

Abstract
CRISPR/Cas9-mediated targeted mutagenesis allows efficient generation of loss-of-function alleles in zebrafish. To date, this technology has been primarily used to generate genetic knockout animals. Nevertheless, the study of the function of certain loci might require tight spatiotemporal control of gene inactivation. Here, we show that tissue-specific gene disruption can be achieved by driving Cas9 expression with the Gal4/UAS system. Furthermore, by combining the Gal4/UAS and Cre/loxP systems, we establish a versatile tool to genetically label mutant cell clones, enabling their phenotypic analysis. Our technique has the potential to be applied to diverse model organisms, enabling tissue-specific loss-of-function and phenotypic characterization of live and fixed tissues.
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)/CRISPR-associated (Cas) system has recently emerged as a powerful tool to generate constitutive loss-of-function alleles, enabling detailed analyses of gene function (Cho et al. 2013; Cong et al. 2013; Hwang et al. 2013b; Mali et al. 2013). Gene disruption is achieved by the activity of two components: a single guide RNA (sgRNA) that contains 20 nt complementary to a DNA target, and a Cas9 endonuclease that catalyzes DNA cleavage at the target site after complex formation with the sgRNA. Following DNA cleavage, mutations are efficiently induced upon imperfect repair by the nonhomologous end-joining (NHEJ) pathway at the targeted sequence. sgRNAs that bind within the coding region of a gene thereby allow disruption of the open reading frame (ORF), leading to loss of gene activity. Pioneer studies in worms (Shen et al. 2014), fruit fly (Port et al. 2014), mouse (Platt et al. 2014), and, more recently, zebrafish (Ablain et al. 2015) have taken advantage of this methodology to induce conditional gene knockouts by driving tissue-specific expression of Cas9. However, in zebrafish, this approach relies on cell-type–specific promoter sequences to control Cas9 expression, usually requiring the screening of several lines to achieve sufficient and nonectopic transgene expression. One alternative and powerful method for cell-specific expression of transgenes in model organisms is the Gal4/UAS binary system (Asakawa and Kawakami 2008), in which transcription is driven by the binding of the Gal4 transcription factor to tandem upstream activation sequences (UAS) placed at the 5′ end of a gene of interest. An important resource of Gal4 transgenic lines has been established by gene- and enhancer-trap screens (Davison et al. 2007; Asakawa et al. 2008; Scott and Baier 2009; Kawakami et al. 2010; Balciuniene et al. 2013) based on random integration of the Gal4 ORF via Tol2 transposition (Kawakami 2004) into the fish genome. In particular, Gal4 driver lines have been identified for many tissues as well as for novel and previously uncharacterized cell types, especially in the nervous system (Scott et al. 2007; Asakawa et al. 2008). More recently, we have developed a simple and efficient method for converting GFP- into Gal4-transgenic lines, significantly expanding the potential resource of Gal4 driver lines (Auer et al. 2014a,b). Finally, Gal4-driven expression of transgenes from UAS promoters is robust and can often be revealed in cells where Gal4 expression itself is difficult to detect. In this report, we show that flexible and efficient gene disruption is achieved as a result of tissue-specific expression of Cas9 via the Gal4/UAS system. In addition, we overcome the challenge of visualizing loss-of-function mutations in specific cells by coexpressing the Cas9 endonuclease with GFP or Cre recombinase, allowing the labeling of potentially mutant cells and thereby facilitating their phenotypic analysis. This step is essential to correlate phenotype with genotype for analysis of gene function, a task that was not addressed with previously published methods.
Results
Design of a vector system for spatiotemporal control of Cas9 activity via Gal/UAS
The generation of conditional knockout models requires the specific targeting of a gene of interest in a given tissue and, ideally, the labeling of targeted cells. In order to develop a flexible tool for efficient spatiotemporal control of gene disruption, we generated a Tol2-based vector ensuring Gal4/UAS-mediated cell-type–specific expression of the Cas9 enzyme and ubiquitous expression of sgRNAs. Due to its strong transactivating properties, the Gal4/UAS system directs effective expression of the Cas9 endonuclease, even in the case of weak tissue-specific Gal4 transcription by an endogenous promoter. Furthermore, this system renders our vector design compatible with many readily available Gal4 transgenic lines generated in previous Gal4 gene- and enhancer-trap screens (including those where the regulatory elements driving Gal4 expression are not known) (Davison et al. 2007; Asakawa et al. 2008; Scott and Baier 2009; Kawakami et al. 2010; Balciuniene et al. 2013). In our vector design, the use of a viral T2A self-cleaving peptide (Provost et al. 2007) to synthesize a GFP reporter from the same transgene (UAS:Cas9T2AGFP) allows Cas9-positive cells to be unambiguously marked by GFP fluorescence. This feature is critical for the analysis of cellular phenotypes resulting from Cas9-induced gene inactivation. Our vector also contains two sgRNA expression cassettes, each driven by the ubiquitous U6-1 promoter (Halbig et al. 2008; Yin et al. 2015) (UAS: Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2) (Fig. 1A). By using two sgRNAs, we sought to increase the probability of generating loss-of-function alleles, since genomic deletions might be induced, in addition to frame-shifts caused by insertions and deletions (indels). To verify that cell-type–specific expression of Cas9 can be driven by Gal4 transcriptional activation, we generated a stable transgenic line with this vector, Tg(UAS: Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2), and crossed it to different Gal4 driver lines (Fig. 1B). The motor neuron-specific Tg(mnx1:Gal4) transgenic line, the enhancer-trap line Tg(s1020t) expressed in the thalamus and spinal cord, and the optic tectum-specific gene-trap line Tg(SA2AzGFF49A) were used as drivers. We compared the expression of the UAS:Cas9T2AGFP with an independent reporter of the Gal4 activity by crossing the same driver lines with a Tg(UAS:RFP;cry:GFP). In the embryos analyzed, GFP was specifically detected in the expected Gal4 transactivation domain, indicating nonectopic Cas9 expression. Modest differences in GFP and RFP expression were observed, the RFP-positive cells being on average more numerous than the GFP-positive ones. This might be due to transgene specific differences likely linked to positional effect of the integration as commonly seen for other zebrafish transgenic lines. Overall, these expression patterns demonstrate that our approach can be used to both express Cas9 nuclease in a tissue-specific manner and to simultaneously label Cas9-expressing cells with GFP.
Figure 1.

Spatiotemporal control of Cas9 activity via the Gal4/UAS expression system. (A) Schematic illustration of the expression vector design. Spatial and temporal control of Cas9 synthesis is achieved by the Gal4/UAS system and monitored by GFP expression. Two constitutively active U6 promoters drive the transcription of single guide RNAs (sgRNAs) specifically targeting a gene of interest. To facilitate cloning of sgRNA target sequences, BsmbI and BsaI restriction sites (red arrows) were introduced. Tol2 allows efficient transgenesis after injection into one-cell stage zebrafish embryos. (B) (Left panel) Confocal imaging of the spinal cord of 2 dpf Tg(mnx1:Gal4) embryos. The upper image shows the Gal4-induced RFP expression of a double transgenic embryo deriving from a cross between Tg(mnx1:Gal4) and Tg(UAS:RFP; cry:GFP) fish. The same pattern is observed in the GFP channel in a double transgenic Tg(mnx1:Gal4) × Tg(UAS:Cas9T2AGFP;U6sgRNA1;U6sgRNA2) embryo (lower image). Scale bar = 100 µm. (Middle panel) Imaging of whole-mount 3 dpf Tg(s1020t) enhancer-trap line. The GFP expression pattern (lower panel) recapitulates RFP (upper panel). Scale bar = 100 µm. (Right panel) Confocal imaging of 5 dpf optic tectum in the double transgenic embryos deriving from a cross of Tg(gSA2AzGFF49A) gene-trap line and Tg(UAS:RFP,cry:GFP) (upper image) and Tg(UAS:Cas9T2AGFP;U6sgRNA1;U6sgRNA2) (lower image). Scale bar = 50 µm. No target sequence was inserted in the vector for the generation of the Tg(UAS:Cas9T2AGFP;U6sgRNA1;U6sgRNA2) line.
