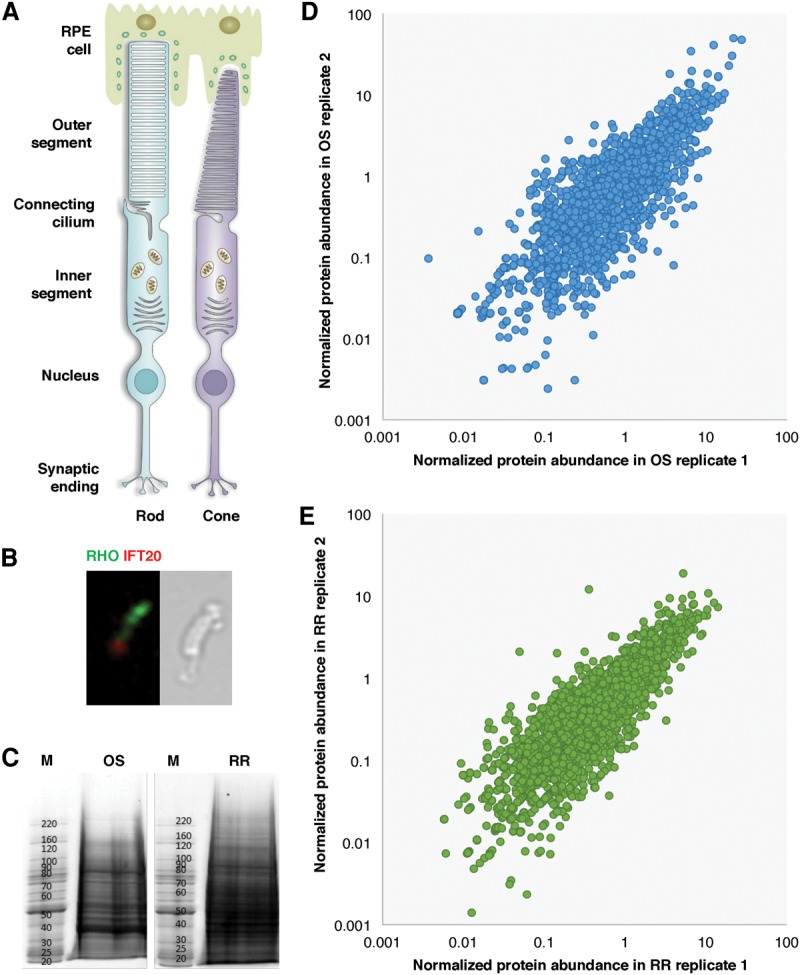
High-quality proteomic data of the OS and the RR were obtained. (A) Schematic diagram of the structure of a rod photoreceptor cell and a cone photoreceptor cell in mouse retina. (B, left) Immunofluorescence of isolated OS preparation stained with antibodies of RHO (green) and IFT20 (red); (right) microscopic analysis of an isolated rod OS. (C) OS protein complex (left) and RR protein complex (right) were electrophoresed. The sizes of the molecular weight markers are indicated in M. (D) Scatter plot of normalized protein abundance for OS proteins between different replicates. The average Pearson correlation between all replicates is 0.85. (E) Scatter plot of normalized protein abundance for RR proteins between different replicates. The Pearson correlation between two replicates is 0.75.
Consistent with its function in light sensing, the visual pigment proteins and other phototransduction components are localized to the OS. In addition, proteins with diverse functions such as vesicle trafficking (Insinna et al. 2010) and microtubule cytoskeleton (Mühlhans et al. 2011) have also been detected. Strikingly, the protein products of many retinal disease genes are enriched in the OS, indicating the crucial role that OS proteins play in visual functions. Given the central role of the OS in photoreceptor function and its relevance to retinal disease, it is highly desirable to obtain a comprehensive list of OS proteins.
Two proteomic studies have been conducted on the OS, and around 2000 proteins have been identified (Liu et al. 2007; Kwok et al. 2008). Since then, both the sensitivity and specificity of mass spectrometry technology have been dramatically improved (Mallick and Kuster 2010; Jimenez and Verheul 2014). Furthermore, current technology allows more accurate label-free proteomic quantification. Given the relatively low detection rate, it is likely that a significant number of proteins in the OS were not detected by previous studies.
In this study, we performed a comprehensive proteomic analysis on purified OS from the mouse retina. In addition, by comparing the OS and the remaining retina (RR), the protein OS enrichment score was derived and can be used to predict protein subcellular localization. Finally, by integrating gene expression and the protein OS enrichment score, a list of highly probable retinal disease genes was identified.
Results
OS and RR proteome profiling
Figure 1A shows a schematic diagram of the structure of photoreceptor cells. The OS is connected to the IS through CC, which is anchored to the basal body. During OS preparation, the OS, CC, and basal body are all separated from the photoreceptor cell body via mechanical shearing. Figure 1B shows by immunofluorescence and light microscopy that the intact rod photoreceptor OS as well as the CC are isolated and marked by RHO and IFT20 staining. The RR is the portion of the retina left after OS separation and was also collected for comparative analysis. The proteomes were extracted from both the OS and the RR and resolved by SDS-PAGE (Fig. 1C). A dense band ∼39 kDa, which corresponds to the molecular weight of RHO in mouse, is consistently detected in the OS fraction while not clearly recognizable from the RR.
