

Abstract
ATP-dependent chromatin remodelers regulate chromatin dynamics by modifying nucleosome positions and occupancy. DNA-dependent processes such as replication and transcription rely on chromatin to faithfully regulate DNA accessibility, yet how chromatin remodelers achieve well-defined nucleosome positioning in vivo is poorly understood. Here, we report a simple method for site-specifically altering nucleosome positions in live cells. By fusing the Chd1 remodeler to the DNA binding domain of the Saccharomyces cerevisiae Ume6 repressor, we have engineered a fusion remodeler that selectively positions nucleosomes on top of adjacent Ume6 binding motifs in a highly predictable and reproducible manner. Positioning of nucleosomes by the fusion remodeler recapitulates closed chromatin structure at Ume6-sensitive genes analogous to the endogenous Isw2 remodeler. Strikingly, highly precise positioning of single founder nucleosomes by either chimeric Chd1-Ume6 or endogenous Isw2 shifts phased chromatin arrays in cooperation with endogenous chromatin remodelers. Our results demonstrate feasibility of engineering precise nucleosome rearrangements through sequence-targeted chromatin remodeling and provide insight into targeted action and cooperation of endogenous chromatin remodelers in vivo.
Due to the intimate wrapping of DNA with histones, nucleosomes present a barrier for factors requiring direct access to DNA. Thus, dynamic regulation of nucleosomes and chromatin structure is paramount for proper control of DNA-dependent processes such as transcription, replication, and DNA repair. A major class of regulatory enzymes are ATP-dependent chromatin remodelers, which can modulate accessibility of underlying DNA sequences to transcription factors, polymerases, and other essential proteins by altering the positions and occupancy of nucleosomes throughout the genome. Despite the importance of chromatin remodelers in genome maintenance, it has been difficult to understand the direct consequences of targeted chromatin remodeling events in vivo.
A central challenge in identifying direct effects of targeted chromatin remodeling on nucleosome positioning has been the inability to disentangle localized changes in chromatin from downstream or indirect secondary effects. In budding yeast, the best-characterized changes in local nucleosome positioning result from targeting of Isw2 by transcriptional repressors such as Ume6 or Tup1/Cyc8 (Goldmark et al. 2000; Fazzio et al. 2001; Kent et al. 2001; Zhang and Reese 2004a,b; Hanlon et al. 2011; Rizzo et al. 2011). On a global scale, Isw2 localizes nonspecifically to nucleosome-depleted regions (NDRs) to affect nucleosomes within gene bodies (Zentner et al. 2013), enriches at nucleosomes flanking transcription start sites (TSSs) to act on proximal (+1) nucleosomes (Yen et al. 2012), yet also binds specific target nucleosomes to facilitate nucleosome sliding into NDRs (Fazzio and Tsukiyama 2003; Gelbart et al. 2005; Whitehouse and Tsukiyama 2006). This discrepancy in genomic Isw2 localization, along with the nonspecific nucleosome sliding activity observed in vitro (Kagalwala et al. 2004; Zofall et al. 2004; Dang et al. 2006; Stockdale et al. 2006), makes it difficult to reconcile how the activity of chromatin remodeling factors such as Isw2 can be utilized to faithfully maintain chromatin structure in vivo.
By swapping the DNA binding domain of the Chd1 chromatin remodeler with sequence-specific DNA binding proteins or monomeric streptavidin, we previously showed that chromatin remodeling activity could be targeted to specific nucleosomes in vitro (McKnight et al. 2011; Patel et al. 2013). This approach allowed us to control nucleosome selection and predict nucleosome sliding directionality and positioning. Here, we extend the strategy of site-specifically targeting nucleosome sliding activity to investigate the direct consequences of precisely altering local chromatin structure in vivo. We show that targeted chromatin remodeling in Saccharomyces cerevisiae leads to highly specific and predictable nucleosome reorganization that gives rise to phased chromatin arrays and context-dependent effects on transcription. Interestingly, we find that nucleosome boundaries for chromatin array formation at target loci are similarly generated by the endogenous Isw2/Ume6 nucleosome positioning system. These results give unique insight into how Isw2, a conserved chromatin remodeling factor, influences nucleosome organization in vivo. Moreover, this work establishes a novel tool for inducing and characterizing targeted chromatin rearrangements in live cells.
