

Abstract
RNA polymerase III (Pol III) is tightly controlled in response to environmental cues, yet a genomic-scale picture of Pol III regulation and the role played by its repressor MAF1 is lacking. Here, we describe genome-wide studies in human fibroblasts that reveal a dynamic and gene-specific adaptation of Pol III recruitment to extracellular signals in an mTORC1-dependent manner. Repression of Pol III recruitment and transcription are tightly linked to MAF1, which selectively localizes at Pol III loci, even under serum-replete conditions, and increasingly targets transcribing Pol III in response to serum starvation. Combining Pol III binding profiles with EU-labeling and high-throughput sequencing of newly synthesized small RNAs, we show that Pol III occupancy closely reflects ongoing transcription. Our results exclude the long-term, unproductive arrest of Pol III on the DNA as a major regulatory mechanism and identify previously uncharacterized, differential coordination in Pol III binding and transcription under different growth conditions.
Cell survival and proliferation are largely controlled by the RAS/MAPK and PI3K/mTORC1 pathways, which couple resource-demanding processes such as de novo synthesis of proteins, lipids, and nucleotides to nutrient availability. Regulation of these signaling pathways ensures cell growth in response to amino acid levels, growth factors, and energy status, as well as cell survival during periods of starvation (Mendoza et al. 2011). So far, many studies investigating the control of protein synthesis through the PI3K/mTORC1 pathway have focused on the ability of mTORC1 to coordinate translation initiation through S6K and 4E-BP (Shimobayashi and Hall 2014). However, mTORC1 regulation of RNA polymerase II (Pol II) and III (Pol III) transcription has recently emerged as a new mechanism to adapt cellular responsiveness to extra- and intracellular cues (Laplante and Sabatini 2013; Grewal 2015).
Reflecting the highly diversified gene-expression programs needed to effect development and cellular adaptation to external conditions, Pol II transcription is regulated in large part on a gene per gene basis, with each gene harboring a promoter structure specifying Pol II recruitment under certain conditions. In addition, Pol II is regulated at the level of transcription elongation; it is found in a “poised” or “paused” state at the transcription start sites (TSSs) of a large number of genes, from which it can rapidly escape into productive elongation, affording the cell with a mechanism for a very rapid and synchronous response to changing conditions (Adelman and Lis 2012). Compared with Pol II, RNA Pol III recognizes a limited number of promoter structures, which can be roughly classified as type I, present in the 5S rRNA genes (RNA5S); type II, present in tRNA genes and SINEs; and type III, present in a small collection of genes with different functions, including the U6 snRNA (RNU6) and 7SK RNA (RN7SK) genes (Orioli et al. 2012). This limited number of promoter structures is thought to reflect a much simpler regulation, with the enzyme responding mostly uniformly to growth, proliferation, or stress cues (Harismendy et al. 2003). Indeed, elevated tRNA levels are a hallmark of many tumors (White et al. 1989; Pavon-Eternod et al. 2009; Zhou et al. 2009) and, in Drosophila, drive larval development and increase in body size (Marshall et al. 2012; Rideout et al. 2012). However, there is increasing evidence that the developmental and proliferative states of a cell are affected not only by global tRNA levels but also by the balance between the supply of different tRNA isoacceptors and the decoding requirements of the mRNAs expressed in a certain tissue or developmental state (Gingold et al. 2014; Schmitt et al. 2014). These studies point to a more subtle regulation of Pol III transcription and, in particular, tRNA synthesis, than suggested by the relatively uniform structure of Pol III promoters.
While several factors contribute to the regulation of Pol III transcription, transition from a permissive to a restrictive transcriptional state and vice versa is largely controlled by a highly conserved Pol III repressor called Maf1 (Pluta et al. 2001). In Saccharomyces cerevisiae, Maf1 can be inactivated by the TORC1-controlled kinase Sch9 (Boguta and Graczyk 2011) and other kinases (Moir et al. 2006) in response to nutrient availability and energy status. Upon TORC1 inhibition by nutrient deprivation or rapamycin treatment, Pol III transcription is repressed in a Maf1-dependent manner, and this parallels with Maf1 dephosphorylation and association with Pol III genes (Oficjalska-Pham et al. 2006; Roberts et al. 2006). In mammalian cells, MAF1 is directly phosphorylated by mTORC1 (Michels et al. 2010; Shor et al. 2010) and becomes hypophosphorylated in response to stress or nutrient withdrawal, which correlates with transcriptional repression of RNU6, RNA5S, and some tRNA genes (Reina et al. 2006; Rollins et al. 2007; Goodfellow et al. 2008; Rohira et al. 2013). Since hypophosphorylated MAF1 binds to Pol III (Reina et al. 2006), and the physical interaction of MAF1 with recombinant Pol III prevents closed complex formation and initiation from target gene promoters in vitro (Vannini et al. 2010), MAF1 is considered as a general repressor of Pol III transcription. Nevertheless, the full extent of transcriptional repression at different subsets of genes, including the metazoan-specific type III promoter genes, remains elusive.
Mammalian MAF1 has also been reported to repress Pol II transcription by specifically associating with the promoter of certain mRNA genes, including TBP, FASN, and EGR1 (Johnson et al. 2007; Palian et al. 2014). Targeting of these genes impacts different signaling pathways but can also affect the transcriptional readout of other polymerases; indeed, down-regulation of TBP, an essential component of the Pol I promoter selectivity factor (SL1) and the Pol III–recruiting factor TFIIIB, has been proposed to indirectly affect transcription of both Pol I and Pol III genes. However, since genome-wide binding profiles for mammalian MAF1 have not been reported, the full extent of MAF1-dependent regulation of protein-coding genes remains to be addressed.
Here, we have developed and applied high-throughput strategies to understand the mechanisms responsible for the adaptation of Pol III transcription to prosurvival and stress cues, as well as the role played by MAF1 in such modulation.
Results
Pol III recruitment adapts heterogeneously to environmental cues
To understand the dynamics of Pol III regulation by metabolic pathways, we analyzed IMR90hTert cells grown in the presence (FBS) or absence (SS) of serum, as well as serum-starved cells restimulated with insulin alone (SS + I), or following addition of rapamycin (SS + R + I), a highly specific inhibitor of mTORC1 (Fig. 1A; Yip et al. 2010). Whereas serum starvation led to dephosphorylation of RPS6S235/236, AKTS473, and MAPK3/MAPK1(ERK1/ERK2)T202/Y204, treatment with insulin resulted in robust rephosphorylation of these three proteins (Fig. 1B, lanes 1–3), suggesting reactivation of both the RAS/MAPK and PI3K/mTORC1 pathways. However, in the presence of rapamycin, insulin specifically failed to trigger phosphorylation of RPS6S235/236, indicating suppression of downstream mTORC1 activity, but not RAS/MAPK signaling, under these conditions (Fig. 1B, lane 4). Consistent with previous studies identifying MAF1 as a substrate of mTORC1 (Michels et al. 2010; Shor et al. 2010), MAF1 was dephosphorylated as a result of serum deprivation and rapidly rephosphorylated after insulin stimulation unless the kinase activity of mTOR was inhibited by rapamycin (Fig. 1B).
Figure 1.