Proteomes from both the OS and the RR were profiled by MS. A total of 3607 proteins (corresponding to 4435 isoforms) were identified in the OS fraction. Most of these proteins were conserved in human with 3442 corresponding homologs (Supplemental Tables S1, S2). To test the quality of our data set, three biological replicates were conducted. As shown in Figure 1D, the expression level of proteins correlated very well between multiple experiments, particularly for proteins with a normalized abundance greater than 1. The average Pearson correlation between replicates was 0.85. As expected, RHO along with other well-studied OS proteins, such as the guanine nucleotide binding protein GNAT1, were detected at high abundance in our data set.
In the RR, 3879 proteins (corresponding to 4779 isoforms) were identified, among which 3733 human homologs were mapped. The RR data set was of a similarly high quality to that of the OS proteome. As shown in Figure 1E, the Pearson correlation between different replicates of the RR proteome was 0.75. Proteins known to localize to the IS were detected. For example, spliceosomal proteins PRPF3, PRPF4, PRPF6, PRPF8, and PRPF31, which are known to be involved in pre-mRNA splicing in the IS, were all identified in the RR data set.
Quality assessment of the OS proteome
The high correlation among different biological replicates as well as the successful cell marker staining suggested the high quality of our data sets. In addition, to systematically evaluate the quality of our data, we further compared the identified protein list with previous similar studies. Earlier proteomic studies were performed on the OS of bovine (Kwok et al. 2008) and adult mouse (Supplemental Table S3; Liu et al. 2007). In the bovine OS proteomic study (Kwok et al. 2008), 516 bovine proteins were identified, and 483 of them were conserved in human with corresponding homologs. In comparison, our OS proteome covered ∼90% (432/483) of the proteins that were identified in the bovine OS proteome. In the previous adult mouse OS study (Liu et al. 2007), 1962 mouse proteins were identified that mapped to 1935 human homologs, the majority (81%) of which overlapped with our study. Notably, Figure 2 shows that of 331 proteins that were identified in both bovine (Kwok et al. 2008) and mouse (Liu et al. 2007) OS data sets, only two proteins were not found in our OS list. Therefore, our data set is largely concordant with previous similar studies.
Figure 2.

Novel OS proteins were identified. (A) Venn diagram of OS proteome identified in previous studies and this study. (B) Venn diagram of known retinal disease genes identified in different studies. All proteins were mapped to human homologs. (C) Distribution of log2 (Spectral Counts) for OS proteins that overlapped with previous OS studies and new proteins identified in this study.
Novel OS proteins were identified
Mostly due to improvements in MS technology, our OS proteome displays a higher sensitivity at detecting proteins with low abundance. As a result, 1771 proteins not found in the previously published studies have been identified in our study. We compared the spectral counts of proteins that overlapped with previous studies and those of the proteins newly identified in this study. For the proteins identified in both our study and previous studies, the average spectral count was 180, whereas for proteins that are only identified in our study, the average spectral count was 59. Figure 2C shows that the distribution of the newly identified proteins tends to be more enriched in lower abundant proteins, which directly suggests the higher sensitivity of our data. Manual inspection of these additional proteins indicated that at least some are indeed expected to localize to the OS according to the function. For example, PDE6G and PDE6H are gamma subunits of cyclic GMP-phosphodiesterase proteins, which function in the phototransduction signaling cascade. Both proteins should localize to the OS, where phototransduction takes place.
To further evaluate the newly identified proteins, gene ontology functional analysis was conducted on the 1771 proteins that were not reported in the previous studies. One interesting finding is that several metabolism-related categories are significantly enriched among the newly identified proteins, including the electron transport chain, cellular respiration, and NADH dehydrogenase activity as shown in Supplemental Table S4. The results seem to be unexpected because these biochemical processes take place in mitochondria, which are localized to the IS. However, a series of studies found that although the OS is devoid of mitochondria, the mitochondrial proteins are present in the OS discs, and extra-mitochondrial aerobic metabolism would account for a quantitatively adequate ATP supply needed for phototransduction (Panfoli et al. 2012, 2013). Our observations are thus consistent with previous studies. Overall, our results provide a more comprehensive list of OS proteins, including not only traditional OS proteins but also unexpected ones, that may shed light on new mechanisms in the OS.
The RR has a distinct protein set
The proteins specialized for the phototransduction process are synthesized in the IS and actively trafficked to the OS. Therefore, the proteome of the OS is expected to be significantly different from the rest of the cell. To test this, we analyzed the proteome of the RR and compared it to the OS proteome. Figure 3A shows that there are 2500 proteins that were identified in both the OS and the RR, and distinct protein sets were also observed with 1107 and 1379 proteins that were unique to the OS and the RR, respectively. To systematically look at functional differences between the OS and RR proteins, gene ontology enrichment analysis was performed on the proteins detected in the OS (Supplemental Table S5) and RR (Supplemental Table S6). In the OS proteome, the most dominant category was proteins associated with the “establishment of localization” with a P-value of 6.50 × 10−49. In the RR proteome, proteins that function in mRNA processing were the most significantly enriched category with a P-value of 1.76 × 10−105. This illustrated the biological features of photoreceptors in that mRNA is processed and translated into proteins in the IS, and proteins are sorted and transported to the OS to perform visual functions.
Figure 3.

Protein OS enrichment score is predictive for protein localization. (A) Venn diagram of proteins identified in the OS and RR. (B) OS enrichment score distribution of all proteins identified in the OS and RR. (C) Functional gene ontology analysis of OS-enriched and RR-enriched proteins. (D) Pie chart showing 84% of the protein localizations predicted by the enrichment score are consistent with literature, and 16% are inconsistent.
Protein OS enrichment score is predictive for protein localization
Given the observation that a clear distinction is observed when comparing the OS and RR proteomes, we tested if it is possible to predict protein localization by comparing the proteome profiling data described above.