Targeting the tyrosinase gene in the optic primordium leads to loss of pigmentation in the retinal pigmented epithelium (RPE)
To test the efficiency of our approach for tissue-specific gene disruption, we targeted the tyrosinase (tyr) gene, which codes for a key enzyme in melanin production (Camp and Lardelli 2001), by including two sgRNAs targeting the tyr in our vector locus (pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2). We previously assessed the mutagenesis rate of each sgRNA by injection of in vitro-transcribed sgRNA with synthetic Cas9 mRNA into one-cell stage wild-type embryos (Supplemental Table S1). Loss-of-function of the tyr locus results in pigmentation defects, offering a clear visual read-out of biallelic gene inactivation (Jao et al. 2013). We used fluorescence-activated cell sorting (FACS) to verify that site-specific cleavage only occurs in GFP-expressing cells and to quantify the mutation rate induced by our vector. The quasi-ubiquitous Tg(rpl5b:Gal4) line (Amsterdam et al. 2004) was chosen to enlarge the number of GFP-positive cells in the assay (Supplemental Fig. S1A). We thereby transiently expressed the pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2 by injection into one-cell stage Tg(rpl5b:Gal4) embryos. After separating the pool of GFP-positive cells from the GFP-negative control cells derived from 200 injected embryos, total DNA was extracted from the two cell populations. Upon PCR amplification and sequencing of single amplicons from the DNA of GFP-positive cells, we detected indel out-of-frame mutations at the tyr target region in 30% (4/12) of the analyzed sequences (Supplemental Fig. S1B). In this cell population, we also observed a 488-bp deletion at the tyr gene resulting from simultaneous targeting of the locus by both sgRNAs. In contrast, no tyr mutations were found in the GFP-negative cells (0/15). These data confirm that locus-specific knockout events are restricted to the cells expressing the fluorescent reporter. Subsequently, we tested whether spatiotemporally regulated tyr loss-of-function can be induced by our vector system. For this purpose, we used a transgenic line expressing Gal4 under the promoter of the zebrafish retinal homeobox gene 2 (rx2) (Heermann et al. 2015), Tg(rx2:Gal4;myl7:GFP). We identified transgenic embryos using the myl7 (myosin, light chain 7, regulatory) promoter, driving heart-specific GFP expression, which was incorporated in the same transgene. Rx2 is active in the optic primordium in the progenitors of the neural retina and the retinal-pigmented epithelium (RPE) (Chuang and Raymond 2001). Thus, transient, cell-specific expression of Cas9 from the pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2 in Tg(rx2:Gal4;myl7:GFP) embryos should promote targeted disruption of the tyr locus uniquely in Gal4-positive cells, leading to pigmentation defects exclusively in the RPE after biallelic gene inactivation. In agreement with the known expression pattern of the rx2 promoter (Supplemental Fig. S2A), we detected sparse green fluorescent cells in the embryonic retina of Gal4-positive larvae, at 24 hours post-fertilization (hpf) (Fig. 2A). Of the embryos analyzed at 5 days post-fertilization (dpf), 15% displayed loss of pigmentation specifically in the RPE (16/107 embryos in three independent experiments) compared to 0% (0/116 in three independent experiments) in embryos injected with a plasmid containing control sgRNA sequences (Fig. 2B,C). We observed a variable degree of pigmentation loss, and all the embryos displaying the phenotype were scored as positives. Importantly, no pigmentation defects were detected outside of the eye-specific Gal4 expression domain. Together, these results indicate that knockout of the tyr gene driven by the rx2 promoter leads to tissue-specific pigmentation defects. Having this proof of principle for our approach, we decided to create a stable integration of the UAS-based transgene to achieve a more robust and more reliable expression and to evaluate the penetrance of the pigmentation phenotype. We generated a Tg(UAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2) transgenic line and crossed it with Tg(rx2:Gal4;myl7:GFP) fish. Gal4-positive embryos were prescreened by myl7:GFP expression and subsequently analyzed to assess tyr inactivation. The rate of larvae displaying RPE pigmentation defects increased to 48% (24/52) (Fig. 2D); therefore, tyr loss-of-function most likely occurred in all double transgenic progeny of the cross, as only half of the Gal4-positive larvae were expected to carry the UAS transgene. This observation shows that stable expression of the Cas9/sgRNA complex dramatically improves the efficiency of gene inactivation, most likely due to higher levels and more widespread expression in retinal progenitor cells (RPCs). Based on these data, we conclude that our vector system is capable of inducing loss-of-function alleles at given genomic loci in a defined spatiotemporal manner. However, at the developmental stage of the phenotypic analysis (5 dpf), GFP fluorescence could no longer be detected since the rx2 promoter is not active in the differentiated RPE. This result reflects the fact that GFP expression is strictly dependent on the temporal activity of the promoter driving Gal4 expression, thus restricting direct detection of potential mutant cells to a limited time window.
Figure 2.

Tissue-specific disruption of the tyrosinase locus induced by the expression of the UAS-based vector in the retinal progenitor cells. (A) Transmitted light and confocal imaging of eye of a 24 hpf Tg(rx2:Gal4;myl7:GFP) embryo injected with pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2 together with Tol2 mRNA. GFP fluorescence indicates Gal4-driven expression of the Cas9 enzyme in the retinal progenitor cells (RPCs) giving rise to the retinal pigmented epithelium (RPE) and to the neural retina. Scale bar = 100 µm. (B) (Lower panel) Specific loss of pigmentation in the RPE of a 5 dpf zebrafish embryo induced by injection of pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2 in Tg(rx2:Gal4; myl7:GFP) at one-cell stage. Arrowhead indicates the eye with lost pigmentation. (Upper panel) Control embryo injected with the same vector containing a control sgRNA sequence (ctlsgRNA). Scale bar = 300 µm. (C) Frontal view of eyes with absent pigmentation (lower panel) and wild-type (upper panel) explanted from the larvae shown in B. (D) Table showing the percentage of Gal4-positive larvae displaying loss of pigmentation in the RPE. (First row) Injection of the pUAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2 into one-cell-stage Tg(rx2:Gal4; myl7:GFP) embryos. (Second row) Cross of Tg(rx2:Gal4; myl7:GFP) fish with Tg(UAS:Cas9T2AGFP;U6:tyrsgRNA1;U6:tyrsgRNA2).
Cre-mediated recombination allows permanent labeling of Cas9-expressing cells
By using the pUAS:Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2 vector, potentially mutated cells are labeled by simultaneous expression of the Cas9 endonuclease and a GFP reporter. Nevertheless, the possibility of phenotypic analysis of the cells expressing the transgene strongly depends on the activity window of the chosen promoter, as it determines when the GFP reporter can be detected. Therefore, the long-term visualization of cells displaying loss-of-function phenotypes that are not self-revealing (unlike the loss of pigmentation induced by mutations on the tyr locus) requires permanent labeling of Cas9-expressing cells and their progeny. To obtain such a labeling, we replaced the GFP ORF with the one of Cre recombinase (Fig. 3A), an enzyme catalyzing site-specific recombination events (Branda and Dymecki 2004; Pan et al. 2005). In order to detect Cre-mediated labeling of Cas9-expressing cells, we used transgenic lines carrying a cassette where a ubiquitous promoter drives constitutive expression of a fluorescent reporter only upon excision of a floxed STOP sequence by Cre recombinase. The tissue-specific Gal4-driven transcription of a UAS:Cas9T2ACre transgene ensures concomitant expression of the Cas9 and Cre enzymes inducing, respectively, double-strand breaks (DSBs) at the target locus and activation of the floxed reporter transgene within the same cell (Fig. 3B). Most importantly, Cre-dependent expression of the fluorescent reporter will persist in all cells derived from a specific Cas9-expressing precursor. This approach, that we name 2C-Cas9 (Cre-mediated recombination for Clonal analysis of Cas9 mutant cells) allows targeted mutagenesis of a cell population and the genetic labeling of the derived mutant cell clones.
Figure 3.

Permanent labeling of Cas9-expressing cells by Cre-mediated recombination. (A) Schematic illustration of the pUAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 expression vector design. The vector contains two U6-sgRNA expression cassettes targeting the same gene. BsaI and BsmBI restriction sites are used for the sgRNA target sequence cloning. UAS elements drive the expression of Cas9 and Cre recombinase linked via the T2A peptide. (B) Microinjection of the pUAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 construct in the double transgenic embryos Tg(Tissue specific promoter:Gal4) × Tg(Ubiquitous promoter:loxP-STOP-loxP-XFP) triggers simultaneous synthesis of Cas9 and Cre in a chosen spatiotemporal pattern, defined by the promoter driving Gal4 expression. Cell-specific Cas9 endonuclease activity induces targeted disruption of the gene of interest and Cre-mediated recombination induces permanent expression of a fluorescent reporter gene (XFP) by deletion of the floxed stop sequence.