Genome-wide analysis of Pol III recruitment reveals differential occupancy profiles under prosurvival and stress conditions. (A,B) IMR90hTert cells were grown in the presence (FBS) or absence (serum-starved, SS) of fetal bovine serum for 8 h. We then added 1 µM insulin to starved cells alone (SS + I) or after treatment with 2 nM rapamycin (SS + R + I), for the indicated times. Whole-cell extracts were subsequently analyzed by immunoblotting for total levels and phosphorylation status of the indicated proteins. The MAF1 hyperphosphorylated forms are indicated by arrows. (C) Pie chart representation of 715 Pol III genes either unbound (226) or bound (489) to polymerase in at least one of the four conditions (FBS, SS, SS + I, or SS + R + I). (D) Pol III occupancy as measured by ChIP-seq, shown for a cluster of tRNA genes on Chromosome 6. Chromatin was prepared from IMR90hTert cells grown as described in A and immunoprecipitated with an antibody against the POLR3D (RPC4) subunit of Pol III. Input DNA signal is shown. Gray shadings indicate nonoccupied genes. tRNA gene names are according to HGNC-approved gene nomenclature (“TR” in front of names is omitted for space constraints). (E,F) MA-plots showing distributions of Pol III occupancy scores [log2 (IP/input)] for 715 loci. In each panel, two comparisons are superimposed: SS versus FBS (blue) and SS + I versus FBS (yellow) in E; SS versus FBS (blue) and SS + R + I versus FBS (pink) in F. Gray dots left of the dotted lines (indicating the cutoff threshold score) represent nonoccupied genes. Small dots are genes that do not change significantly (P > 0.005) in each pairwise comparison. The horizontal line indicates the zero-fold change.
To assess adaptation of Pol III recruitment to prosurvival and stress stimuli, we determined genome-wide Pol III occupancy under the different growth conditions described in Figure 1A using an antibody against the Pol III subunit POLR3D (RPC4) (Canella et al. 2010). After validation of Pol III binding at both type II (TRI-TAT2-1) and type III (RNU6-1) promoter genes by locus-specific ChIP-qPCR (Supplemental Fig. S1A), we performed Pol III ChIP-seq in IMR90hTert cells. From a set of 110–170 million aligned reads per condition (Supplemental Table S1, column I), we generated Pol III enrichment profiles at a compilation of 622 tRNA genes (Chan and Lowe 2009), as well as other previously identified (Renaud et al. 2014) loci. ChIP-seq scores for Pol III enrichment over a window extending 150 bp upstream of and downstream from the gene body were calculated as the mean log2 (IP/input) of two experiments per condition. The analysis identified 489 Pol III–bound loci in actively dividing cells (FDR 0.1%; see Methods), including tRNA genes, 5S rRNA (RNA5S) genes, and type III promoter genes such as RNU6, RN7SK, RPPH1, and others (Supplemental Table S2). Consistent with previous reports (Canella et al. 2010; Moqtaderi et al. 2010; Oler et al. 2010), Pol III was found only at 68% of annotated Pol III genes (Fig. 1C); an 80-kb region on Chromosome 6 is shown in Figure 1D as an example, where three tRNA genes with no significant binding (gray shading) are interspersed with Pol III–occupied loci.
Comparing Pol III occupancy scores with a DNase I hypersensitive site (DHS) (Thurman et al. 2012) and histone modification (Hawkins et al. 2010) data sets generated in the same cell type revealed that unoccupied genes display lower chromatin accessibility and are generally devoid of open chromatin marks (H3K4me3, H3K9ac, H3K27ac, H4K20me1) compared with their highly occupied counterpart (Supplemental Fig. S1B,C), whereas no striking difference was observed for repressive marks (H3K27me3, H3K9me3). Similarly to previous results in mouse liver cells (Canella et al. 2012), highly occupied and unoccupied genes displayed distinct promoter sequences (Supplemental Fig. S1D). While we cannot establish a cause and effect relationship, these analyses and the observation that Pol III is absent from 32% of Pol III genes in all tested conditions (Fig. 1C) suggest that inactive genes are in a lasting repressed state as a result of genetic and epigenetic features.
The mTORC1 target MAF1 controls Pol III recruitment genome-wide
Unlike unoccupied genes, the large majority of Pol III–bound genes displayed metabolic adaptation of Pol III recruitment, as illustrated by MA-plots comparing ChIP-seq score distribution among different growth conditions. Serum withdrawal resulted in decreased (P < 0.005) occupancy at ∼90% of the 489 genes identified above as Pol III bound, which was rapidly rescued, though not to the starting level, 90 min after insulin stimulation (Fig. 1E). Importantly, this recovery was not observed in cells previously treated with rapamycin (Fig. 1F), suggesting that, in IMR90hTert cells, insulin control of Pol III recruitment requires mTORC1.
To determine the role of human MAF1 in the regulation of Pol III recruitment, we performed Pol III ChIP-seq assays in serum-starved IMR90hTert cells transfected with a nontarget siRNA or two different MAF1-specific siRNAs (#1 and #2). Efficient knockdown of MAF1 by either siRNA (Fig. 2A) resulted in significantly higher enrichment of Pol III at occupied loci versus the control (Fig. 2B). Nonoccupied loci (Fig. 2B, gray) were mostly unaffected by MAF1 depletion, further emphasizing that these loci are lastingly repressed in this cell type. Taken together, these data show that MAF1 reduces genome-wide Pol III occupancy under serum-starved conditions and that this decrease correlates with the MAF1 phosphorylation state (see Fig. 1B).
Figure 2.
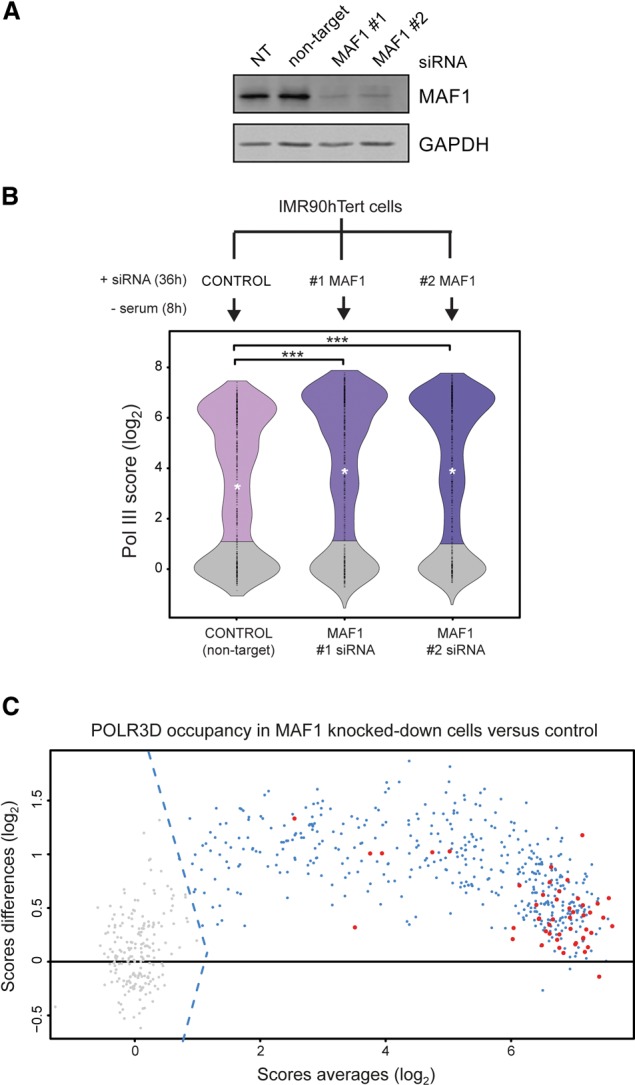
Pol III recruitment is controlled genome-wide by MAF1 in an mTORC1-dependent manner. (A) Depletion of MAF1 by RNAi. Immunoblots of IMR90hTert cells that were not transfected (NT) or were transfected with a control (nontarget) siRNA or with two MAF1-specific siRNAs (#1 and #2), and serum-starved for 8 h. (B) Violin plots showing distributions of Pol III ChIP-seq scores from IMR90hTert cells transfected as described in A. Nonoccupied genes are in gray. The median is in each case represented by a white asterisk. Data were analyzed with Wilcoxon pairs tests. (***) P < 0.001. (C) MA-plot comparing Pol III occupancy scores in MAF1 knockdown cells (mean of #1 and #2 MAF1 siRNAs) versus the control (#NEG siRNA). Gray dots left of the dotted lines (indicating the cutoff threshold score) represent nonoccupied genes. The horizontal line indicates the zero-fold change. Red dots indicate stable genes as defined in Pol III ChIP-seq experiments.