Results
Strategy for targeting chromatin remodeling activity in vivo
By replacing the native DNA binding domain with foreign, sequence-specific binding domains, we previously showed that the Chd1 remodeler could be sequence-targeted and move nucleosomes directionally in vitro (McKnight et al. 2011; Patel et al. 2013). We sought to demonstrate the feasibility of introducing an engineered chromatin remodeling system in a living organism to disrupt nucleosome positions. Such a method would allow us to interrogate downstream changes in chromatin structure and transcriptional regulation in response to isolated, specific changes in nucleosome positioning. Given its strong connections to endogenous chromatin remodeling, the well-studied Ume6 repressor was chosen as an in vivo system for designer remodeler targeting. The DNA binding region of Ume6 contains a Zn2Cys6 DNA binding domain that interacts specifically with URS1 DNA sequences but lacks the ability to repress transcription (Anderson et al. 1995; Kadosh and Struhl 1997). This Ume6 DNA binding domain was used to replace the native Chd1 DNA binding domain, generating the chimeric Chd1-Ume6 remodeler (Fig. 1A). Consistent with other chimeric remodelers (McKnight et al. 2011), recombinantly purified Chd1-Ume6 robustly and specifically repositioned nucleosomes containing a URS1 sequence in vitro (Fig. 1B). Based on our previous in vitro studies (McKnight et al. 2011), one expectation was that Chd1-Ume6 would move nucleosomes toward Ume6 binding motifs until the recruitment motif was occluded by nucleosomal DNA. Such nucleosome rearrangements could conceivably substitute for Isw2 in the absence of a functional Isw2/Ume6 interaction. Conversely, in the presence of a fully repressed wild-type system, these rearrangements could result in a chromatin structure where Ume6 can no longer bind, potentially leading to disruption of transcriptional repression (Fig. 1C).
Figure 1.

Strategy for targeting of chromatin remodeling activity in vivo. (A) Schematic of Chd1-Ume6 fusion protein. A fragment of S. cerevisiae Chd1 (residues 118–1014) was fused to the DNA binding domain of S. cerevisiae repressor Ume6 (residues 764–836) and cloned into the p416-ADH expression vector (Mumberg et al. 1995) for stable yeast transformation. (B) Sequence-directed nucleosome sliding by Chd1-Ume6 in vitro. Nucleosomes (50 nM) containing a URS1 site (TGGCGGCT) 8 bp from the edge of an end-positioned, FAM-labeled mononucleosome with 80 bp of extranucleosomal DNA were incubated with 10 nM of Chd1-Ume6 (top) or Chd1118-1014 lacking a DNA binding domain (middle) for the indicated times, and nucleosome positions were separated by 6% native PAGE. Under these conditions, nucleosome sliding required both the Ume6 DNA binding domain (middle) and the URS1 binding site (bottom). (C) Summary of strains used and expected outcomes for chromatin remodeling by Chd1-Ume6 on transcription and nucleosome positions at genomic target sequences.
Genome-wide targeting of chromatin remodeling activity results in specific and predictable chromatin changes
In the absence of functional Isw2 or Ume6 protein, chromatin structure is generally open (large NDRs) at promoters containing Ume6 binding sites (Goldmark et al. 2000; Fazzio et al. 2001; Kent et al. 2001). To determine if Chd1-Ume6 could substitute for this endogenous chromatin remodeling system and promote closed chromatin (smaller NDRs), the fusion protein was introduced into yeast strains lacking UME6 or ISW2, and nucleosome dyad positions were mapped by MNase-seq (Fig. 2). Compared with the Δume6 parent strain background, expression of Chd1-Ume6 altered chromatin structure near known Ume6 binding motifs of Ume6-responsive genes (Fig. 2A). These alterations of nucleosome positions were not seen when the ATPase-dead variant Chd1D513N-Ume6 was introduced, suggesting that the observed changes in nucleosome positions were due to ATP-dependent chromatin remodeling by Chd1-Ume6.