Visualizing 2C-Cas9-mediated gene inactivation
In order to explore the possibility of directly visualizing protein loss, we sought to induce eye-specific loss-of-function at the parvalbumin 6 (pvalb6) locus, coding for a calcium-binding protein expressed in a subpopulation of amacrine cells (ACs) in the inner nuclear layer (INL) and retinal ganglion cell layer (GCL) of the differentiated retina (Fig. 4A). The pvalb6 gene is the only paralog expressed in mature retinal neurons as reported by the ZFIN in situ database (Sprague et al. 2008). This gene represents an ideal target for unbiased detection of locus disruption, since its expression can be easily revealed by immunohistochemistry with a commercially available antibody (Nevin et al. 2008). To achieve pvalb6 inactivation, we first generated a pUAS:Cas9T2ACre;U6:pvalb6sgRNA1;U6:pvalb6sgRNA1 vector. The mutagenesis rate of the sgRNA used was tested in transient injection in wild-type embryos prior to cloning in the 2C-Cas9 vector (Supplemental Table S1). As a single efficient sgRNA could be identified for this gene, the same target sequence was inserted downstream from each of the two U6 promoters. We chose the rx2:Gal4 driver to express this UAS construct targeting pvalb6 in multipotent retinal progenitor cells as the rx2 promoter is active during early eye field development (Chuang and Raymond 2001; Heermann et al. 2015). It has been previously reported that one single RPC can give rise to clones including all of the different retinal cell types spanning all retinal layers (Livesey and Cepko 2001). Thus, a loss-of-function mutation, when induced at the level of a RPC, is expected to propagate to a clone of cells derived from the progenitor. To obtain clonal labeling of mutant retinal cells, we injected the pUAS:Cas9T2ACre;U6:pvalb6sgRNA1;U6:pvalb6sgRNA1 into one- cell stage embryos of a cross of Tg(rx2:Gal4;myl7:GFP) and Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 fish. The latter reporter line allows visualization of Cre-mediated recombination by switching from lacZ to GFP expression. This cross enables the detection in the retina of radial columns of GFP-positive cells arising from RPCs having expressed the Cas9 during the activation time of the rx2 promoter. Discrete clones of GFP-positive Cas9-expressing cells were analyzed in retinas of 5 dpf larvae after double immunohistochemistry for GFP and Pvalb. For each clone, we counted the total number of cells labeled by the GFP reporter, excluding photoreceptors, and the number of cells simultaneously stained for GFP and Pvalb. This second population represents the subset of amacrine cells deriving from Cas9-expressing RPCs that escaped pvalb6 biallelic inactivation. We observed that the total number of cells was not affected in the examined clones (15 ± 1 in controls vs. 16 ± 1 in pvalb6 sgRNA clones; P = 0.9, Wilcoxon Mann–Whitney test), while the number of cells showing colocalization of Pvalb and GFP signals was considerably reduced by the targeting of pvalb6 (1.9 ± 0.3 in controls vs. 0.8 ± 0.2 in pvalb6 sgRNA clones; P < 0.005, Wilcoxon Mann–Whitney test) (Fig. 4B). Overall, in control retinas the number of GFP-positive cells expressing Pvalb represented 12% of each clone on average (n = 23 clones; 345 cells analyzed). On the contrary, in clones where the pvalb6 gene was targeted, only 4% of GFP-positive cells were also positive for Pvalb (n = 27 clones; 435 cells analyzed), representing cells that escaped biallelic pvalb6 inactivation (Fig. 4C,D). Altogether, pvalb6 targeting led to a reduction of two thirds of the pvalb6-expressing ACs. These results confirm that 2C-Cas9 can mediate gene loss-of-function and that antibody staining can efficiently reveal protein loss at a cellular resolution, offering a clear read-out of the proportion of cells carrying biallelic mutations within the analyzed population.
Figure 4.

Targeting of the pvalb6 locus leads to loss of Pvalb expression in ACs. (A) Schematic of a clone of cells deriving from a single RPC. The black frame defines the position of the amacrine cell population. (ONL) Outer nuclear layer, (INL) inner nuclear layer, (GCL) ganglion cell layer. (B) GFP-positive Cas9-expressing cells in the retinal section of a 5 dpf double transgenic Tg(rx2:Gal4; myl7:GFP) × Tg (-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 embryos transiently expressing the 2C-Cas9 vector containing control sgRNAs (upper panel) or pvalb6-specific sgRNAs (lower panel). The same sgRNA target sequence was inserted downstream from each U6 promoter (U6:pvalb6sgRNA1). The number of Pvalb- and GFP-double-positive cells (black dots) is reduced if pvalb6 is targeted compared to control. Scale bar = 50 µm. (C) Quantification of the percentage of Pvalb-positive cells per GFP-positive clone. Data are represented as mean ± SEM. (***) P-value < 0.001 following Wilcoxon Mann–Whitney test. (D) Table showing the percentage of Pvalb-positive cells per GFP-positive clone in the retinal sections of double transgenic Tg(rx2:Gal4; myl7:GFP) × Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 larvae microinjected with the 2C-Cas9 plasmid containing control sgRNAs (first row) or pvalb6-targeting sgRNAs (second row).
atoh7 gene inactivation in retinal progenitors inhibits determination of retinal ganglion cells (RGCs) in clonally derived cell populations
We next wanted to test the utility of the 2C-Cas9 system in the analysis of a loss-of-function phenotype in cell clones. We chose to target the atonal bHLH transcription factor 7 (atoh7) gene because a characterized loss-of-function allele (lakritz) shows a clearly identifiable retinal phenotype (Kay et al. 2001). The lak mutation results in the loss of retinal ganglion cell (RGC) specification during eye development. Because rx2 expression precedes atoh7 activation, mutations induced by rx2-driven Cas9 may give rise to atoh7 mutant cells in RPCs before endogenous atoh7 is expressed. We therefore transiently expressed a pUAS:Cas9T2ACre;U6:atoh7sgRNA1;U6:atoh7sgRNA2 vector in double transgenic embryos deriving from a cross of Tg(rx2:Gal4;myl7:GFP) and Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 fish to induce targeted mutations at the atoh7 locus in RPCs. The mutagenesis efficiency of the sgRNAs used was assessed prior to insertion in the transgenesis vector (Supplemental Table S1) in transient injection experiments. Clones of GFP-positive cells were analyzed in sections of differentiated retinas of 5 dpf larvae (Fig. 5A–C). For this analysis, we chose to quantify only spatially well-separated clones that could be unambiguously identified, ranging from 10 to 55 cells per clone. We observed that the total number of cells per clone was not affected (25 ± 3 in controls vs. 24 ± 2 in atoh7 sgRNA clones; P = 0.91, Wilcoxon Mann–Whitney test), while the number of RGCs was significantly reduced in atoh7 targeted clones (6.1 ± 1.2 in controls vs. 2.4 ± 0.5 in atoh7 sgRNA clones; P < 0.01, Wilcoxon Mann–Whitney test). On average, in control retinas the RGC population represented 23% of the cells in each clone (57/247 cells analyzed). In contrast, in potentially atoh7-depleted clones only 9% of GFP-positive cells corresponded to differentiated RGCs (48/532 cells analyzed) (Fig. 5D). In the retinas of embryos injected with the control plasmid, all clones of cells (10/10) contained RGCs, representing, in the majority of clones (7/10), >20% of the GFP-positive cell population (Fig. 5E). In the retinal sections of embryos where the atoh7 locus was targeted, one third of the examined clones (7/22) displayed complete absence of RGCs, while only in a few cases (2/22 clones) the percentage of specified RGCs per clone was higher than 20% (Fig. 5E). These observations indicate that atoh7 loss-of-function is effectively induced by our vector system in rx2-expressing retinal progenitors.
Figure 5.

Clonal deletion of atoh7 leads to reduction in RGCs differentiation. Targeting of the atoh7 gene. Atoh7 is a transcription factor essential for the differentiation of retinal ganglion cells (RGCs). The pUAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 construct, with control or atoh7-specific sgRNAs, was injected in double transgenic embryos Tg(rx2:Gal4; myl7:GFP) × Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2. The latter transgene drives Gal4 expression in retinal progenitor cells. (A) (Upper panel) Differentiated RGCs are detected in wild-type clones injected with the DNA construct containing control sgRNAs. (Lower panel) GFP-labeled mutant clones show reduction of RGCs in the retinal section of a 5 dpf larva, as expected from atoh7 loss-of-function. Quantification of RGCs was done according to soma location in the ganglion cell layer (GCL), and displaced amacrine cells were distinguished by Parvalbumin counterstaining. Scale bar = 100 µm. (B) Higher magnification of the wild-type (upper panel) and atoh7 (lower panel) mutant retinal cell layers. RGCs are indicated by asterisks. (C) A schematic of labeled clones in the zebrafish retinal layers. (ONL) Outer nuclear layer, (INL) inner nuclear layer, (GCL) ganglion cell layer. (D) Quantification of the percentage of RGCs per clone derived from Cas9-expressing cells. Data are represented as mean ± SEM. (***) P-value < 0.001 following Wilcoxon Mann–Whitney test. (E) Table showing the percentage of RGCs per GFP-positive clone in the retinal sections of double transgenic Tg(rx2:Gal4; myl7:GFP) × Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 embryos microinjected with the 2C-Cas9 vector containing control sgRNAs (first row) or atoh7-specific sgRNAs (second row).