Although Pol III recruitment showed metabolic adaptation at the majority of sites, it did not significantly change at 49 active loci (see Pol III gene stability sheet in Supplemental Table S2), which we therefore refer to as “stable” in terms of Pol III occupancy after serum starvation (Fig. 1E,F, small blue dots). These genes included representatives of most tRNA isotypes and type III promoter genes and showed only moderate sensitivity to MAF1 knockdown (Fig. 2C, red dots), indicating that both environmental cues and MAF1 have little effect on their levels of resident Pol III. While this observation might reflect productive transcription from a subset of genes to sustain basal cellular activities during periods of starvation, it also raised the possibility that Pol III could occupy a gene in an unproductive state.
Long-term, unproductive arrest of Pol III on the DNA can be generally excluded in human fibroblasts
To address the activity of DNA-bound Pol III in serum-starved cells, we optimized 5-ethynyl uridine (EU) metabolic labeling for selective high-throughput quantification of newly synthesized EU-labeled small RNAs (Fig. 3A, neusRNA-seq). Cells grown under normal or stress conditions were provided for 1–8 h with EU, which was incorporated into nascent Pol III RNAs in a time-dependent manner, as shown by Northern blotting (Fig. 3B). EU-labeled RNAs arising from type II (tRNAs) and type III (RNY3) promoter genes were clearly detected after 4 h in normally growing cells (Fig. 3C, lanes 1–4), and consistent with repression of Pol III recruitment upon serum starvation, expression of EU-labeled RNAs was diminished in the absence of serum (Fig. 3C, cf. lanes 4 and 8). Importantly, treatment with actinomycin D, an inhibitor of transcription, abolished EU labeling (Fig. 3C, lanes 9–12), confirming that the nucleotide analog was incorporated transcriptionally. Given these results, we prepared neusRNA-seq libraries from three biological replicates of serum-fed and serum-starved IMR90hTert cells, either metabolically labeled with EU or mock-labeled with DMSO for 5 h. After size selection of <400-nt RNAs, EU was biotinylated with click chemistry for selective enrichment of pulse-labeled RNAs on streptavidin beads. A dsDNA library was then generated from the RNA on beads and analyzed by single-end sequencing on a HiSeq Illumina platform. Spearman correlation of independent replicates revealed high reproducibility of the data (r ≥ 0.98) (Supplemental Fig. S2A), and expression levels were calculated as the mean score of three EU-labeled samples normalized to the DMSO controls (neusRNA-seq scores) (Supplemental Table S2). Consistent with transcriptional repression under stress conditions, we observed a significant decrease (Student's t-test, P < 2 × 10−16) in neusRNA-seq scores after serum starvation (Fig. 3D) and a correlation with ChIP-seq scores in both conditions (r = 0.52) (Fig. 3E). Strikingly, Pol III genes were particularly depleted in the high ChIP-seq and low neusRNA-seq scores region (bottom right corner of the graphs in Fig. 3E), indicating that the presence of unproductive, arrested Pol III can be excluded at the large majority of loci, including those with stable (Fig. 3E, red dots) Pol III occupancy in starved cells.
Figure 3.

neusRNA-seq reveals correlation between Pol III enrichment and expression during stress conditions. (A) Outline of the neusRNA-seq protocol. (B,C) Northern blot analysis showing time-dependent, cotranscriptional incorporation of 5-ethynyl uridine (EU) into Pol III–transcribed RNAs. Probes identify EU-labeled RNAs from the RN7SL1 and RNY3 genes or multiple tRNAs from a collection of proline (tRNA-Pro), aspartate (tRNA-Asp), and valine (tRNA-Val) isoacceptor tRNA genes. In B, DMSO (8 h) or 0.25 mM EU was added to cell media for the indicated times. In C, RNA from IMR90hTert cells grown in the presence or absence of serum (5 h) was pulse labeled with 0.25 mM EU for 5 h. 5 μg/mL actinomycin D (actD) was added where noted. (I) Twenty percent input RNA; (AP) streptavidin bead affinity purified EU-labeled RNAs. (D) Scatterplot comparing levels of EU-labeled Pol III–transcribed RNAs in different growth conditions (+ serum vs. −serum). (E) Scatterplots showing correlation of ChIP-seq and neusRNA-seq scores at Pol III–enriched loci for both serum-grown (upper panel) and serum-starved (lower panel) IMR90hTert cells. Red dots indicate genes defined as “stable” in Pol III ChIP-seq experiments. The Pearson correlation coefficient (r) is given. (F) Heatmaps showing correlation of Pol III binding (left) with gene transcription levels measured by neusRNA-seq (right). Pol III genes are split by promoter type and ranked by Pol III occupancy scores in the FBS sample. The heatmaps show sequencing read intensity centered on the annotated 5′ of genes (which for tRNA genes correspond to the 5′-end of the mature tRNA) and extending ±1.5 kb for ChIP-seq or −0.2/+0.5 kb for neusRNA-seq. Negative scores are set to zero.
Pol III occupancy correlates with unprocessed RNA levels
Alignment of tags relative to the annotated 5′-ends of Pol III genes further confirmed a general overlap between ChIP-seq and RNA-seq signals (Fig. 3F) and also revealed density of reads in the regions upstream of and downstream from gene bodies. This reflects map-ability of both mature and unprocessed neusRNAs still containing precursor sequences extending past the annotated mature ends (Phizicky and Hopper 2010). To distinguish between unprocessed and processed tRNAs, we separated neusRNA-seq reads uniquely assigned to pre-tRNAs (i.e., containing intron, 5′-leader, or 3′-trailer sequences) from reads corresponding to mature tRNAs (i.e., ending exactly at annotated 5′- or 3′-ends or spanning exon–exon junctions). The most significant correlation with ChIP-seq scores was found for unprocessed tRNA reads, consistent with the idea that quantification of pre-tRNAs closely reflects ongoing transcription (Supplemental Fig. S2B). This was confirmed by Northern analysis of unlabeled tRNAs in serum-fed and serum-starved cells; while mature tRNAs levels showed little to no change after 8 h of starvation, most pre-tRNAs levels were reduced (Supplemental Fig. S2C, middle and lower panels), in line with the ChIP-seq and neusRNA-seq signals under the same conditions (Supplemental Fig. S2C, upper panels). Strikingly, one of the genes showing stable Pol III occupancy in the ChIP-seq experiments (TRY-GTA2-1) also displayed stable expression of the corresponding pre-tRNA. Taken together, these results show that Pol III enrichment is a very good proxy of ongoing Pol III transcription, and generally exclude the presence of DNA-bound but arrested, unproductive Pol III in IMR90hTert cells.
MAF1 preferentially localizes at Pol III–enriched loci across the genome
Our results show that reduced Pol III occupancy in serum-starved IMR90hTert cells is dependent on MAF1. Previous work in yeast has indicated that Maf1 specifically associates genome-wide with Pol III genes under repressive conditions (Roberts et al. 2006), but no such studies have been performed in mammalian cells. Indeed, our own attempts at defining MAF1 binding sites across the genome by ChIP-seq were unsuccessful (data not shown). We reasoned that since MAF1 alone cannot bind nucleic acids (Desai et al. 2005) and since binding of MAF1 to Pol III prevents its recruitment to the preinitiation complex in vitro (Vannini et al. 2010), any interaction between MAF1 and Pol III–transcribed genes might be highly transient in nature. We therefore decided to use DamIP-seq (Xiao et al. 2012), a technique that, similarly to DamID-seq (Luo et al. 2011), relies on the fusion of a potential chromatin binding protein to the Eserichia coli DNA adenine methyltransferase (Dam). When expressed in vivo, the Dam moiety is tethered to its partner's DNA binding site, where it methylates N6-adenosine within the “GATC” recognition motif, leaving a covalent and unique mark that is normally absent in eukaryotic DNA (van Steensel et al. 2001; Greil et al. 2006). Instead of using wild-type Dam, we fused our proteins of interest to DamK9A, a mutant with reduced enzymatic activity and looser recognition requirements (Horton et al. 2006). DamK9A can methylate the first nucleotide of the more frequent “ATC” target site, which is present within 150 bp of 98% of Pol III genes.