Figure 2.

Chd1-Ume6 organizes chromatin at genomic Ume6 target sequences. (A) MNase-seq signal of nucleosome dyads (opaque) and nucleosome footprints (transparent) for the indicated backgrounds at genomic instances of Ume6 recognition motifs. The presence of catalytically active Chd1-Ume6 led to shifted nucleosome dyad peaks (red asterisks) adjacent to predicted Ume6 recognition motifs (blue lines). The Chd1D513N-Ume6 indicates a catalytically inactive Chd1-Ume6 fusion protein containing a Walker B mutation (Hauk et al. 2010). (B, left) Differences in nucleosome dyad signal between Δume6 and Δume6 +[Chd1-Ume6] strains for a 400-bp window centered at permutations of a core GGCGGC motif. Groups 1, 2, and 3 correspond to variations of WNGGCGGCWW, NNGGCGGCNN, and disrupted GGCGGC motifs, respectively. Peak nucleosome movement occurs at the sequence WNGGCGGCWW (asterisk). (Right) Signal differences from GGCGGC-based motifs are compared with other known transcription factor binding motifs from the JASPAR database (Mathelier et al. 2014). Black and red indicate where nucleosome signal increased and decreased, respectively, upon addition of Chd1-Ume6. All ordered motif identities are provided in Supplemental Data File 1. (C) Difference in nucleosome dyad signal between Δume6 and Δume6 +[Chd1-Ume6] strains at all intergenic instances of the WNGGCGGCWW motif, clustered by direction of nucleosome repositioning (left) with average dyad signal within each cluster for individual strains (right).
The Ume6 DNA binding domain interacts with URS1 elements containing a core GGCGGC motif (Anderson et al. 1995). To determine the extent of targeted Chd1-Ume6 remodeling, we evaluated differences in nucleosome positions genome-wide at intergenic regions containing permutations of this core URS1 motif (Fig. 2B; Supplemental Data File 1). Nucleosome positioning differences between the Δume6 and Δume6+[Chd1-Ume6] strains were maximized at the sequence WNGGCGGCWW, which closely resembles previously published Ume6 binding motifs (Park et al. 1992; Harbison et al. 2004; Gordan et al. 2011). Significant differences in nucleosome positions were not observed at sequences lacking the core GGCGGC or at any other known transcription factor binding motif, demonstrating high specificity of the Chd1-Ume6 fusion remodeler. Additionally, no positioning was observed when the catalytically inactive Chd1D513N-Ume6 construct was introduced into cells (Supplemental Fig. S1).
Our previous in vitro studies showed that Chd1 fusion proteins directionally reposition nucleosomes toward DNA recruitment sites and can support flexible linkages between Chd1 and the foreign binding domain (McKnight et al. 2011; Nodelman and Bowman 2013). Consistent with this, in yeast intergenic regions, Chd1-Ume6 stimulated nucleosome positioning toward and on top of WNGGCGGCWW motifs (Fig. 2C). On average, recruitment motifs were buried by ∼20 bp, placing the motif ∼50 bp from the dyad of the positioned nucleosome. The orientation of the recruitment motif did not affect the direction of positioning, suggesting flexibility between the Ume6 DNA binding domain and the rest of Chd1. The direction of nucleosome movement did not correlate to the location of neighboring TSSs and positioning was also observed within coding regions, at transcription termination sites (TTSs), and at locations lacking known genomic features (Supplemental Fig. S2; Supplemental Data File 2). Interestingly, repositioning was only observed for nucleosomes with dyads located between 50 and 150 bp from the recruitment motif in the Δume6 strain (Fig. 2C, clusters 1 and 3). Nucleosomes with dyads closer than 50 bp or farther than 150 bp were not significantly shifted, suggesting that the motif either was inaccessible due to burial on a nucleosome or was out of reach. Similar results were obtained in an Δisw2 strain background (Supplemental Fig. S3), indicating that observed nucleosome shifts were not due to endogenous Isw2 remodeling activity. These results agree well with previous in vitro results of other Chd1 fusion proteins, which were found to have a finite “reach” and move nucleosomes ∼20 bp on top of recruitment motifs, irrespective of motif orientation (McKnight et al. 2011; Nodelman and Bowman 2013; Patel et al. 2013). Together, these results verify the successful engineering and introduction of targeted, directional ATP-dependent nucleosome sliding in vivo.