Analysis of genetic chimeras by combining tissue-specific loss-of-function with Brainbow single-cell labeling
To gain insights into complex loss-of-function phenotypes, it is necessary to understand if the biological effect of gene inactivation is cell-autonomous or non-cell-autonomous. Our method can be applied to generate genetic mosaics in which the phenotype of single mutant cells can be analyzed in a wild-type background, thus allowing the evaluation of cell-autonomous gene function. To this end, we combined our conditional knockout strategy with the Brainbow technology (Livet 2007; Pan et al. 2013), in which Cre recombinase stochastically activates the expression of different fluorescent proteins, allowing the differential labeling of single mutant cells in a wild-type tissue. In the UAS:Brainbow transgene (Tg[UAS:Br1.0L]s1997t) (Robles et al. 2013), Cre recombinase sites separate the cDNAs of the fluorescent proteins tdTomato, Cerulean, and YFP. When crossing a transgenic line carrying this cassette to cell-type–specific Gal4 lines, the Gal4 transactivator leads to the expression of tdTomato, in the absence of Cre-mediated recombination. In contrast, if Cre recombinase expression is induced, transcription of either cerulean or YFP is triggered (Fig. 6A). We first verified that transient expression of the pUAS:Cas9T2ACre;U6:sgRNA1;U6:sgRNA2 construct induces stochastic recombination of the UAS:Brainbow allele by injecting the plasmid into one-cell stage Tg(rpl5b:Gal4) or Tg(mnx1:Gal4) × Tg(UAS:Br1.0L)s1997t double transgenic embryos. Mosaic expression of fluorescent proteins was detected, as expected, in Gal4-positive cells (Fig. 6B) in agreement with the known Gal4 expression patterns of the two promoters (Fig. 1B; Supplemental Fig. S2B). We next used the 2C-Cas9 vector system to generate genetic chimeras where tdTomato- fluorescent cells are wild-type, while Cerulean- or YFP-labeled cells represent potentially mutant cells. In order to achieve this differential labeling, we injected the 2C-Cas9 vector in double transgenic embryos (carrying tissue-specific Gal4 driver and UAS:Brainbow). In our construct, Cas9 and Cre expression occur from the same transcript; therefore, Gal4-expressing cells that received the Cas9T2ACre plasmid will recombine the UAS:Br1.0L allele, inducing expression of cerulean or YFP and will potentially be knockout for the target gene. On the contrary, Gal4-expressing cells that did not receive the Cas9T2ACre plasmid will be marked by tdTomato fluorescence and wild-type (Fig. 6A).
Figure 6.
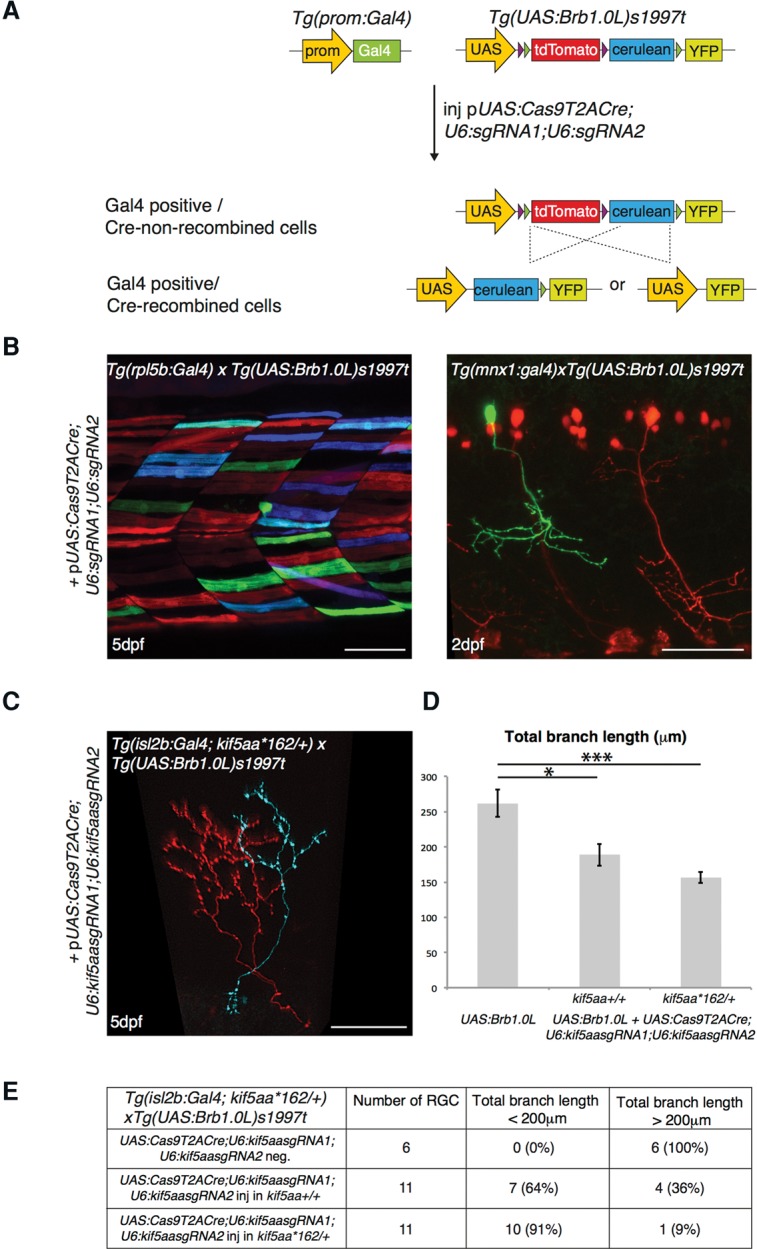
Labeling of mutant single cells by combining the 2C-Cas9 and the Brainbow methodologies. (A) Microinjection of the pUAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 construct in the double transgenic embryos Tg(promoter:Gal4) × Tg(UAS:Brb1.0L)s1997t leads to synthesis of both Cas9 and Cre. In Gal4-expressing cells, Cas9 promotes targeted cleavage of the chosen genomic locus. At the same time, Cre activity induces stochastic recombination of the UAS:Brb1.0L cassette, thus promoting a switch from the default tdTomato to YFP or Cerulean fluorescence in the same cells. (B) Recombination of the UAS:Brb1.0L cassette in a tissue-specific pattern induced by injection of the pUAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 plasmid. No target sequence was inserted in the vector prior to injection. (Left panel) Multicolor labeling of muscle cells in the Tg(rpl5b:Gal4) transgenic line. (Right panel) Single YFP motor neuron labeled by expression of UAS:Cas9T2ACre;U6sgRNA1;U6sgRNA2 transgene in the double transgenic embryos Tg(mnx1:Gal4) × Tg(UAS:Brb1.0L)s1997t. tdTomato marks motor neurons that do not express the UAS transgenic cassette. Scale bar = 50 µm. (C) Double transgenic Tg(isl2b:Gal4; kif5aa*162/+-) × Tg(UAS:Brb1.0L)s1997t larva, injected with pUAS:Cas9T2ACre;U6:Kif5aasgRNA1;U6:Kif5aasgRNA2. isl2b:Gal4 drives the expression of UAS:Brb1.0L transgene in RGC axons in the optic tectum. Note that the axon marked by cerulean expression, and therefore by Cre recombinase activity, shows reduced arbor size as expected from the phenotype of kif5aa−/− RGCs transplanted in wild-type host (Auer et al. 2015). Scale bar = 30 µm. (D) Quantification of total branch length in tdTomato-positive wild-type and potentially mutant Cerulean- or YFP-positive RGC axons (analyzed in kif5aa+/+: [*] P-value < 0.05 following Wilcoxon Mann–Whitney test, and kif5aa*162/+: [***] P-value < 0.001 following Wilcoxon Mann–Whitney test). Data are represented as mean ± SEM. (E) Table describing the percentage of RGCs displaying a wild-type-like (axonal length > 200 µm) and a mutant-like (axonal length < 200 µm) phenotype in the tectum of double transgenic Tg(isl2b:Gal4; kif5aa*162/+-) × Tg(UAS:Brb1.0L)s1997t larva, injected with pUAS:Cas9T2ACre;U6:Kif5aasgRNA1;U6:Kif5aasgRNA2. (First row) tdTomato-positive cells (UAS:Cas9T2ACre;U6:Kif5aasgRNA1;U6:Kif5aasgRNA2-negative). (Second row) Cerulean- or YFP-positive neurons (UAS:Cas9T2ACre;U6:Kif5aasgRNA1;U6:Kif5aasgRNA2-positive) in kif5aa+/+fish. (Third row) Cerulean- or YFP-positive neurons (UAS:Cas9T2ACre;U6:Kif5aasgRNA1;U6:Kif5aasgRNA2-positive) in kif5aa*162/+fish.