We generated IMR90hTert stable cell lines expressing HA-tagged MAF1-DamK9A and EGFP-DamK9A under the control of the weak MoMuLV promoter and validated the expression of full-length chimeric proteins by RT-PCR and immunoblotting of both whole-cell extracts and α-HA immunoprecipitates (Fig. 4A,B). The EGFP-DamK9A control, for which nuclear enrichment was confirmed by fluorescence microscopy (Supplemental Fig. S3A), is essential to normalize for local chromatin accessibility and stochastic DNA methylation. We first assessed methylation enrichment over the control by measuring differential sensitivity to DpnII or Sau3AI digestion at several Pol III loci. Genomic DNA (gDNA) from serum-starved MAF1-DamK9A and EGFP-DamK9A–expressing cell lines was digested with either enzyme and analyzed by qPCR with tDNA-specific primers bracketing a DpnII/Sau3AI site. Since adenomethylated GATC is protected from digestion by DpnII but not by Sau3AI, which cleaves its GATC target regardless of methylation, an amplification product is expected only at methylated loci after DpnII digestion (Fig. 4C, upper panel). The assay revealed significantly higher methylation at Pol III–bound loci (TRL-TAA1-1, TRR-TCG5-1, and TRF-GAA1-6) for both MAF1-DamK9A clones over the EGFP-DamK9A controls; notably, a higher signal in the clone (MAF1-A) expressing higher levels of MAF1-DamK9A was detected, as expected for MAF1-driven tethering of DamK9A at these genes (Fig. 4C, lower panel, cf. MAF1-A and MAF1-B clones). Interestingly, no difference between the controls and MAF1-DamK9A–expressing cells was observed at a Pol III–unbound locus (TRN-GTT20-1), suggesting that Pol III is required to recruit MAF1 at tRNA genes.
Figure 4.

DamIP-seq shows MAF1 recruitment at Pol III–bound genes. (A) Schematic representation of MAF1-DamK9A and EGFP-DamK9A chimeric constructs depicting the position of PCR primers. Two stable IMR90hTert clonal cell lines expressing each chimera (MAF1-A, MAF1-B and EGFP-A, EGFP-B) were generated, and mRNA expression was validated via RT-PCR. The left and right blot panels show reactions containing (+RT) or not containing (−RT) reverse transcriptase. (B) Western blotting of whole-cell extracts or α-HA immunoprecipitates from the cell lines described in A. (C) Genomic DNA from MAF1-DamK9A–expressing and EGFP-DamK9A–expressing IMR90hTert cell lines was extracted after 8 h of serum starvation, digested with either DpnII or Sau3A, and analyzed by qPCR with tDNA-specific primers (upper panel). The qPCR signal (lower panel) was normalized against an intergenic region on Chr21 and is shown as DpnII versus Sau3A ratio in arbitrary units (A.U.). Data are mean ± SD (n = 3). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001 (as calculated by Student's t-test). PCR primers and tDNA HUGO nomenclature are indicated in Supplemental Table S4. (D) Score versus average plots showing MAF1 association within 400-bp bins across the genome. The DamIP-seq score, defined as the log2 (MAF1tag count/EGFPtag count), is plotted on the x-axis, and the log2 of the mean MAF1 and EGFP DamIP-seq normalized tag counts is plotted on the y-axis, in each case for nonoverlapping 400-bp bins across the genome. The dotted line indicates the cutoff, set at the lowest score giving an FDR < 0.005. Pol III loci–containing bins are represented as red or blue triangles. (E) Meta-sample profiles depicting sequence coverage in a ±2-kb window surrounding the annotated 5′-ends of isolated (i.e., separated from each other by at least 2 kb) tRNA genes, type III promoter genes, and gene-free regions. MAF1 DamIP-seq scores per nucleotide are normalized for total tag count and calculated as the log2 (MAF1/EGFP). Signal for the +serum and −serum experiments is indicated in orange and blue, respectively. In the left panel, the dotted lines show Pol III–unbound tDNAs; the solid lines, Pol III–bound tDNAs. (F) Overlap between MAF1 (blue dots) and Pol III associated loci (boxes) in serum-starved IMR90hTert cells. Genes are ranked by Pol III SS ChIP-seq scores and split into Pol III unbound, bound-stable, and bound-unstable classes according to FBS versus SS ChIP-seq fold-change (Fig. 1C, blue). Only genes having at least one ATC within the gene body are considered.
To extend the analysis of MAF1 target sites to the genomic scale, we performed DamIP-seq with MAF1-DamK9A and EGFP-DamK9A clones. gDNA from cells grown in complete or serum-deprived medium was sonicated, denatured, and immunoprecipitated with an α-N6-methyladenosine antibody. Following second-strand synthesis, enriched methylated DNA was used for library preparation and single-end sequencing with the HiSeq Illumina platform. To reduce the effects of clone-to-clone variability and to increase signal, a single meta-sample was generated by pooling together four data sets per condition (biological replicates of MAF1-A + MAF1-B and of EGFP-A + EGFP-B). The analysis identified about 1500 (in serum-starved cells) and 8000 (in serum-grown cells) 400-bp bins across the genome having a positive score and a FDR < 0.005 (see Methods; Supplemental Table S3). However, the large majority of bins had low scores and were positioned within, or in proximity of, repeat elements. We also observed low levels of MAF1-DamK9A methylation at 264 Pol II genes (Supplemental Table S3), but we did not identify enrichment at specific features such as TSSs or promoters (Supplemental Fig. S3B). Much higher scores were observed for bins overlapping with Pol III genes (Fig. 4D, triangles), indicating preferential binding of MAF1 to these loci. Tag profiling around the TSS of “isolated” tRNA genes, i.e., tDNAs separated from one another by at least 2 kb, and of type III promoter genes further revealed the accumulation of reads in proximity of the gene body compared with the genomic surroundings or with random intergenic regions (Fig. 4E). MAF1 interaction was mainly observed at Pol III–bound tRNA genes, whereas only a weak association was seen at Pol III–unbound tRNA genes. This distribution appeared not to be biased by the relative ATC content, as many Pol III genes displayed low scores but high ATC frequencies (Supplemental Fig. S3C). Overall, higher MAF1 scores were detected upon serum starvation at both type II and type III promoter genes, consistent with enhanced MAF1 recruitment at Pol III loci under starvation conditions (Fig. 4E, left and middle panel).
Given the preferential binding of MAF1 to Pol III loci, we compared Pol III chromatin association profiles generated by ChIP-seq with those of MAF1 obtained by DamIP-seq. We calculated MAF1 DamIP-seq scores for Pol III genes and identified 64 loci with highly significant (P < 0.005) MAF1 enrichment and at least one ATC within the gene body; of these loci, 52 were bound in serum-starved cells (and some of them, also in serum-replete cells), and the remaining 12 exclusively in serum-replete cells (note that only one locus, a tRNA pseudogene, had high MAF1 score but no ATC) (Supplemental Table S3). We then compared the 52 MAF1 targets in serum-starved cells with genes previously defined as “unbound,” “stable,” or “unstable” based on the different Pol III occupancy responses to serum starvation. Strikingly, MAF1 was found to bind exclusively to Pol III–occupied loci and to preferentially localize to genes with high Pol III occupancy scores (Fig. 4F) located in a genetic environment enriched in open chromatin marks (Supplemental Fig. S3D). Most MAF1-bound genes were unstable, but we also observed MAF1 binding to some stable genes, with a similar proportion of MAF1-bound genes in both categories (14% of unstable genes and 10% of stable genes) (Fig. 4F).