It is well established that Ume6-directed recruitment of Isw2 leads to local reorganization of chromatin structure (Goldmark et al. 2000; Fazzio et al. 2001; Kent et al. 2001). To directly compare the endogenous Isw2/Ume6 system to the Chd1-Ume6 fusion remodeler, we determined nucleosome positions of the parent wild-type strain by MNase-seq (Fig. 3). As expected and similar to the Chd1-Ume6 fusion remodeler, comparison of the wild type with the Δume6 strain revealed that Isw2/Ume6 preferentially moved nucleosomes with adjacent WNGGCGGCWW motifs and did not target chromatin with sequences lacking the full GGCGGC motif or at other transcription factor binding sites (Fig. 3A). As for sequence-directed sliding by Chd1-Ume6, this movement appeared to be specific to motif location and not to the relative location of a nearby TSS, as precise positioning was not observed with respect to nearby TSSs, and nucleosome positioning was also achieved near TTSs and featureless motifs (Supplemental Fig. S4; Supplemental Data File 2). Similar results were seen when the wild-type strain was compared with Δisw2 at variations of the Ume6 motif. Notably, however, deletion of ISW2 resulted in significant chromatin changes at many other motif-proximal nucleosomes, consistent with a role of additional transcription factors in targeting Isw2 (Supplemental Fig. S5).
Figure 3.

Genome-wide nucleosome positioning by Isw2/Ume6 at Ume6 target sequences. (A) Difference in nucleosome dyad signal at permutations of Ume6 recognition motifs for wild-type and Δume6 strains as in Figure 2B. Black and red indicate where remodeling by endogenous Isw2/Ume6 caused increases and decreases in nucleosome signal, respectively. Asterisk indicates WNGGCGGCWW motif. Ordering is the same as in Figure 2B. (B) The difference in nucleosome dyad signal between wild-type and Δume6 strains at all intergenic instances of the WNGGCGGCWW motif, clustered by direction of nucleosome repositioning (left) with average dyad signal within each cluster for each strain (right) as in Figure 2C. (C) Comparison of nucleosome positioning by Chd1-Ume6 and endogenous Isw2/Ume6 at Ume6 binding motifs. Each trace reveals the average sites of gain (positive) or loss (negative) of nucleosome dyad signal when Δume6 is compared with Chd1-Ume6 (red) or Isw2/Ume6 (blue) at Ume6 binding motifs from clusters 1 (left) or 3 (right). (D) Schematic representation of the range and direction of nucleosome positioning for Isw2/Ume6 (blue) or Chd1-Ume6 (red) at Ume6 binding sites. In comparing the wild type with the Δume6 or Δisw2 backgrounds, nucleosomes situated up to 200 bp from the Ume6 recognition sequence in a Δume6 or Δisw2 strain were shifted by Isw2 to a final dyad position ∼100 bp from the recognition motif. Likewise, nucleosomes with dyads situated up to 150 bp from the motif center in a Δume6 or Δisw2 background were shifted by Chd1-Ume6 to a final dyad position ∼50 bp from the motif (corresponding to burial of the motif by ∼20 bp). (E) Example loci demonstrating differences in nucleosome distributions for Isw2/Ume6 or Chd1-Ume6 in wild-type or Δume6 +[Chd1-Ume6] strains compared with a Δume6 strain. Nucleosome dyad signal (opaque) and associated nucleosome footprints (transparent) are shown for each strain with the predicted nucleosome movements by Isw2/Ume6 (blue) or Chd1-Ume6 (red) shown above, and the expected final dyad positions are represented by asterisks.