To confirm that cell-autonomous gene function can be examined by this method, we targeted the kif5aa gene, coding for the motor protein Kinesin family member 5A, a (Campbell and Marlow 2013; Auer et al. 2015). We selected this gene because we have recently shown that kif5aa inactivation results in the reduction of axon arbor complexity via a cell-autonomous mechanism: kif5aa−/− cells transplanted into wild-type host, after differentiation into RGCs, showed a severe reduction in axon arbor total length (Auer et al. 2015). We decided to apply the 2C-Cas9 system to inactivate the kif5aa gene in single RGCs while differentially labeling wild-type and potentially mutant cells in the same embryos. We used Tg(isl2b:Gal4) larvae to express the Gal4 transactivator in RGCs and drive 2C-Cas9-mediated gene disruption in this neuronal population. To further increase the efficiency of the knockout strategy, we used Tg(isl2b:Gal4) embryos heterozygous for an existing loss-of-function mutation in the kif5aa locus (kif5aa*162/+). We hypothesized that, if only one wild-type allele needs to be targeted by the Cas9/sgRNA complex, a loss-of-function phenotype would be generated with a higher probability. We therefore cloned two sgRNAs targeting the kif5aa locus (Auer et al. 2014b) (Supplemental Table S1) in the 2C-Cas9 vector and injected the resulting pUAS:Cas9T2ACre;U6:kif5aasgRNA1;U6:kif5aasgRNA2 in double transgenic embryos Tg(isl2b:Gal4; kif5aa*162/+) × Tg(UAS:Br1.0L)s1997t. In the injected embryos, YFP- or Cerulean-fluorescent RGCs (potentially kif5aa mutant) showed a decrease of total branch length compared to tdTomato-expressing RGCs (wild-type). As expected, an overall reduction of axonal length was detected in YFP or Cerulean RGCs compared to tdTomato RGCs, and it was more severe in kif5aa*162/+ than in kif5aa+/+ embryos (tdTomato-expressing RGCs = 261.8 ± 19.3 µm, n = 6; cerulean- or YFP-expressing RGCs in kif5aa+/+ = 188.8 ± 15.7 µm, n = 11, P < 0.05, Wilcoxon Mann–Whitney test; cerulean- or YFP-expressing RGCs in kif5aa*162/+ = 156.6 ± 7.7 µm, n = 11, P <0 .001, Wilcoxon Mann–Whitney test) (Fig. 6D). In addition, we noted the presence of two distinct RGC phenotypes in the Cre-labeled neurons: a wild-type-like (axonal length >200 µm) and a mutant-like (axonal length <200 µm). Remarkably, the number of cells showing a mutant-like phenotype was more abundant in the kif5aa*162/+ fish (91% of analyzed axons, n = 11) than in the kif5aa+/+ embryos (64% of analyzed axons, n = 11) (Fig. 6E). This observation confirms that the activation of the 2C-Cas9 system in a heterozygous genetic background leads to an increase of the proportion of mutant cells.
Our results demonstrate the utility of this method to generate genetic mosaic embryos and allow the labeling of multiple axons with different genotypes in distinct color combinations.
Discussion
The constant improvement of CRISPR/Cas9-mediated technologies over the past years is revolutionizing reverse genetic approaches in model organisms. Using the Gal4/UAS system for the first time in a vertebrate model to drive Cas9 expression, our method expands the possibility of spatiotemporally regulated gene knockout, allowing researchers to tap into the resource of existing Gal4 driver lines. The generation of a stable Tg(UAS:Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2) transgenic line, represents an advantage in the study of in vivo gene function in diverse cell types and tissues, as tissue-specific gene inactivation can be obtained simply by crossing to already established Gal4 driver lines. Although this transgenic expression system has been shown to experience somatic mosaicism from epigenetic silencing (Akitake et al. 2011), this has not diminished its great popularity and use in zebrafish and other organisms. Indeed, in some applications mosaic gene expression can even be a positive feature, allowing the analysis of the effect of gene inactivation in a sparse manner in single cells or cell clones. However, even when tissue-specific gene inactivation is achieved, the visualization of mutant cells is essential to the phenotypic analysis, an integral element that was not possible with previously published approaches.
In our work, we tested two methods to identify cells carrying loss-of-function alleles. In both cases, the use of a T2A self-cleaving peptide allows for the stoichiometric expression of the Cas9 and a reporter gene that are translated as a single polypeptide from the same mRNA (Provost et al. 2007; Kim et al. 2011). First, we used a GFP reporter whose expression is linked to that of the Cas9 to allow for efficient visualization of Cas9-expressing cells. Cas9T2AGFP transcription being dependent on transactivation by Gal4, the analysis of gene loss-of-function can be performed during the time frame of Gal4 expression, which is detected by GFP fluorescence. In some cases, the phenotypic evaluation of mutant cells may be required after Gal4 expression has terminated. For instance, when targeting the tyr gene with a Gal4 driver active in retinal progenitors, we could observe cells lacking pigmentation in larvae, but they did not express GFP, and therefore, absence of pigmentation could not be strictly correlated to inactivation of the tyrosinase gene in the unpigmented cells. The second method that we tested, 2C-Cas9, addresses the time frame issue by enabling both conditional generation of mutant cells and their permanent lineage tracing. In this assay, we used Cre activity to permanently label the population of Cas9-expressing cells, making it possible to analyze the phenotype resulting from targeted gene disruption after Cas9 expression has ceased. Importantly, if Cre-mediated recombination of a floxed allele results in the expression of a fluorescent protein driven by a constitutive promoter, the label would be transmitted from the Cas9-expressing founder cell to its progeny and remain detectable, thus generating a clone of labeled mutant cells. By using this method in retinal progenitor cells, we successfully disrupted the pvalb6 and atoh7 genomic loci, thus modifying molecular identity and cell-fate determination of their progeny, respectively.
To expand further the possibility to analyze tissue-specific gene inactivation, we took advantage of the flexibility of the pUAS:Cas9T2ACre;U6:sgRNA1;U6:sgRNA2 construct to achieve labeling of single mutant cells in an otherwise wild-type animal. So far, generation of genetic chimeras in biological systems has mainly been possible in the mouse and the fruit fly, greatly contributing to the preeminence of these animal models in genetic studies (Perrimon 1998; Wijgerde et al. 2002). In these models, the use of recombination-based approaches such as Cre/LoxP has been accepted as a gold standard technique for conditional mutagenesis. Until now, a similar approach has not been available in zebrafish. The phenotypic analysis of single mutant cells in a wild-type genetic background has relied on technically challenging transplantation experiments limited to embryonic development, which could not be temporally controlled. For this reason, genetic chimera experiments allow the investigation of only early phenotypes and cannot be used to examine the role of genes and pathways that are used repeatedly during development. Our multicolor labeling approach, that we achieved by combining the 2C-Cas9 system with the Brainbow technology (Livet 2007; Pan et al. 2013), permits the analysis of tissue-specific phenotypes of gene inactivation at a single cell resolution and enables the direct comparison of potentially mutant cells with their wild-type counterpart in the same animal. Cre/LoxP-based approaches in zebrafish, if properly developed, could also circumvent these limitations but would require much longer generation time and challenging homologous recombination-based genomic manipulations. Furthermore the efficiency of Cre-mediated recombination is well known to depend strongly on the Cre driver line used and the target locus, and potential mosaic inactivation needs to be taken into account also in this case (Branda and Dymecki 2004).