Upon close examination of the MAF1-bound genes, we noticed that 91% of them contained a GATC sequence, suggesting that DamK9A may still retain a preference for a G in the first position upstream of the ATC motif, a possibility raised previously (Xiao et al. 2012). Since only 32% of the remaining Pol III–occupied genes (with no detectable MAF1) contain a GATC, a number of binding events might have been lost due to the lesser availability of potential Dam sites. This possibility, however, does not affect the conclusions that (1) MAF1 preferentially binds to Pol III–occupied genes, as we have no example of MAF1 binding to Pol III–devoid loci, and (2) the proportions of MAF1-bound genes are similar in the stable and unstable categories, as they remain similar even if we consider only GATC-containing genes (25% of unstable and 30% of stable). Taken together, these results show enhanced association of MAF1 to a subset of Pol III–bound genes in response to serum starvation.
Discussion
An unexpectedly nuanced program for regulation of Pol III recruitment in response to environmental cues
In yeast, Pol III occupancy is uniformly reduced in stationary phase compared with exponentially growing cells (Harismendy et al. 2003). The present work reveals that in mammalian cells, individual Pol III loci show a broad range of adaptation to growth conditions; this is far from the uniform response one might expect from the action of MAF1, a protein targeting the polymerase rather than individual promoters. Our ChIP-seq analyses in IMR90hTert cells reveal that about one-third of Pol III loci tend to have poor promoters and are generally devoid of histone marks associated with actively transcribed chromatin. These genes do not bind Pol III in any of the growth conditions tested, consistent with persistent silencing. The remaining genes bind Pol III but can be divided into two broad classes that differ greatly in their response to environmental cues. Class I (unstable genes) is the largest and contains genes whose Pol III occupancy varies according to cell growth conditions, being higher in favorable conditions (FBS and SS + I) and much lower in stress conditions (SS, SS + R + I). Exposure of serum-starved cells to insulin causes a general increase in Pol III occupancy on these genes, and this effect is inhibited in the presence of rapamycin, revealing that insulin regulation of mammalian Pol III occupancy is strictly dependent on mTORC1 activity. Class II (stable genes) includes 49 genes with generally high Pol III occupancy scores that do not vary significantly upon serum starvation. Despite their small number, stable genes encode for most tRNA isotypes and some RNAs involved in diverse functions, such as pre-mRNA splicing, tRNA and rRNA processing, and the control of Pol II transcription elongation.
Pol III occupancy is a proxy of ongoing transcription
In mammalian cells, Pol II is often bound near the TSS of coding genes (Guenther et al. 2007; Danko et al. 2013) in a poised or paused state from which it can rapidly escape into productive elongation in response to environmental cues (Adelman and Lis 2012). The identification of stable loci, showing high Pol III occupancy even upon serum starvation, raised the possibility that Pol III is also in an arrested, unproductive state at selected genes, perhaps to rapidly re-enter transcription when conditions are favorable. Although stalling of Pol III at a few loci has been suggested based on a comparison of ChIP-seq and RNA-seq profiles (Raha et al. 2010), it has not been shown experimentally, in part because standard techniques to evaluate transcriptional activity, such as GRO-seq (Core et al. 2008), are not customized to the short (∼70–300 bp) Pol III genes. By using neusRNA-seq to quantitatively assess Pol III transcription during the response of human fibroblasts to serum deprivation, we reveal a correlation between Pol III occupancy and active transcription. While it remains possible that, during its transcription cycle, Pol III is transiently poised within the preinitiation complex or stalled in a post-recruitment step, the absence of genes with high Pol III occupancy and low RNA output in serum-starved conditions strongly argues against the presence of arrested, unproductive Pol III and confirms the existence of a group of Pol III genes that remain actively transcribed under adverse conditions. Adaptation and continued synthesis of certain RNAs may have evolved to coordinate the Pol II and Pol III machineries with changing conditions and ensure cell survival during periods of stress. Indeed, in higher eukaryotes the relative amount of tRNA isoacceptors in the overall pool has been reported to coordinate, through an unknown mechanism, with the translational needs of the cell and to adapt to match the codon usage of protein coding genes selectively expressed in a certain tissue or proliferative state (Gingold et al. 2014; Schmitt et al. 2014).
A model for MAF1 repression of transcribing Pol III
In yeast, Maf1 has been shown to be increasingly recruited to Pol III genes after rapamycin treatment (Oficjalska-Pham et al. 2006; Roberts et al. 2006). To assess the contribution of mammalian MAF1 to the differential stress response of Pol III genes, we mapped genome-wide MAF1 binding sites in normal and serum-starved IMR90hTert cells. Despite a number of studies showing MAF1 association with individual Pol III genes and Pol II promoters by ChIP-qPCR experiments (Johnson et al. 2007; Goodfellow et al. 2008; Rohira et al. 2013), an analysis on the genomic scale has not been reported in mammalian cells. The unavailability of MAF1 ChIP-seq binding profiles might be explained by the transient nature of the interaction between MAF1 and Pol III transcribed genes, which would make ChIP-seq signals difficult to detect. To identify MAF1 genomic binding sites, we therefore turned to the DamIP-seq method, which does not depend on the stable association of proteins and DNA at the time of cross-linking and provides a way to mark transient protein–DNA interaction sites in vivo. DamIP-seq revealed a strong preference for MAF1 localization at Pol III–bound loci, and we detected significant MAF1 enrichment at 64 type II and type III promoter genes. This number is almost certainly an underestimation due to the relatively low sequence coverage of the DamIP-seq experiment and the fact that DamK9A methylation may depend on the availability of potential GATC targets, which are absent in a large fraction of Pol III loci.
Besides Pol III repression, MAF1 has been reported to repress Pol II transcription by direct binding to specific Pol II promoters, including those of the TBP, EGR1 (Johnson et al. 2007), and FASN (Palian et al. 2014) genes. Our results failed to identify MAF1 interaction with these promoters, even though they all include a GATC motif in proximity of the TSS. Although we cannot exclude that MAF1 binding goes undetected at the TBP, EGR1, and FASN genes because of a sequence context that restricts DamK9A targeting, the very low MAF1 scores observed at Pol II genes do not provide evidence for MAF1 acting as a Pol II transcription factor.
Our DamIP-seq analysis reveals that MAF1 occupancy increases at Pol III loci after serum starvation, correlating with decreased MAF1 phosphorylation and a decreased Pol III occupancy. Moreover, siRNA-mediated down-regulation of MAF1 results in higher Pol III occupancy in serum-starved condition. These observations strongly suggest that MAF1 mediates, at least in part, the down-regulation of Pol III occupancy after serum starvation. The finding that MAF1 is preferentially associated with Pol III–bound genes enriched in open chromatin marks is consistent with MAF1 targeting of actively transcribed genes and reveals that MAF1 acts as a repressor of ongoing Pol III transcription rather than as a long-term silencing factor. Unexpectedly, MAF1 was also detected on five stable genes whose Pol III occupancy score did not change significantly after serum starvation. The negligible effect of MAF1 on stable genes might be explained by their relatively high Pol III content. Since protein abundance studies in human cell lines estimate that MAF1 is four- to 10-fold less abundant than most Pol III–specific subunits (Beck et al. 2011; Kulak et al. 2014), it is conceivable that highly occupied genes have a lesser sensitivity to MAF1 repression than do genes displaying average Pol III occupancy. A second possibility is that facilitated recycling contributes to the stability of stable Pol III loci during stress conditions, given that MAF1 association with the polymerase itself does not inhibit elongation of transcriptionally engaged Pol III (Vannini et al. 2010) or facilitated recycling (Čabart et al. 2008). Additionally, Pol III activity may be affected by other players modulating the MAF1 effect, such as the previously proposed casein kinase 2 (Boguta and Graczyk 2011), or by putative MAF1 binding partners brought in close proximity of Pol III genes. For example, MAF1 has recently been shown to interact with PCNA (Cooper et al. 2015), a member of the DNA sliding clamp family with the ability to inhibit EP300 acetyltransferase activity (Hong and Chakravarti 2003). It is conceivable that MAF1-delivered PCNA could repress transcription of Pol III, which contains a PCNA binding site in the POLR3E (Rpc5) subunit (Gilljam et al. 2009), by preventing histone acetylation in the vicinity of Pol III genes. This mechanism might counteract the reported MYC-TRAPP–mediated acetylation and activation of Pol III genes' transcription in prosurvival conditions (Kenneth et al. 2007).