The protein OS enrichment score was calculated as the ratio of likelihoods for differential expression for each protein and is represented by the Enrichment (OS/RR) score from QSpec (Choi et al. 2008) as described in Methods. A positive enrichment score indicates that the corresponding protein is more abundant in the OS than the RR. The distribution of enrichment scores for all proteins identified is shown in Figure 3B. To compare the differences of OS-enriched and RR-enriched proteins, the proteins with the highest 20% of enrichment scores and those with the lowest 20% of enrichment scores were compared by functional gene ontology analysis as shown in Figure 3C and Supplemental Tables S7, S8. These statistically overrepresented functional modules revealed underlying molecular mechanisms of the OS and the RR. For example, proteins involved in visual function are prominent in the OS-enriched group, especially proteins in the phototransduction pathway, which are enriched as high as 10-fold with a P-value of 1.07 × 10−13. In contrast, the RR enriched proteins are mainly involved in RNA processing and nucleic acid metabolic processes.
To test if the enrichment score is informative for predicting protein subcellular localization, we examined the protein OS enrichment scores of a list of 124 well-documented proteins. Supplemental Table S9 and Figure 3D show that 84% have consistent localization information between our prediction and literature records, indicating the enrichment score is highly informative. For the 16% inconsistent ones, it is likely due to limitations in OS preparation in which proteins localized to the subretinal space between photoreceptor and retinal pigment epithelium (RPE) cells are copurified. It is also possible that the inconsistency of some proteins can be due to incorrect records in literature. The immunolocalization has several limitations, such as variable specificity, low sensitivity, and epitope masking. Overall, from this estimation, our protein localization prediction accuracy at least reached 84%.
Retinal disease–associated proteins are enriched in the OS
It has been reported that the OS proteome is enriched in known human retinal disease genes. To test if this enrichment is also observed in our expanded OS protein list, we checked for the presence of 215 known retinal disease genes (RetNet, https://sph.uth.edu/retnet/) in our data set. Strikingly, 56% (121/215) of known disease genes have been identified in our OS data set. This is a 3.8-fold enrichment over the whole human genome background with a P-value of 2.65 × 10−49. In comparison to previous OS proteomic studies, our new OS data set not only covered 93% (94/101) of the disease gene proteins reported in these studies, but also included 27 unreported ones (Fig. 2B). For example, SDCCAG8 and INPP5E are two transition zone proteins (Otto et al. 2010; Luo et al. 2012), and mutations in either gene can give rise to Bardet-Biedl syndrome, including a retinal degeneration phenotype. Both genes were missed in the previous reports and were identified in our new data set.
Another interesting observation is that retinal disease genes also have the higher OS enrichment scores. In Figure 4A, retinal disease proteins show a significant shift to positive OS enrichment scores compared to the background. Indeed, among the 121 known retinal disease genes, 106 have positive enrichment scores.
Figure 4.

Integrative method was applied to predict novel retinal disease genes. (A) The enrichment score distribution of all retinal disease and background proteins identified in the OS and RR proteomes. (B) The attribute importance, measured by ReliefF score, of all attributes used during machine learning. (C) The prediction score distributions for background genes, known retinal disease genes, and candidate disease genes. (D) The average prediction scores of background genes, known retinal disease genes, and candidate disease genes. All other genes identified in the OS and RR proteome are used as background.
Novel retinal disease gene prediction
The enrichment of retinal disease proteins in the OS suggested that the enrichment score might be able to facilitate novel disease gene discovery. Here, we used machine learning methods to rank all genes identified in the OS and the RR by the possibility that the gene is linked to human retinal disease.
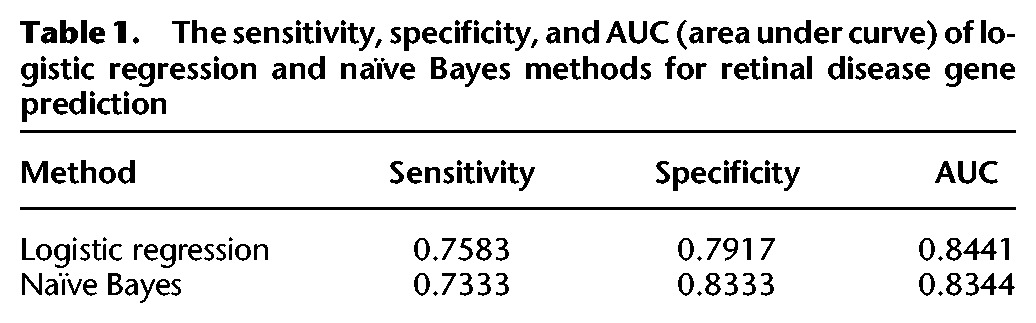
Compared to transcriptomic studies, current MS-based proteomics is less sensitive. To compensate for this, we supplemented our predictive model training data by incorporating RNA-seq data on the mouse retina. The attributes used for machine learning included the protein OS enrichment score, the protein abundance in the OS and the RR, and the RNA expression level. Both naïve Bayes and logistic regression classification methods were applied. The sensitivity and specificity are shown in Table 1. Overall, the logistic regression had a higher sensitivity around 0.758, whereas the naïve Bayes had a higher specificity of about 0.833. The impact of each attribute to the overall prediction results was calculated using the ReliefF algorithm (Kononenko 1994). Figure 4B shows the most important attribute was the protein OS enrichment score. The prediction scores were combined from both machine learning methods and ranged from 0 to 1, indicating the probability that a given gene is linked to human retinal disease. To evaluate the prediction results, a list of 64 candidate retinal disease genes was generated. These candidate genes either showed a retinal degeneration phenotype in mutant mice based on the MGI database (Blake et al. 2014) or have already been identified as candidate retinal disease genes in literature or public databases (Supplemental Table S10). In Figure 4C, the distribution of prediction scores for known retinal disease genes, candidate retinal disease genes, and background genes are clearly separated. The average prediction score is 0.74 for retinal disease genes, 0.69 for candidate disease genes, and 0.29 for background genes. Both known and candidate disease gene groups showed significantly higher scores than background genes with P-values of 1.74 × 10−41 and 2.53 × 10−18, respectively, under a one-tail t-test (Fig. 4D).