Although the 2C-Cas9 system is meant to allow the phenotypic evaluation of mutant cells, different parameters may influence the interpretation of phenotypes arising from the targeting of a specific locus with this method. To unambiguously follow the fate of mutant cells (or clones of cells), it would be necessary to activate a reporter gene exclusively in these populations. However, in the zebrafish model system, the tools to obtain this kind of tracking are still missing. As an alternative, we propose to mark potentially mutant cells by coexpressing the Cas9 enzyme together with GFP or Cre reporters. By using this approach, not all the analyzed cells will be mutant and each mutant cell will have independently induced mutations (a percentage of these might be silent in-frame mutations). If an antibody recognizing the protein encoded by the targeted gene is available, a single staining could identify all the cells carrying protein null-mutations. Indeed, we could reveal pvalb6 loss-of-function in the zebrafish neural retina by staining the population of Cas9-expressing cells with an antibody directed against this protein. In other cases, a molecular evaluation of the proportion of truly mutant cells within the analyzed population would be ideally needed to facilitate the statistical analysis of the behavior of the Cas9-expressing cells. A possibility to estimate the rate of mutation in the targeted tissue would be the separation of the marked Cas9-expressing cells by fluorescence-activated cell sorting (FACS). To be efficient, this procedure requires a highly pure and concentrated single-cell suspension, and it might be prone to contamination with debris or nonfluorescent cells. In addition, many transgenic lines contain fluorescent transgenesis markers (such as myl7:GFP or cry:GFP), which are wild-type fluorescent cells limiting the accuracy of the FACS for the evaluation of the mutagenesis efficiency.
In addition, the strength of the Gal4 driver used is essential in determining the efficiency of the 2C-Cas9 system, and its variability may result in differences in gene disruption. For instance, when using the Tg(rpl5:Gal4) line to target the tyr locus, we detected a mutagenesis rate that appeared lower than when using the rx2:Gal4 driver, where we observed phenotypes suggesting a highly efficient gene disruption. This result suggests that, in driver lines displaying low expression of the Gal4 transactivator, gene inactivation may be insufficient to observe phenotypic effects of loss-of-function. Therefore strong Gal4 carrier lines need to be used in order to achieve high mutagenesis rates. The intra-cellular levels of Cas9 expression provided by the 5×UAS of our vector system were sufficient to induce gene inactivation in cell clones or single cells in our experiments. Nevertheless, further improvements in the Cas9 expression cassette could increase Cas9 protein levels. Recently, noncoding elements of the zebrafish genome have been used to increase expression of transgenes and have been incorporated in UAS vectors (Horstick et al. 2015). The addition of such features to our vector design may have a strong impact on the expression of the Cas9, thus leading to a more penetrant tissue-specific loss-of-function. In addition to strong and stable Cas9 expression, the generation of a high rate of biallelic mutations requires highly active sgRNAs that will be facilitated by recently reported efficiency predictions (Moreno-Mateos et al. 2015). Furthermore, the design of sgRNAs recognizing genomic loci coding for essential domains of a protein would increase the possibility of inducing a null phenotype, even in the case of in-frame mutations (Shi et al. 2015). Importantly, we also show that one simple and powerful way to increase the efficiency of gene disruption is to inject the pUAS:Cas9T2ACre;U6:sgRNA1;U6:sgRNA2 in a heterozygous mutant background, where one allele is already constitutively mutated.
In this report, we present for the first time in zebrafish a strategy to track the fate of individual genetically modified cells within a wild-type whole animal environment, thus allowing detection of cell-autonomous defects resulting from mutations. The genetic lineage tracing of potentially mutant cells allows the phenotypic analysis of cell populations of interest until adulthood, broadening the use of the 2C-Cas9 in fields ranging from stem cells and regeneration to cancer biology and aging. Finally, because none of the tools that we generated are restricted to zebrafish, similar experiments are readily possible in virtually any organism where transgenesis and DNA injection are feasible.
Methods
Fish lines and husbandry
For this study, the transgenic and mutant lines used are listed in Supplemental Table S2. Zebrafish strains were maintained according to standard protocols (Westerfield 2000).
Molecular cloning
The Cas9T2AGFP fragment was generated by inserting a PCR-amplified fragment containing the T2AGFP sequence from the pCas9_GFP plasmid (Addgene, #44719) into the pCS2-nCas9n plasmid (Addgene, #47929). Primers used (5′ to 3′) were bglII-Cas9-T2A fwd: GGTGAGATCTCCTAAGAAGAAGAGAAAGGTGAGGTCCGGCGGCGGAG, and GFP XbaI rev: AGCTTCTAGATTACTTGTACAGCTC. BglII and XbaI enzymes were used to digest both plasmid and PCR product prior to ligation.
The U6:sgRNA1 sequence was synthetized as G-block from IDT. BsmbI restriction sites were introduced to clone the 20-bp target sequence at the predicted transcription start site (+1). The Gibson assembly kit (NEB) was used to clone the U6:sgRNA1 and the Cas9T2AGFP fragments into a pminiTol2 vector (Balciunas et al. 2006) containing a 5×UAS cassette and digested with EcoRI and ClaI. Primers used were (5′ to 3′): U6 fwd: GCAATAAACCTTGTACAAAGTGGGGGATC, and U6 rev: GAGCTCGAATTAATTCATAATTGAAAAAAAGCACCGAC to amplify the U6:sgRNA fragment, and Cas9-T2A-GFP fwd: CTGAATAGGGAATTGGGGCCACCATGGCTTCTCCA, and Cas9-T2A-GFP rev: TTGTACAAGGTTTATTGCAGCTTATAATGGTTACAAATAAAG to amplify the Cas9T2AGFP fragment.
The second U6:sgRNA2 cassette was designed with SalI overhangs for insertion into the p(UAS:Cas9T2AGFP;U6:sgRNA1) plasmid linearized with the same enzyme. BsaI sites were used to clone the 20-nt sgRNA target sequence at the +1 position.
The Cas9T2ACre fragment was synthetized by fusion of individual PCR products using Phusion High-Fidelity DNA Polymerase (Thermo Scientific). The Cre sequence was amplified from the pCR8GW-Cre-FRT-kan-FRT 2 plasmid (Suster et al. 2011). Primers used (5′ to 3′) were T2A-Cre fwd: GAGGAAGTCTTCTAACATGCGGTGACGTGGAGGAGAATCCCGGCCCAATGGCCAATTTACTGACCGTACAC, and Cre-XbaI rev: CGATTCTAGACTAATCGCCATCTTCCAGC. The Cas9T2A fragment was amplified from the pUAS:Cas9T2AGFP;U6:sgRNA1;U6sgRNA2. Primers used (5′ to 3′) were Cas9 KpnI fwd: GGTCGGTACCGCACTGATCAAGAAATAC, and T2A rev: GTCACCGCATGTTAGAAGACTTCC. Both fragments were fused, amplified, and inserted into the pUAS:Cas9T2AGFP;U6:sgRNA1;U6sgRNA2 digested with KpnI and XbaI. The sequence of all constructs was verified by sequencing.
Generation of stable transgenic lines
The Tg(UAS:Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2) and Tg(UAS:Cas9T2AGFP; U6:tyrsgRNA1;U6:tyrsgRNA2) were generated by injecting the previously described plasmids into one-cell stage wild-type embryos. The rx2:Gal4 vector was generated by three-way-Gateway cloning recombining a 5′ entry vector carrying the rx2 promoter fragment (Heermann et al. 2015) with a Gal4 middle entry, a polyA 3′entry vector, and a myl7:eGFP-Tol2 destination vector (Kwan et al. 2007). To generate the rpl5b:Gal4 vector, we cloned a 5700-bp promoter fragment of the ribosomal gene rpl5b in a 5′ entry vector and recombined it with a Gal4 middle entry, a polyA 3′ entry vector, and a Tol2 destination vector (Kwan et al. 2007). To generate the Tg(-3.5ubb:loxP-lacZ-loxP-eGFP)cn2 transgenic line (also known as cn2Tg or Hulk), we used a three-way-Gateway cloning system. p5E -3.5ubb:loxP-lacZ-loxP was generated by inserting the result of digesting the iZEG plasmid (Novak et al. 2000) with XbaI and XhoI and making blunt ends into the p5E ubi (Mosimann et al. 2011) after being linearized with BamHI and blunting the ends. pDest cry:GFP was generated by inserting the result of digesting the p1 cry:GFP (Love et al. 2011) with SacII and making blunt ends into the pDestTol2pA2 (Kwan et al. 2007) that was linearized with BglII. The -3.5ubb:loxP-lacZ-loxP-eGFP; cry:GFP construct was generated combining these four plasmids into a single one using Gateway LR Clonase II, Life Technologies: p5E -3.5ubb:loxP-lacZ-loxP, pME GFP (Kwan et al. 2007), p3E pA (Kwan et al. 2007), and pDest cry:GFP. Stable transgenic lines were generated injecting plasmid DNA with Tol2 mRNA transposase and founders identified by genetic crossing and transgene transmission. The cry:GFP cassette contained in this transgene allows the selection of transgenic embryos due to the GFP expression in the developing lens under the control of the gamma-crystallin promoter.