Importantly, our DamIP-seq analysis revealed not only that MAF1 association with Pol III genes increases after serum starvation but also that MAF1 association with Pol III genes is not limited to stress conditions. While we observed increased MAF1 targeting of active Pol III genes after serum starvation, MAF1 was detected at numerous genes in normally growing cells (Fig. 4D,E). In agreement with these results, MAF1 knock-down by RNAi results in increased Pol III transcription from a RNU6 promoter even in the absence of stress (Reina et al. 2006), and liver from Maf1−/− mice fed ad libitum displays increased Pol III occupancy compared with liver from wild-type mice (Bonhoure et al. 2014). Our work indicates that, even under favorable growth conditions, mammalian Pol III genes, like yeast Pol III genes, which were found significantly occupied by Maf1 even before rapamycin treatment (Oficjalska-Pham et al. 2006; Roberts et al. 2006), are targeted by MAF1, possibly to partially repress transcription and avoid overproliferation. MAF1 is thus not only an acute but also a chronic repressor of Pol III transcription.
Our results lead to the model summarized in Figure 5, which clarifies the mechanisms responsible for genome-wide modulation of Pol III transcription in response to nutrient availability in human cells. When nutrients are available and mTORC1 is active, a large fraction of MAF1 is hyperphosphorylated and most Pol III is engaged in transcription. Stress-induced MAF1 dephosphorylation results in increased targeting of gene-bound Pol III, which would then fall off the template and fail to reinitiate, with a consequent decrease in the transcriptional readout. On “stable” genes, MAF1 binding would have little or no consequence, possibly because of stoichiometric constraints or additional regulators.
Figure 5.

Model depicting MAF1-dependent regulation of Pol III transcription under different growth conditions. The tRNA molecules in red and green derive from unstable and stable genes, respectively.
In the absence of a mechanism blocking the polymerase on the DNA, binding of MAF1 to Pol III genes might suffice as an on/off switch for rapid transcriptional regulation of the majority of loci. Since pausing of Pol II has arguably evolved as a means to facilitate synchronous and rapid transcriptional activation by bypassing a number of rate-limiting steps (Adelman and Lis 2012), it is reasonable to think that maintaining Pol III on the DNA in an unproductive state in unfavorable conditions is not required, given that de novo recruitment of the enzyme is comparatively a much simpler process.
Methods
Cell culture and transfections
IMR90hTert (Canella et al. 2010) cells were cultured at 37°C under 5% CO2 in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin unless otherwise specified. For ChIP-qPCR, ChIP-seq, and immunoblotting, 70%–80% confluent IMR90hTert cells were serum-starved for 8 h and restimulated for 90 min with 1 µM insulin (Sigma, I9278) directly or after treatment with 2 nM rapamycin (Calbiochem, 553211) for 45 min. For neusRNA-seq and Northern blotting of labeled RNAs, 70%–80% confluent IMR90hTert cells were serum-starved for 5 h and labeled with 0.25 mM 5-EU (Life Technologies, E-10345) or DMSO for 5 h. Where indicated, Lipofectamine RNAiMax (Life Technologies, 13778-075) was used to transfect IMR90hTert cells with silencer select MAF1-specific siRNAs (Ambion, catalog 4427037, id. s38693 #siRNA1 and s38691 #siRNA2) or a nontarget siRNA (Ambion, catalog 4390843); 36 h after transfection cells were serum-starved for 8 h where indicated. To generate stable cell lines for DamIP-seq, IMR90hTert cells were transfected with the relevant plasmids at a 1:2.5 DNA:FugeneHD (Promega, E2312) ratio according to the manufacturer's instructions and selected with 1 µg/mL of puromycin.
ChIP-seq and ChIP-qPCR
Chromatin immunoprecipitation was carried out as previously described (Canella et al. 2010) using 8 µL per 107 IMR90hTert cells of an anti-POLR3D antibody. The detailed protocol and modifications are described in Supplemental Methods.
neusRNA-seq
IMR90hTert cells grown in a 10-cm dish as indicated were collected by centrifugation (6 min, 600g, 4°C) and washed twice with 1× PBS. Cell pellets were resuspended in 700 µL of TRIzol (Life Technologies, 15596026), homogenized with 140 µL of chloroform, and centrifuged for 15 min at 12,000g and 4°C. To selectively enrich in RNAs <400 nt, including most Pol III–transcribed RNAs, the aqueous phase was diluted with 1 volume of 48% ethanol before proceeding to RNA extraction with the RNeasy kit (Qiagen, 74104). Two micrograms of the small RNA-enriched fraction was used for biotinylation and bead-purified with the click-iT Nascent RNA capture kit (Life Technologies, C10365) according to the manufacturer's instructions. RNA on beads was resuspended in RNase-free water, heated 5 min at 70°C, and used for first-strand cDNA synthesis with random hexamers and SuperScript Vilo master mix (Life Technologies, 11755050) as described by the supplier. After heating the beads for 5 min at 85°C, the supernatant containing single-stranded cDNA was purified with NucleoSpin columns (Macherey-Nagel, 740609) and NTC buffer. The cDNA was used for library generation with the TruSeq RNA sample prep v2 kit (Illumina, RS-122-2001) according to the manufacturer's instructions, but with NucleoSpin columns and NTC buffer when required instead of AmpureXP beads. In brief, samples were diluted with 8 µL of FSM before second-strand synthesis, end repair, 3′-adenylation, and adapter ligation. DNA was PCR amplified, size selected (150–500 bp) on a 4% agarose gel, and eluted in 26 µL of EB buffer before multiplex (three indexes per lane) sequencing with the Illumina HiSeq 2000 platform. This library preparation strategy has the advantages to both prevent potential loss of Pol III transcripts with 3′cyclic phosphates (Lund and Dahlberg 1992) and preserve small cDNA fragments (>20 nt) that might arise from premature RT termination at modified nucleotides (Motorin et al. 2007). For read size distribution, see Supplemental Figure S4.