Table 1.
The sensitivity, specificity, and AUC (area under curve) of logistic regression and naïve Bayes methods for retinal disease gene prediction
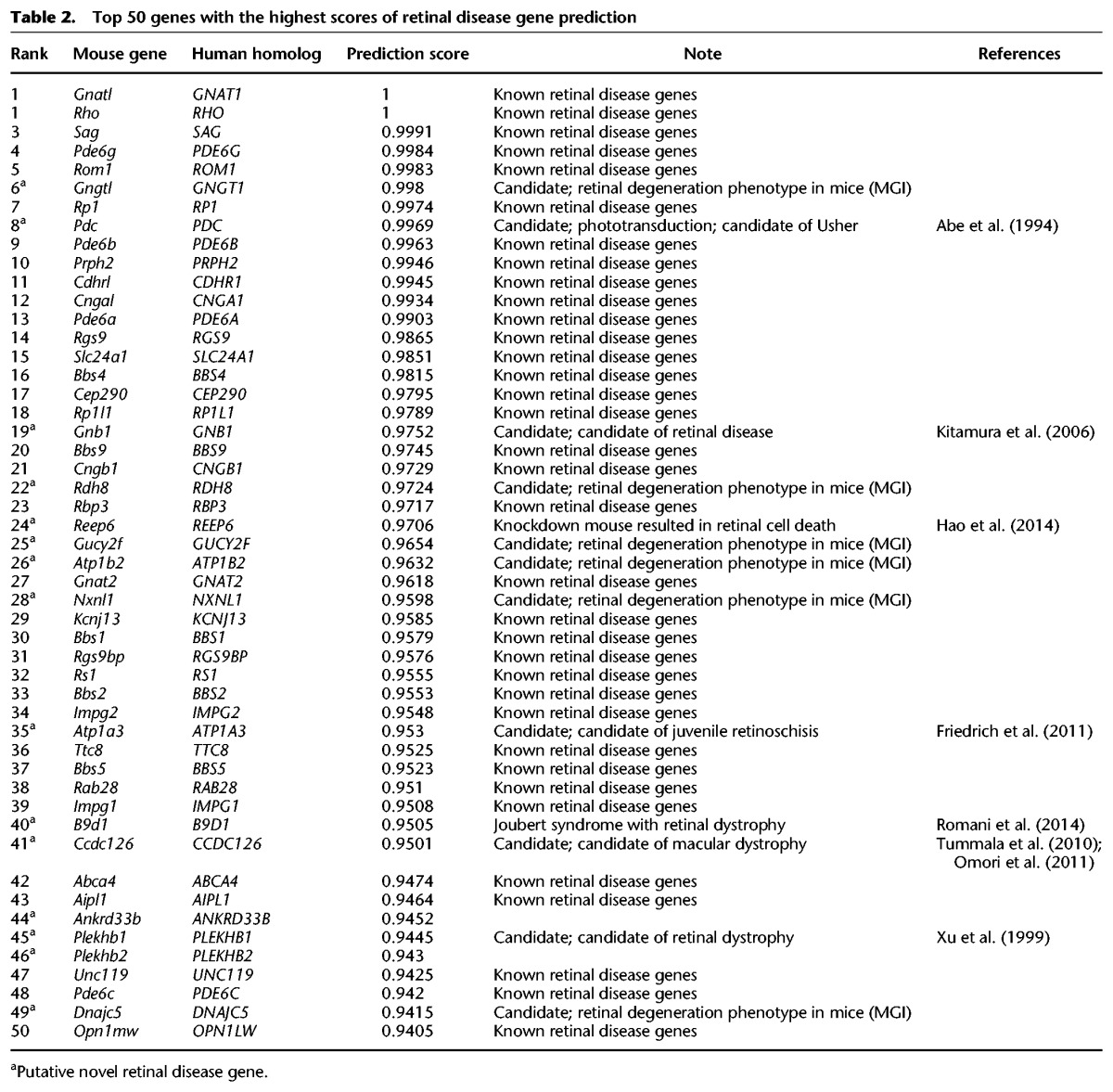
To further investigate the utility of our prediction tool, we performed detailed analysis of the top 50 ranked predicted retinal disease genes (Table 2). Among them, 35 genes were previously known retinal disease genes recorded in the RetNet database (https://sph.uth.edu/retnet/). Interestingly, in the remaining 15 genes, one gene, B9D1, which had a prediction score of 0.95, was recently identified as a novel gene causing Joubert syndrome, and the patients showed retinal dystrophy phenotype (Romani et al. 2014). Furthermore, seven genes showed retinal degenerative phenotypes when mutated in mice. For example, the sixth-ranked gene, GNGT1, has a prediction score of 0.998. This gene encodes the gamma subunit of transducin. Homozygous null mice display gradual retinal photoreceptor degeneration with a loss of rod photoreceptors by 6 mo of age (Lobanova et al. 2008). Another interesting example is REEP6, with a prediction score of 0.97. Although it is not included in the MGI database, this gene has been recently identified as a key functional target of the NRL-centered transcriptional regulatory network in rod photoreceptors, and knockdown of Reep6 in both mouse and zebrafish resulted in death of retinal photoreceptor cells (Hao et al. 2014). Finally, five genes have been proposed as candidate retinal disease genes in previous literature based on studies in model organisms (Abe et al. 1994; Xu et al. 1999; Kitamura et al. 2006; Tummala et al. 2010; Friedrich et al. 2011; Omori et al. 2011). Therefore, for the set of 15 predicted novel disease genes, only two genes, ANKRD33B and PLEKHB2, currently lack substantial experimental evidence support, indicating a minimal accuracy at 87% (13/15).