Injections
To test the mutagenesis efficiency of sgRNAs, a mixture of sgRNA and Cas9 mRNA was injected into one-cell stage zebrafish embryos. The final concentration was 75 ng/µL for sgRNA and 150 ng/µL for Cas9 mRNA. For Tol2-mediated transgenesis, p(UAS:Cas9T2AGFP;U6:sgRNA1;U6sgRNA2) or p(UAS:Cas9T2ACre;U6:sgRNA1;U6sgRNA2) were co-injected at a concentration of 30 ng/µL with the Tol2 transposase mRNA (50 ng/µL) into the selected Tg(Promoter:Gal4) lines. Genomic DNA was extracted from either single embryos or pools of embryos and then used for PCR and DNA sequencing experiments.
Whole-embryos DNA extraction
For genomic DNA extraction, pools of 25 embryos at 5 dpf were digested for 1 h at 55°C in 0.5 mL lysis buffer (10 mM Tris, pH 8.0, 10 mM NaCl, 10 mM EDTA, and 2% SDS) with proteinase K (0.17 mg/mL, Roche Diagnostics). To check for frequency of indel mutations, target genomic loci were PCR-amplified using Phusion High-Fidelity DNA polymerase (Thermo Scientific). PCR amplicons were subsequently cloned into the pCR-bluntII-TOPO vector (Invitrogen). Plasmid DNA was isolated from single colonies and sent for sequencing. Mutant alleles were identified by comparison with the wild-type sequence.
Cryosections
Embryos at the stage of 5 dpf were fixed in 4% paraformaldehyde in PBS (pH 7.4) for 2 h at room temperature and subsequently cryoprotected overnight in a 30% sucrose/0.02% sodium azide/PBS solution. Embryos were transferred to plastic molds and embedded in OCT after removal of the sucrose. The blocks were placed on dry ice before sectioning. The sections were cut with a thickness of 14 µm and mounted on Fisherbrand Superfrost plus slides (No. 12- 550-15).
Immunohistochemistry
Cryosections of 5 dpf retinas were washed twice in 1× PBS/0.1% Tween-20 (PBS-T) solution. Subsequently, they were incubated 1 h at room temperature in 10% normal goat serum (Invitrogen) in PBS-T blocking solution followed by overnight incubation with 1/500 dilution of chicken primary anti-GFP (Genetex) or mouse anti-Parvalbumin antibody (Millipore). The Alexa Fluor 488 secondary antibody goat anti-chicken IgG or the Alexa Fluor 568 secondary antibody goat anti-mouse IgG (1/500, Molecular Probes) and a 1/500 dilution of DAPI (50 µg/µL) in blocking solution were added for 2 h at room temperature. After five washings in wash buffer (1× PBS/0.1% Tween-20) coverslips were placed on the slides after addition of Vectashield drops. Slides were left at room temperature for 1 h before microscopy analysis.
sgRNAs and Cas9 mRNA generation
sgRNA sequences (listed in Supplemental Table S1) were cloned into the BsaI-digested pDR274 (Addgene, #42250) vector. The sgRNAs were synthetized by in vitro transcription (using the Megascript T7 transcription kit #AM1334, Ambion). After transcription, sgRNAs were purified using an RNAeasy Mini Kit (Qiagen). The quality of purified sgRNAs was checked by electrophoresis on a 2% agarose gel. As a control, we used RFP-specific sgRNAs, whose sequence is not present in the fish genome (Supplemental Table S1). Cas9 mRNA was generated as described previously (Hwang et al. 2013a).
FACS
Fluorescent Cas9-expressing cells were isolated from wild-type cells by fluorescence-activated cell sorting. 3 dpf embryos were dissociated as previously described by Manoli and Driever (2012). Cell sorting was performed on FACS Aria (BD Biosciences), and data were analyzed using FACSDiva version 6.1.2 (BD Biosciences). The GFP fluorescence was detected in the gfpBlue-B-530/30-A channel. GFP-positive and -negative cells were sorted in lysis buffer provided in the NucleoSpin Tissue Kit (Macherey-Nagel), and genomic DNA was extracted with the same kit. PCR on target genomic loci and DNA sequencing were performed as described above.
Microscopy
Low magnification images were acquired with a Leica MZ FLIII stereomicroscope (Leica) equipped with a Leica DFC310FX digital camera (Leica). Whole-eye pictures were taken with a Leica upright wide-field epifluorescence microscope using a 20× oil immersion objective. A Zeiss LSM 780 confocal microscope (Zeiss) was used for confocal microscopy, employing a 40× water immersion or 10× objective. Z-volumes were acquired with a 1- to 2-μm resolution, and images were processed using ImageJ, Adobe Photoshop, and Adobe Illustrator software.
Data access
The sequences of mutant tyr clones from this study have been submitted to the NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession numbers KU751772–KU751776. The sequences and the maps relative to the plasmids pUAS:Cas9T2AGFP;U6:sgRNA1;U6:sgRNA2 and pUAS:Cas9T2ACre;U6:sgRNA1;U6:sgRNA2 are available in Addgene (https://www.addgene.org). Plasmid numbers are #74009 and #74010.
Acknowledgments
We thank Allison Bardin, Allison Mallory, Yohanns Bellaiche, Christoph Gebhardt, Shahad Albadri, Celine Revenu, and Valerie Bercier for critical reading of the manuscript. We also thank Herwig Baier, Estuardo Robles, Joachim Wittbrodt, Koichi Kawakami, Claire Wyart, Nadine Peyriéras, and Georges Lutfalla for sharing reagents and transgenic lines. We thank all members of the Del Bene lab for fruitful discussions. We thank the Developmental Biology Curie imaging facility (PICT-IBiSA@BDD, Paris, France, UMR3215/U934) member of the France-BioImaging national research infrastructure for their help and advice with confocal microscopy and the Flow Cytometry and Cell Sorting Platform at Institute Curie for their expertise. The Del Bene laboratory “Neural Circuits Development” is part of the Laboratoire d'Excellence (LABEX) entitled DEEP (ANR -11-LABX-0044), and of the École des Neurosciences de Paris Ile-de-France network. V.D.D. was supported by a doctoral fellowship of the Fondation Pierre-Gilles de Gennes pour la recherche and Fondation Recherche Medicale (FRM) 4th year doctoral fellowship. F.D.S. was supported by a doctoral fellowship of the Curie International PhD program. T.O.A. was supported by a Boehringer Ingelheim Fonds PhD fellowship. H.S. was funded through FPU12/03007. This work has been supported by an ATIP/AVENIR program starting grant (F.D.B.), ERC-StG #311159-Zebratectum (F.D.B.), ANR-II-INBS-0014 (J.P.C.), CNRS, INSERM, Institut Curie (F.D.B), and Muséum National d'Histoire Naturelle (J.P.C) core funding, the Fundación CNIC Carlos III (N.M.), the Fundación ProCNIC (N.M.), the Spanish Ministry of Economy and Competiveness (Tercel and BFU2011-25297) (N.M.), and ERC Starting grant 337703 – zebraHeart (N.M.).
Author contrubutions: V.D.D., F.D.S., T.O.A, J.P.C., and F.D.B. conceived and designed experiments. V.D.D., F.D.S., and N.T. performed molecular cloning experiments. V.D.D. and F.D.S. performed microinjections, immunohistochemistry, and imaging experiments. T.O.A. generated essential transgenic lines. V.D.D. and F.D.S. analyzed the data. V.D.D., F.D.S., J.P.C., and F.D.B. wrote the manuscript with inputs from all the coauthors. H.S. and N.M. provided essential unpublished reagents (Tg[-3.5ubb:loxP-lacZ-loxP-eGFP]cn2 transgenic line).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.196170.115.