DamIP-seq and digestion assay
IMR90hTert cells expressing HA-tagged MAF1-DamK9A or EGFP-DamK9A were serum-starved for 8 h where indicated, and gDNA was extracted with the DNeasy kit (Qiagen, 69504) following the supplier's instructions (including RNase A treatment). For the digestion assay, 200 ng of gDNA was digested with 5 U of either DpnII (NEB, R0543S) or Sau3A (NEB, R0169L), purified on column (MN, 740609), and analyzed by qPCR with primers listed in Supplemental Table S4. DamIP-seq was as previously described (Xiao et al. 2010, 2012) with modifications. In brief, 10 µg of gDNA was sheared to 100–300 bp with a Covaris S220 focused-ultrasonicator in 6 × 16 mm µTUBE (Covaris, catalog 520045). The DNA was boiled for 10′ at 98°C, cooled for 10′ in ice, diluted with 1 volume of ice-cold 2X DamIP buffer (20 mM Tris-Cl at pH 7.4, 300 mM NaCl, and 0.2% NP40), and incubated O/N at 4°C under rotation with 4 µg of anti-N6-methyladenosine antibody (Millipore, 1 µg/µL, ABE572). Antibody–DNA complexes were pulled-down with 25 µL of Dynabeads protein-A (Life Technologies, 10002D) by 1-h incubation at room temperature. Beads were washed four times in 1× DamIP buffer and resuspended in 300 µL of elution buffer (50 mM Tris-Cl at pH 8, 10 mM EDTA, and 0.5% SDS) with 40 µg of proteinase K. After 3-h incubation at 48°C, immunoprecipitated single-stranded DNA (ssDNA) was extracted with phenol chloroform, ethanol precipitated, and resuspended in 17 µL of EB buffer. Approximately 100 ng of ssDNA was used for library generation with the TruSeq RNA sample prep v2 kit (Illumina, RS-122-2001) as described for the neusRNA-seq protocol, except that 200- to 300-bp DNA fragments were size-selected on a 4% agarose gel before PCR amplification.
Data analysis
For ChIP-seq, we mapped the sequence tags obtained after ultra-high-throughput sequencing onto the UCSC genome hg19 as previously described (Renaud et al. 2014), allowing up to 500 multiple matches across the genome. Data were scaled to the mean of total tag count, and Pol III scores were calculated for bins including the annotated gene body ±150 bp as the mean log2 (IPtag count/INPUTtag count) of two replicates per condition. Cutoffs to define Pol III–bound genes were calculated as previously described (Canella et al. 2010), except that for tags sequenced multiple times, we included up to 100 copies instead of 50. Two-by-two comparisons between ChIP-seq scores from different growth conditions were done using the Limma package (Smyth 2005). For comparison of Pol III ChIP-seq scores with epigenetic marks and DHS, raw tags from publicly available data sets were downloaded from the Sequence Read Archive (SRA) and then mapped with the procedure described above. As the input was not always available, the gene scores were calculated using only the IP tags. The combined A-box and B-box score was calculated as follows: By using MEME (Bailey et al. 2009) on the 25% most highly occupied tRNA genes, we extracted a position-specific scoring matrix (PSSM) for both the A-box and B-box regions. We then calculated the E-value for each Pol III gene using MAST with the two PSSMs generated above. The plotted scores are given as −log10 (E-value).
For neusRNA-seq, sequenced tags were first cleared of adapters with Cutadapt (Martin 2011), leaving tags with sizes ranging between 19 and 99 nt. The tags were then mapped in three sequential steps and categorized as either mature or precursors as previously described (Bonhoure et al. 2015). Each tag contribution to the gene score was weighted according to its length, its number of matches on the transcriptome, and the number of times it was sequenced. A cutoff for tags with multiple matches was calculated as a function of the tag length with the following approach. By using the reference list of human loci shown in Supplemental Table S2, we generated a list of all possible tags with a length varying between 19 and 99. For each length, we identified the tag with the maximum number of matches in the Pol III loci reference and applied this number as the cutoff for the neusRNA-seq sequenced tags of this specific length; any tag of this length with a larger number of possible matches in the entire genome was discarded. For any tag with a possible number of matches in the entire genome below or equal to the cutoff number, the tag contribution to the gene score was calculated as the number of times it was sequenced divided by the number of matches in the Pol III loci reference. To build scores, we first calculated for each gene the mean of three negative DMSO control replicates and then subtracted this value to the gene tag count of every EU-labeled sample. The mean score of three EU-labeled replicates was then calculated for each condition (with or without serum) and used for the analysis described in the text.
For DamIP-seq, the tags were trimmed with Cutadapt (Martin 2011), and the mapping, scoring, and cutoff procedures used for ChIP-seq were applied. Supplemental Figure S5 shows that the correlations among MAF1 DamIP-seq samples were relatively high, whereas those among the EGFP control DamIP-seq samples were low, as expected. Because of low sequence coverage, we generated a meta-sample consisting of sequenced tags from four experiments per condition (two replicates per clone, two clones per condition). The data were scaled to the mean of total tags count before calculation of scores. The whole genome was split into 400-bp bins, and bin scores were calculated as the mean log2 (MAF1tag count/EGFPtag count) for each condition. Bins having (1) more than eight tags in the MAF1-expressing clones and (2) a positive score were plotted. Bins having a score bigger than a certain cutoff, chosen as the lowest score giving a FDR of 0.005 on the whole genome and calculated as previously described (Renaud et al. 2014), were associated to the corresponding genomic feature (Supplemental Table S3). For Pol III genes, scores were calculated as above, but on a region centered on the gene body ±150 bp.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE73936.
Acknowledgments
We thank Donatella Canella for her help in setting up ChIP-seq experiments and Diane Buczynski-Ruchonnet for preliminary results on Pol III insulin sensitivity. High-throughput sequencing was performed at the Lausanne Genomic Technologies Facility. Alignments were executed at the Vital-IT (www.vital-it.ch) Center for High-Performance Computing of the Swiss Institute of Bioinformatics. This work was supported by the University of Lausanne and the Swiss National Science Foundation (SNSF) grants CRSI33_125230 and 31003A_132958.
Author contributions: A.O. and N.H. designed the study. A.O. designed and performed the experiments. V.P. designed and executed the computational analyses. P.L. cultured IMR90hTert cells. A.O., V.P., and N.H. contributed to data analysis and interpretation. A.O. and N.H. wrote the primary manuscript with contributions from V.P.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.201400.115.
Freely available online through the Genome Research Open Access option.