Table 2.
Top 50 genes with the highest scores of retinal disease gene prediction
Discussion
We have designed a novel cell compartment proteomic profiling strategy that allows the systematic, accurate prediction of protein localization and function. In total, 3607 proteins from the OS of photoreceptors have been identified, including 1771 novel proteins. In parallel, 3879 proteins have been found in the RR of the mouse retina. The protein subcellular localizations were predicted by comparing the OS and RR proteomic profiles, achieving an overall accuracy of 84%. Finally, machine learning models were developed by integrating protein OS enrichment scores, RNA expression levels, and protein abundance to predict a gene's likelihood to cause retinal disease. High specificity and sensitivity were achieved and validated by both literature search and animal model studies. Therefore, our study not only dramatically increased the known proteome of photoreceptor cells, but also demonstrated a new approach and created a rich resource for subsequent functional studies and novel retinal disease gene identification.
Compared to previous studies (Liu et al. 2007; Kwok et al. 2008), our methodology was more sensitive at detecting proteins with a low abundance, primarily due to recent improvement in MS technology. The Q Exactive tandem mass spectrometer used in this study allows fast acquisition of high-resolution higher-energy collisional dissociation (HCD) tandem mass spectra, generating more sensitive and precise proteomic results (Michalski et al. 2011; Gallien et al. 2012; Jones et al. 2013). Our OS proteome data set covered >81% of the reported OS proteins and also included 1771 novel proteins that have not been identified previously in OS proteomic studies. Among these novel OS proteins, we found a set of metabolic proteins in the OS, which added to growing evidence of a metabolic mechanism in the OS. These proteins may also be responsible for retinal degenerative diseases. For example, one such protein identified in our OS data set was NMNAT1, an enzyme synthesizing NAD+ and is involved in nuclear NAD+ homeostasis necessary for both metabolism and cell signaling. Mutations in this gene have been shown to cause the severe retinal degenerative disease Leber congenital amaurosis, but the underlying pathophysiology remains unclear (Chiang et al. 2012; Falk et al. 2012; Koenekoop et al. 2012).
Compared to previous studies, ∼10%–19% of proteins were not identified in our OS proteome. After investigation, the discordance between studies is likely due to the following reasons. First, the ciliary rootlet that extends into the IS can be included in the OS fraction when isolating the OS by mechanical shearing, and thus, the OS proteome may contain some proteins that are mainly localized to the IS but are captured during this process. Interestingly, among 367 proteins found in the previous studies’ adult mouse OS proteome but not in our OS list, 53% were found in our RR protein list. For example, several translation initiation factors (EIF2B5, EIF2S2, EIF2B4, EIF2B3, EIF5B, and EIF3M) were found in the previous OS protein lists but only identified in our RR proteome. As inferred by the structure of photoreceptor cells and the function of these proteins, they are more likely to be localized to the IS. The second possible reason for discordant findings is the dynamic movement of proteins between the IS and OS. The protein composition of the IS and OS is under a constant flux with proteins being transported to and from the OS by vesicles and microtubules. For instance, the IFT protein complex constantly moves between the IS and OS along the CC, and is therefore possible to be detected in either the OS, IS, or even both at any given time (Rosenbaum and Witman 2002; Xu et al. 2015). Lastly, the animal models used to generate the three data sets are different. The protein expression profile of an animal can vary at different ages, and some proteins are not conserved through different species.
The methodology presented here illustrates the usefulness of MS-based proteomics for the elucidation of the protein components of a certain subcellular structure. A proteomic analysis at the subcellular level represents an analytical approach that combines classical biochemical fractionation methods and tools for the comprehensive identification of proteins in a high-throughput way. This strategy is capable of screening for both known and unknown proteins and can assign them to a particular subcellular structure. Protein localization information is usually determined either by microscopy or cell fractionation combined with protein blotting techniques, both of which are intrinsically low throughput and limited to known proteins. With the help of subcellular enrichment information from proteomic data, we can accurately provide a less biased, quantitative, and high-throughput approach for measuring the protein subcellular distribution. The profiling of rod-specific and cone-specific proteins also suggested that >95% of the photoreceptor cells in mice are rods, which is consistent with previous knowledge from a differential interference contrast optics study (Jeon et al. 1998).