References
- Ablain J, Durand EM, Yang S, Zhou Y, Zon LI. 2015. A CRISPR/Cas9 vector system for tissue-specific gene disruption in zebrafish. Dev Cell 32: 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akitake CM, Macurak M, Halpern ME, Goll MG. 2011. Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol 352: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amsterdam A, Nissen RM, Sun Z, Swindell EC, Farrington S, Hopkins N. 2004. Identification of 315 genes essential for early zebrafish development. Proc Natl Acad Sci 101: 12792–12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa K, Kawakami K. 2008. Targeted gene expression by the Gal4-UAS system in zebrafish. Dev Growth Differ 50: 391–399. [DOI] [PubMed] [Google Scholar]
- Asakawa K, Suster ML, Mizusawa K, Nagayoshi S, Kotani T, Urasaki A, Kishimoto Y, Hibi M, Kawakami K. 2008. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proc Natl Acad Sci 105: 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer TO, Duroure K, Concordet JP, Del Bene F. 2014a. CRISPR/Cas9-mediated conversion of eGFP- into Gal4-transgenic lines in zebrafish. Nat Protoc 9: 2823–2840. [DOI] [PubMed] [Google Scholar]
- Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F. 2014b. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24: 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer TO, Xiao T, Bercier V, Gebhardt C, Duroure K, Concordet JP, Wyart C, Suster M, Kawakami K, Wittbrodt J, et al. 2015. Deletion of a kinesin I motor unmasks a mechanism of homeostatic branching control by neurotrophin-3. eLife 4 10.7554/eLife.05061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, Wang X, Hackett PB, Largaespada DA, McIvor RS, et al. 2006. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet 2: e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciuniene J, Nagelberg D, Walsh KT, Camerota D, Georlette D, Biemar F, Bellipanni G, Balciunas D. 2013. Efficient disruption of Zebrafish genes using a Gal4-containing gene trap. BMC Genomics 14: 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branda CS, Dymecki SM. 2004. Talking about a revolution: the impact of site-specific recombinases on genetic analyses in mice. Dev Cell 6: 7–28. [DOI] [PubMed] [Google Scholar]
- Camp E, Lardelli M. 2001. Tyrosinase gene expression in zebrafish embryos. Dev Genes Evol 211: 150–153. [DOI] [PubMed] [Google Scholar]
- Campbell PD, Marlow FL. 2013. Temporal and tissue specific gene expression patterns of the zebrafish kinesin-1 heavy chain family, kif5s, during development. Gene Expr Patterns 13: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232. [DOI] [PubMed] [Google Scholar]
- Chuang JC, Raymond PA. 2001. Zebrafish genes rx1 and rx2 help define the region of forebrain that gives rise to retina. Dev Biol 231: 13–30. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison JM, Akitake CM, Goll MG, Rhee JM, Gosse N, Baier H, Halpern ME, Leach SD, Parsons MJ. 2007. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev Biol 304: 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbig KM, Lekven AC, Kunkel GR. 2008. Zebrafish U6 small nuclear RNA gene promoters contain a SPH element in an unusual location. Gene 421: 89–94. [DOI] [PubMed] [Google Scholar]
- Heermann S, Schutz L, Lemke S, Krieglstein K, Wittbrodt J. 2015. Eye morphogenesis driven by epithelial flow into the optic cup facilitated by modulation of bone morphogenetic protein. eLife 4 10.7554/eLife.05216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstick EJ, Jordan DC, Bergeron SA, Tabor KM, Serpe M, Feldman B, Burgess HA. 2015. Increased functional protein expression using nucleotide sequence features enriched in highly expressed genes in zebrafish. Nucleic Acids Res 43: e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JR. 2013a. Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One 8: e68708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK. 2013b. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol 31: 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao LE, Wente SR, Chen W. 2013. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci 110: 13904–13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. 2004. Transgenesis and gene trap methods in zebrafish by using the Tol2 transposable element. Methods Cell Biol 77: 201–222. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Abe G, Asada T, Asakawa K, Fukuda R, Ito A, Lal P, Mouri N, Muto A, Suster ML, et al. 2010. zTrap: zebrafish gene trap and enhancer trap database. BMC Dev Biol 10: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Finger-Baier KC, Roeser T, Staub W, Baier H. 2001. Retinal ganglion cell genesis requires lakritz, a zebrafish atonal homolog. Neuron 30: 725–736. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, Kim MK, Shin BA, Choi SY. 2011. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 6: e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB. 2007. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236: 3088–3099. [DOI] [PubMed] [Google Scholar]
- Livesey FJ, Cepko CL. 2001. Vertebrate neural cell-fate determination: lessons from the retina. Nat Rev Neurosci 2: 109–118. [DOI] [PubMed] [Google Scholar]
- Livet J. 2007. [The brain in color: transgenic “Brainbow” mice for visualizing neuronal circuits]. Med Sci (Paris) 23: 1173–1176. [DOI] [PubMed] [Google Scholar]
- Love NR, Thuret R, Chen Y, Ishibashi S, Sabherwal N, Paredes R, Alves-Silva J, Dorey K, Noble AM, Guille MJ, et al. 2011. pTransgenesis: a cross-species, modular transgenesis resource. Development 138: 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339: 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli M, Driever W. 2012. Fluorescence-activated cell sorting (FACS) of fluorescently tagged cells from zebrafish larvae for RNA isolation. Cold Spring Harb Protoc 2012 10.1101/pdb.prot069633. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. 2015. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12: 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, Zon LI. 2011. Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development 138: 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevin LM, Taylor MR, Baier H. 2008. Hardwiring of fine synaptic layers in the zebrafish visual pathway. Neural Dev 3: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. 2000. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis 28: 147–155. [PubMed] [Google Scholar]
- Pan X, Wan H, Chia W, Tong Y, Gong Z. 2005. Demonstration of site-directed recombination in transgenic zebrafish using the Cre/loxP system. Transgenic Res 14: 217–223. [DOI] [PubMed] [Google Scholar]
- Pan YA, Freundlich T, Weissman TA, Schoppik D, Wang XC, Zimmerman S, Ciruna B, Sanes JR, Lichtman JW, Schier AF. 2013. Zebrabow: multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development 140: 2835–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrimon N. 1998. Creating mosaics in Drosophila. Int J Dev Biol 42: 243–247. [PubMed] [Google Scholar]
- Platt RJ, Chen S, Zhou Y, Yim MJ, Swiech L, Kempton HR, Dahlman JE, Parnas O, Eisenhaure TM, Jovanovic M, et al. 2014. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159: 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Chen HM, Lee T, Bullock SL. 2014. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc Natl Acad Sci 111: E2967–E2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provost E, Rhee J, Leach SD. 2007. Viral 2A peptides allow expression of multiple proteins from a single ORF in transgenic zebrafish embryos. Genesis 45: 625–629. [DOI] [PubMed] [Google Scholar]
- Robles E, Filosa A, Baier H. 2013. Precise lamination of retinal axons generates multiple parallel input pathways in the tectum. J Neurosci 33: 5027–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EK, Baier H. 2009. The cellular architecture of the larval zebrafish tectum, as revealed by Gal4 enhancer trap lines. Front Neural Circuits 3: 13 10.3389/neuro.04.013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott EK, Mason L, Arrenberg AB, Ziv L, Gosse NJ, Xiao T, Chi NC, Asakawa K, Kawakami K, Baier H. 2007. Targeting neural circuitry in zebrafish using GAL4 enhancer trapping. Nat Methods 4: 323–326. [DOI] [PubMed] [Google Scholar]
- Shen Z, Zhang X, Chai Y, Zhu Z, Yi P, Feng G, Li W, Ou G. 2014. Conditional knockouts generated by engineered CRISPR-Cas9 endonuclease reveal the roles of Coronin in C. elegans neural development. Dev Cell 30: 625–636. [DOI] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. 2015. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol 33: 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague J, Bayraktaroglu L, Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Haendel M, Howe DG, Knight J, et al. 2008. The Zebrafish Information Network: The zebrafish model organism database provides expanded support for genotypes and phenotypes. Nucleic Acids Res 36: D768–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suster ML, Abe G, Schouw A, Kawakami K. 2011. Transposon-mediated BAC transgenesis in zebrafish. Nat Protoc 6: 1998–2021. [DOI] [PubMed] [Google Scholar]
- Westerfield M. 2000. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio), 4th ed University of Oregon Press, Eugene, OR. [Google Scholar]
- Wijgerde M, McMahon JA, Rule M, McMahon AP. 2002. A direct requirement for Hedgehog signaling for normal specification of all ventral progenitor domains in the presumptive mammalian spinal cord. Genes Dev 16: 2849–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L, Maddison LA, Li M, Kara N, LaFave MC, Varshney GK, Burgess SM, Patton JG, Chen W. 2015. Multiplex conditional mutagenesis using transgenic expression of Cas9 and sgRNAs. Genetics 200: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]