References
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R. 2011. The quantitative proteome of a human cell line. Mol Syst Biol 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguta M, Graczyk D. 2011. RNA polymerase III under control: repression and de-repression. Trends Biochem Sci 36: 451–456. [DOI] [PubMed] [Google Scholar]
- Bonhoure N, Bounova G, Bernasconi D, Praz V, Lammers F, Canella D, Willis IM, Herr W, Hernandez N, Delorenzi M, et al. 2014. Quantifying ChIP-seq data: a spiking method providing an internal reference for sample-to-sample normalization. Genome Res 24: 1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoure N, Byrnes A, Moir RD, Hodroj W, Preitner F, Praz V, Marcelin G, Chua SC, Martinez-Lopez N, Singh R, et al. 2015. Loss of the RNA polymerase III repressor MAF1 confers obesity resistance. Genes Dev 29: 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čabart P, Lee J, Willis IM. 2008. Facilitated recycling protects human RNA polymerase III from repression by Maf1 in vitro. J Biol Chem 283: 36108–36117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canella D, Praz V, Reina JH, Cousin P, Hernandez N. 2010. Defining the RNA polymerase III transcriptome: genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res 20: 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canella D, Bernasconi D, Gilardi F, LeMartelot G, Migliavacca E, Praz V, Cousin P, Delorenzi M, Hernandez N, Deplancke B, et al. 2012. A multiplicity of factors contributes to selective RNA polymerase III occupancy of a subset of RNA polymerase III genes in mouse liver. Genome Res 22: 666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37: D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SE, Hodimont E, Green CM. 2015. A fluorescent bimolecular complementation screen reveals MAF1, RNF7 and SETD3 as PCNA-associated proteins in human cells. Cell Cycle 14: 2509–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core L, Waterfall J, Lis J. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL. 2013. Signaling pathways differentially affect RNA polymerase II initiation, pausing, and elongation rate in cells. Mol Cell 50: 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai N, Lee J, Upadhya R, Chu Y, Moir RD, Willis IM. 2005. Two steps in Maf1-dependent repression of transcription by RNA polymerase III. J Biol Chem 280: 6455–6462. [DOI] [PubMed] [Google Scholar]
- Gilljam KM, Feyzi E, Aas PA, Sousa MML, Müller R, Vågbø CB, Catterall TC, Liabakk NB, Slupphaug G, Drabløs F, et al. 2009. Identification of a novel, widespread, and functionally important PCNA-binding motif. J Cell Biol 186: 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, Kooistra SM, Christophersen NS, Christensen LL, Borre M, Sørensen KD, et al. 2014. A dual program for translation regulation in cellular proliferation and differentiation. Cell 158: 1281–1292. [DOI] [PubMed] [Google Scholar]
- Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gérard M, Lefebvre O, White RJ. 2008. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J Mol Biol 378: 481–491. [DOI] [PubMed] [Google Scholar]
- Greil F, Moorman C, van Steensel B. 2006. DamID: mapping of in vivo protein–genome interactions using tethered DNA adenine methyltransferase. Methods Enzymol 410: 342–359. [DOI] [PubMed] [Google Scholar]
- Grewal SS. 2015. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochim Biophys Acta 1849: 898–907. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130: 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harismendy O, Gendrel C-G, Soularue P, Gidrol X, Sentenac A, Werner M, Lefebvre O. 2003. Genome-wide location of yeast RNA polymerase III transcription machinery. EMBO J 22: 4738–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. 2010. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6: 479–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong R, Chakravarti D. 2003. The human proliferating cell nuclear antigen regulates transcriptional coactivator p300 activity and promotes transcriptional repression. J Biol Chem 278: 44505–44513. [DOI] [PubMed] [Google Scholar]
- Horton JR, Liebert K, Bekes M, Jeltsch A, Cheng X. 2006. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol 358: 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SS, Zhang C, Fromm J, Willis IM, Johnson DL. 2007. Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases. Mol Cell 26: 367–379. [DOI] [PubMed] [Google Scholar]
- Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ. 2007. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci 104: 14917–14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. 2014. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods 11: 319–324. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. 2013. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126: 1713–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Dahlberg J. 1992. Cyclic 2′,3′-phosphates and nontemplated nucleotides at the 3′ end of spliceosomal U6 small nuclear RNA's. Science 255: 327–330. [DOI] [PubMed] [Google Scholar]
- Luo SD, Shi GW, Baker BS. 2011. Direct targets of the D. melanogaster DSXF protein and the evolution of sexual development. Development 138: 2761–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, Rideout EJ, Grewal SS. 2012. Nutrient/TOR-dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J 31: 1916–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Mendoza MC, Er EE, Blenis J. 2011. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36: 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N. 2010. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol 30: 3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir RD, Lee J, Haeusler RA, Desai N, Engelke DR, Willis IM. 2006. Protein kinase A regulates RNA polymerase III transcription through the nuclear localization of Maf1. Proc Natl Acad Sci 103: 15044–15049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, Wang J, Raha D, White RJ, Snyder M, Weng Z, Struhl K. 2010. Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells. Nat Struct Mol Biol 17: 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Muller S, Behm-Ansmant I, Branlant C. 2007. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425: 21–53. [DOI] [PubMed] [Google Scholar]
- Oficjalska-Pham D, Harismendy O, Smagowicz WJ, Gonzalez de Peredo A, Boguta M, Sentenac A, Lefebvre O. 2006. General repression of RNA polymerase III transcription is triggered by protein phosphatase type 2A-mediated dephosphorylation of Maf1. Mol Cell 22: 623–632. [DOI] [PubMed] [Google Scholar]
- Oler AJ, Alla RK, Roberts DN, Wong A, Hollenhorst PC, Chandler KJ, Cassiday PA, Nelson CA, Hagedorn CH, Graves BJ, et al. 2010. Human RNA polymerase III transcriptomes and relationships to Pol II promoter chromatin and enhancer-binding factors. Nat Struct Mol Biol 17: 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orioli A, Pascali C, Pagano A, Teichmann M, Dieci G. 2012. RNA polymerase III transcription control elements: themes and variations. Gene 493: 185–194. [DOI] [PubMed] [Google Scholar]
- Palian BM, Rohira AD, Johnson SAS, He L, Zheng N, Dubeau L, Stiles BL, Johnson DL. 2014. Maf1 is a novel target of PTEN and PI3K signaling that negatively regulates oncogenesis and lipid metabolism. PLoS Genet 10: e1004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. 2009. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res 37: 7268–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. 2010. tRNA biology charges to the front. Genes Dev 24: 1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta K, Lefebvre O, Martin NC, Smagowicz J, Stanford DR, Ellis SR, Anita K, Sentenac A, Boguta M, Smagowicz WJ, et al. 2001. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol Cell Biol 21: 5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M. 2010. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci 107: 3639–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina JH, Azzouz TN, Hernandez N. 2006. Maf1, a new player in the regulation of human RNA polymerase III transcription. PLoS One 1: e134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud M, Praz V, Vieu E, Florens L, Washburn MP, L'Hôte P, Hernandez N. 2014. Gene duplication and neofunctionalization: POLR3G and POLR3GL. Genome Res 24: 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout EJ, Marshall L, Grewal SS. 2012. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci 109: 1139–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DN, Wilson B, Huff JT, Stewart AJ, Cairns BR. 2006. Dephosphorylation and genome-wide association of Maf1 with Pol III-transcribed genes during repression. Mol Cell 22: 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohira AD, Chen C-Y, Allen JR, Johnson DL. 2013. Covalent small ubiquitin-like modifier (SUMO) modification of Maf1 protein controls RNA polymerase III-dependent transcription repression. J Biol Chem 288: 19288–19295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins J, Veras I, Cabarcas S, Willis I, Schramm L. 2007. Human Maf1 negatively regulates RNA polymerase III transcription via the TFIIB family members Brf1 and Brf2. Int J Biol Sci 3: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt BM, Rudolph KLM, Karagianni P, Fonseca NA, White RJ, Talianidis I, Odom DT, Marioni JC, Kutter C. 2014. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA–tRNA interface. Genome Res 24: 1797–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimobayashi M, Hall MN. 2014. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 15: 155–162. [DOI] [PubMed] [Google Scholar]
- Shor B, Wu J, Shakey Q, Toral-Barza L, Shi C, Follettie M, Yu K. 2010. Requirement of the mTOR kinase for the regulation of Maf1 phosphorylation and control of RNA polymerase III-dependent transcription in cancer cells. J Biol Chem 285: 15380–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G. 2005. limma: linear models for microarray data. In Bioinformatics and computational biology solutions using R and Bioconductor (ed. Gentleman R, et al. ), pp. 397–420. Springer, New York. [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, Delrow J, Henikoff S. 2001. Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet 27: 304–308. [DOI] [PubMed] [Google Scholar]
- Vannini A, Ringel R, Kusser AG, Berninghausen O, Kassavetis GA, Cramer P. 2010. Molecular basis of RNA polymerase III transcription repression by Maf1. Cell 143: 59–70. [DOI] [PubMed] [Google Scholar]
- White RJ, Stott D, Rigby PW. 1989. Regulation of RNA polymerase III transcription in response to F9 embryonal carcinoma stem cell differentiation. Cell 59: 1081–1092. [DOI] [PubMed] [Google Scholar]
- Xiao R, Roman-Sanchez R, Moore DD. 2010. DamIP: a novel method to identify DNA binding sites in vivo. Nucl Recept Signal 8: e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Sun D, Ayers S, Xi Y, Li W, Baxter JD, Moore DD. 2012. Research resource: the estrogen receptor α cistrome defined by DamIP. Mol Endocrinol 26: 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. 2010. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell 38: 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Goodenbour JM, Godley LA, Wickrema A, Pan T. 2009. High levels of tRNA abundance and alteration of tRNA charging by bortezomib in multiple myeloma. Biochem Biophys Res Commun 385: 160–164. [DOI] [PMC free article] [PubMed] [Google Scholar]