Compared to cell-wide RNA and protein profiling, subcellular protein profiling and protein localization offers additional critical information about the functionality of the cell. In our study, we have created a highly accurate retinal disease gene prediction model by integrating protein OS enrichment information with gene expression levels. Although using mouse retina to study human disease could have some bias, fortunately, a vast majority of the known human retinal disease genes are also conserved in mice, both at the sequence and functional levels. With the prediction model, we were able to prioritize likely retinal disease genes, and our top predicted genes are well supported by both a literature search and animal model studies. Such analysis is highly valuable, particularly considering the challenges of rare genetic disease analysis in the next generation sequencing (NGS) era. The causes of inherited diseases can be genetically heterogeneous, and the mutations identified can be extremely rare. It is therefore unlikely that geneticists will observe the large number of patients needed to establish a strong genotype–phenotype correlation, and it is also not feasible to test every candidate gene with an animal model. Because of these challenges, it is of great importance to prioritize candidate disease genes before further validation. After a prioritized candidate gene list has been generated, these candidates can be screened for mutations in an NGS-based molecularly characterized patient cohort in a high-throughput fashion. Our approach is of wide general interest as it can be applied to other diseases. For example, the stereocilia are isolatable from the hair cells in the ear (Shepherd et al. 1989), and characterizing proteins in stereocilia can facilitate the discovery of hearing-loss disease genes. The integration of gene expression data with a subcellular enrichment profile in the affected tissue can provide a rich and valuable resource for candidate disease gene prioritization and ultimately lead to a better understanding of disease pathogenesis.
Methods
Experimental animals
All animals were handled according to NIH guidelines, and all experiments were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Wild-type C57BL/6J mice were obtained from the Jackson laboratory. In the OS proteomic experiments, three biological replicates were performed independently, which included 24, 26, and 26 retinas from mice at age postnatal day 15 (P15), respectively. We selected P15 as the optimal time point since most OSs are fully developed at this time (Zhang et al. 2011). We also performed the OS proteomic experiment on 40 retinas from mice at age P13. As no major differences were found between P13 and P15, we combined the results to increase the sensitivity of protein detection. For the RR proteomic experiments, two biological replicates were performed independently using 30 and 30 retinas, respectively.
OS isolation
Intact OSs of mouse retinas were isolated taking advantage of the fragility of the CC as previously described with modification (Papermaster and Dreyer 1974; Gilliam et al. 2012). Briefly, fresh mouse retinas were dissected and collected in sample buffer containing 24% (w/v) sucrose, 10 mM MOPS pH7.4, 30 mM NaCl, 60 mM KCl, and 2 mM MgCl2. The OS was isolated using mechanical shearing to break the CC and separated from the remaining retinal components via centrifugation at 4000 rpm for 1 min. Crude OS-containing solution was subjected to sucrose gradient ultracentrifugation, and the fraction containing purified OSs was collected. After the OS isolation, the remaining fractions were gathered as the RR and treated in the same way for downstream MS analysis. RHO and IFT20 were used as markers for the OS and the cilia, respectively.
SDS/PAGE and trypsin digestion
Thirty micrograms total protein was extracted from both the OS fraction and the RR. Extracted protein was resolved by SDS-PAGE, and the gel was sliced into 10 bands for in-gel trypsin digestion overnight as previously described (Malovannaya et al. 2010). Peptides were extracted twice with 100% acetonitrile and dried completely in a vacuum concentrator. MS experiments were performed with a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher) equipped with an HPLC system.
MS/MS data analysis
An in-house pipeline was used to identify peptides and map peptides to proteins. The database used for protein identification was the mouse reference sequence protein database (RefSeq) from NCBI (downloaded August 2013), and duplicate entries were combined before database searching. To enhance protein identification, sample-specific customized protein sequence databases were derived from in-house matched RNA-seq data as previously described (Ramakrishnan et al. 2009; Wang et al. 2012). The database searching results were combined. The maximum missed cleavage site was set to one and limited to trypsin cleavage sites. The precursor ion mass error tolerance was set to 20 ppm, and the fragment ion mass tolerance used was 0.5 Da. Both static and dynamic modifications were considered. The maximum variable PTMs per peptide was set to three. To optimize protein identification, we applied a very stringent filter at 0.1% PSM FDR and required a minimum of two distinct peptides for each protein.
Quantitative analyses and enrichment score calculation
We used the total number of MS/MS spectra taken on peptides, namely the absolute spectral count, as the basis for protein quantification. The spectral count shows linear correlation with protein concentration over a wide range (Liu et al. 2004), and the absolute spectral count quantification method is simple, practical, and has been extensively used in previous quantitative proteomic studies (Zhang et al. 2006; Li et al. 2010). The protein abundance was calculated by the absolute spectral counts of the protein Sabsolute, normalized by protein length L, and total spectra Stotal per million. For genes with multiple isoforms, we calculated protein abundance as the average abundance of all isoforms as follows:
QSpec (Choi et al. 2008) was used to calculate differential protein expression between OSs and the RR. The protein OS enrichment score was calculated as log2(OS/RR) using QSpec and represented as Enrichment (OS/RR).
Functional analysis of OS enriched and RR enriched groups
The OS-enriched and RR-enriched proteins were defined by the protein OS enrichment score and mapped to human homologs according to the Mouse Genome Informatics (MGI) database (Blake et al. 2014). Functional analysis was performed using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt) (Zhang et al. 2005; Wang et al. 2013). Functional annotations were based on Gene Ontology, including the biological process, molecular function, and cellular component. The statistical significance of functional categories was represented by an adjusted P-value with a cutoff of 0.05, and the top ten most significant functional categories were reported.
Literature mining for protein localization of retinal disease genes
To test the power of enrichment score for predicting protein localization, we performed literature mining and manually annotated the subcellular localization information of known retinal disease proteins. Briefly, relevant publications for all retinal disease genes were identified by searching literature databases. Solid evidence was required for protein localization, meaning the information was stated clearly in the paper or was inferred from high-quality immunostaining figures.
Machine learning for candidate retinal disease gene prioritization
To prioritize candidate retinal disease genes, we performed machine learning on all proteins identified in the retina (Supplemental Fig. S1). The per-gene attributes used for predictive model training included the normalized protein abundance in the OS and the RR, the protein OS enrichment score, and the RNA expression level in the mouse retina. The training sets included equal numbers of positive and negative controls. Positive controls were known retinal disease genes from RetNet (https://sph.uth.edu/retnet/) (Supplemental Table S11), and negative controls were randomly selected from genes that are known to cause other diseases without a retinal degenerative phenotype according to the Online Mendelian Inheritance in Man (OMIM) database (http://omim.org/) (Supplemental Table S12). In order to get robust results, we performed 10 rounds of training with different negative controls. Two machine learning classification algorithms, naïve Bayes and logistic regression, were used, and the final prediction score for a gene was taken as the average of the two methods across 10 different training sets. A fivefold cross-validation was used to estimate the sensitivity and specificity of the predictions. Furthermore, a list of 64 candidate retinal disease genes, which were never included in the positive training data set, was generated for true validation (Supplemental Table S10). These candidate genes either show a retinal degeneration phenotype in mutant mice based on the MGI database or have already been identified as candidate retinal disease genes in literature.
Data access
The proteomic data from this study have been submitted to the ProteomeXchange Consortium (Vizcaíno et al. 2014) via the PRIDE partner repository (http://www.ebi.ac.uk/pride/archive) under accession number PXD003441. The data are also available on the FTP site of HGSC (ftp://ftp.hgsc.bcm.edu/chen_retina_proteomics/).
Acknowledgments
This study is supported by grants from the Retinal Research Foundation, Foundation Fighting Blindness (BR-GE-0613-0618-BCM), and the National Eye Institute (R01EY022356, R01EY018571, R01EY020540).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.198911.115.
References
- Abe T, Kikuchi T, Shinohara T. 1994. The sequence of the human phosducin gene (PDC) and its 5′-flanking region. Genomics 19: 369–372. [DOI] [PubMed] [Google Scholar]
- Aebersold R, Mann M. 2003. Mass spectrometry-based proteomics. Nature 422: 198–207. [DOI] [PubMed] [Google Scholar]
- Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. 2003. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426: 570–574. [DOI] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE, Mouse Genome Database Group. 2014. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res 42(Database issue): D810–D817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Chen Y, Yi X, Wu R, Gan H, Shi Y, et al. 2012. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet 44: 972–974. [DOI] [PubMed] [Google Scholar]
- Choi H, Fermin D, Nesvizhskii AI. 2008. Significance analysis of spectral count data in label-free shotgun proteomics. Mol Cell Proteomics 7: 2373–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, Audo I, Mackay DS, Zeitz C, Borman AD, et al. 2012. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet 44: 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich U, Stöhr H, Hilfinger D, Loenhardt T, Schachner M, Langmann T, Weber BH. 2011. The Na/K-ATPase is obligatory for membrane anchorage of retinoschisin, the protein involved in the pathogenesis of X-linked juvenile retinoschisis. Hum Mol Genet 20: 1132–1142. [DOI] [PubMed] [Google Scholar]
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. 2012. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics 11: 1709–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam JC, Chang JT, Sandoval IM, Zhang Y, Li T, Pittler SJ, Chiu W, Wensel TG. 2012. Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 151: 1029–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H, Veleri S, Sun B, Kim DS, Keeley PW, Kim JW, Yang HJ, Yadav SP, Manjunath SH, Sood R, et al. 2014. Regulation of a novel isoform of Receptor Expression Enhancing Protein REEP6 in rod photoreceptors by bZIP transcription factor NRL. Hum Mol Genet 23: 4260–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinna C, Baye LM, Amsterdam A, Besharse JC, Link BA. 2010. Analysis of a zebrafish dync1h1 mutant reveals multiple functions for cytoplasmic dynein 1 during retinal photoreceptor development. Neural Dev 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon CJ, Strettoi E, Masland RH. 1998. The major cell populations of the mouse retina. J Neurosci 18: 8936–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez CR, Verheul HM. 2014. Mass spectrometry-based proteomics: from cancer biology to protein biomarkers, drug targets, and clinical applications. Am Soc Clin Oncol Educ Book: e504–e510. [DOI] [PubMed] [Google Scholar]
- Jones KA, Kim PD, Patel BB, Kelsen SG, Braverman A, Swinton DJ, Gafken PR, Jones LA, Lane WS, Neveu JM, et al. 2013. Immunodepletion plasma proteomics by tripleTOF 5600 and Orbitrap elite/LTQ-Orbitrap Velos/Q exactive mass spectrometers. J Proteome Res 12: 4351–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura E, Danciger M, Yamashita C, Rao NP, Nusinowitz S, Chang B, Farber DB. 2006. Disruption of the gene encoding the β1-subunit of transducin in the Rd4/+ mouse. Invest Ophthalmol Vis Sci 47: 1293–1301. [DOI] [PubMed] [Google Scholar]
- Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, Chen Y, Li Y, Fishman GA, Genead M, et al. 2012. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet 44: 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko I. 1994. Estimating attributes: analysis and extensions of RELIEF. Mach Learn 784: 171–182. [Google Scholar]
- Kwok MC, Holopainen JM, Molday LL, Foster LJ, Molday RS. 2008. Proteomics of photoreceptor outer segments identifies a subset of SNARE and Rab proteins implicated in membrane vesicle trafficking and fusion. Mol Cell Proteomics 7: 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gray W, Zhang H, Chung CH, Billheimer D, Yarbrough WG, Liebler DC, Shyr Y, Slebos RJ. 2010. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res 9: 4295–4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Sadygov RG, Yates JR III. 2004. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 76: 4193–4201. [DOI] [PubMed] [Google Scholar]
- Liu Q, Tan G, Levenkova N, Li T, Pugh EN Jr, Rux JJ, Speicher DW, Pierce EA. 2007. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics 6: 1299–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanova ES, Finkelstein S, Herrmann R, Chen YM, Kessler C, Michaud NA, Trieu LH, Strissel KJ, Burns ME, Arshavsky VY. 2008. Transducin γ-subunit sets expression levels of α- and β-subunits and is crucial for rod viability. J Neurosci 28: 3510–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo N, Lu J, Sun Y. 2012. Evidence of a role of inositol polyphosphate 5-phosphatase INPP5E in cilia formation in zebrafish. Vision Res 75: 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick P, Kuster B. 2010. Proteomics: a pragmatic perspective. Nat Biotechnol 28: 695–709. [DOI] [PubMed] [Google Scholar]
- Malovannaya A, Li Y, Bulynko Y, Jung SY, Wang Y, Lanz RB, O'Malley BW, Qin J. 2010. Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc Natl Acad Sci 107: 2431–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski A, Damoc E, Hauschild JP, Lange O, Wieghaus A, Makarov A, Nagaraj N, Cox J, Mann M, Horning S. 2011. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol Cell Proteomics 10: M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlhans J, Brandstätter JH, Gießl A. 2011. The centrosomal protein pericentrin identified at the basal body complex of the connecting cilium in mouse photoreceptors. PLoS One 6: e26496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafi D, Engel AH, Palczewski K. 2009. Structure of cone photoreceptors. Prog Retin Eye Res 28: 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Katoh K, Sato S, Muranishi Y, Chaya T, Onishi A, Minami T, Fujikado T, Furukawa T. 2011. Analysis of transcriptional regulatory pathways of photoreceptor genes by expression profiling of the Otx2-deficient retina. PLoS One 6: e19685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto EA, Hurd TW, Airik R, Chaki M, Zhou W, Stoetzel C, Patil SB, Levy S, Ghosh AK, Murga-Zamalloa CA, et al. 2010. Candidate exome capture identifies mutation of SDCCAG8 as the cause of a retinal-renal ciliopathy. Nat Genet 42: 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panfoli I, Calzia D, Ravera S, Morelli AM, Traverso CE. 2012. Extra-mitochondrial aerobic metabolism in retinal rod outer segments: new perspectives in retinopathies. Med Hypotheses 78: 423–427. [DOI] [PubMed] [Google Scholar]
- Panfoli I, Calzia D, Bruschi M, Oneto M, Bianchini P, Ravera S, Petretto A, Diaspro A, Candiano G. 2013. Functional expression of oxidative phosphorylation proteins in the rod outer segment disc. Cell Biochem Funct 31: 532–538. [DOI] [PubMed] [Google Scholar]
- Papermaster DS, Dreyer WJ. 1974. Rhodopsin content in the outer segment membranes of bovine and frog retinal rods. Biochemistry 13: 2438–2444. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan SR, Vogel C, Prince JT, Li Z, Penalva LO, Myers M, Marcotte EM, Miranker DP, Wang R. 2009. Integrating shotgun proteomics and mRNA expression data to improve protein identification. Bioinformatics 25: 1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, Ruta R, Mancini F, Mazza T, Castellana S, et al. 2014. Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis 9: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. 2002. Intraflagellar transport. Nat Rev Mol Cell Biol 3: 813–825. [DOI] [PubMed] [Google Scholar]
- Sadowski PG, Dunkley TP, Shadforth IP, Dupree P, Bessant C, Griffin JL, Lilley KS. 2006. Quantitative proteomic approach to study subcellular localization of membrane proteins. Nat Protoc 1: 1778–1789. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Barres BA, Corey DP. 1989. “Bundle blot” purification and initial protein characterization of hair cell stereocilia. Proc Natl Acad Sci 86: 4973–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tummala P, Mali RS, Guzman E, Zhang X, Mitton KP. 2010. Temporal ChIP-on-Chip of RNA-Polymerase-II to detect novel gene activation events during photoreceptor maturation. Mol Vis 16: 252–271. [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, et al. 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol 32: 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Slebos RJ, Wang D, Halvey PJ, Tabb DL, Liebler DC, Zhang B. 2012. Protein identification using customized protein sequence databases derived from RNA-Seq data. J Proteome Res 11: 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Duncan D, Shi Z, Zhang B. 2013. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res 41(Web Server issue): W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Ladak R, Swanson DA, Soltyk A, Sun H, Ploder L, Vidgen D, Duncan AM, Garami E, Valle D, et al. 1999. PHR1 encodes an abundant, pleckstrin homology domain-containing integral membrane protein in the photoreceptor outer segments. J Biol Chem 274: 35676–35685. [DOI] [PubMed] [Google Scholar]
- Xu M, Yang L, Wang F, Li H, Wang X, Wang W, Ge Z, Wang K, Zhao L, Li H, et al. 2015. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum Genet 134: 1069–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. 2005. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33(Web Server issue): W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. 2006. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res 5: 2909–2918. [DOI] [PubMed] [Google Scholar]
- Zhang X, Serb JM, Greenlee MH. 2011. Mouse retinal development: a dark horse model for systems biology research. Bioinform Biol Insights 5: 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]