Like the Chd1-Ume6 fusion remodeler, the endogenous Isw2/Ume6 system positioned nucleosomes in a highly predictable manner, yet with distinct characteristics. Nucleosome dyads initially located between 100 and 200 bp from URS1 motifs in Δume6 or Δisw2 strains are were selectively repositioned in wild-type cells toward these Ume6 binding motifs. The endogenous Isw2/Ume6 system maintained nucleosome dyads ∼100 bp from the URS1 motifs and, similar to Chd1-Ume6, failed to move nucleosomes that were closer than this remodeled position (Fig. 3C–E). Given the ∼73-bp nucleosomal footprint on either side of the dyad, these preferred nucleosome positions indicate that URS1 motifs were maintained ∼30 bp outside the nucleosome. Thus, unlike Chd1-Ume6, the final nucleosome positions established by Isw2/Ume6 did not result from burial of recruitment URS1 motifs on the nucleosome (Fig. 3C–E), suggesting the presence of additional control mechanisms for precise placement of nucleosomes by Isw2 and Ume6.
Nucleosome repositioning by Chd1-Ume6 leads to mild transcriptional repression
Ume6-repressed genes are known to be regulated through recruitment of Isw2 and Rpd3, which are thought to reduce binding of activators and/or transcriptional machinery in promoters by establishing repressive chromatin structure through nucleosome repositioning and histone deacetylation, respectively (Kadosh and Struhl 1997, 1998a,b; Rundlett et al. 1998; Goldmark et al. 2000). As expected, deletion of UME6 results in derepression of genes containing Ume6 binding sites, likely due to loss of repressive chromatin structure mediated by both Isw2 and Rpd3 (Fig. 4A; Supplemental Fig. S6). Since Chd1-Ume6 is able to position nucleosomes at promoters containing Ume6 binding sequences, we speculated that Chd1-Ume6 may partially repress transcription at target genes by potentially blocking access of DNA to transcriptional machinery. We did not expect full repression because Chd1-Ume6 should not lead to deacetylation of chromatin typically catalyzed by Ume6-recruited Rpd3 (Supplemental Fig. S6C). To determine the extent that nucleosome positioning by Chd1-Ume6 might affect transcriptional repression in a Δume6 strain, we measured transcript abundance by RNA-seq and characterized transcription differences at genomic loci containing Ume6 recognition motifs (Fig. 4B,C).
Figure 4.

Nucleosome positioning by Chd1-Ume6 is mildly repressive. (A) Differences in transcription between the wild type and Δume6 at all genes (black) or at genes with a WNGGCGGCWW motif within 500 bp of the open reading frame (red). Transcript abundance was calculated using Cufflinks (Trapnell et al. 2012). (FPKM) Fragments per kilobase of transcript per million fragments mapped. Green diagonal indicates the y = x axis. Points above the diagonal are repressed in wild-type cells. (B) Differences in transcription (log2 FPKM) in Δume6 cells with or without addition of Chd1-Ume6 at all genes (black) or genes with associated WNGGCGGCWW motifs (red). Green diagonal indicates the y = x axis. Points above the diagonal are repressed upon introduction of Chd1-Ume6. (C) Example loci showing nucleosome positioning (upper panels), RNA signal (lower panels), and relative transcript abundance from RNA-seq data (associated numbers) at indicated genes for the wild-type, Δume6, and Δume6 + [Chd1-Ume6] and Δume6 + [Chd1D513N-Ume6] strains. Blue lines represent associated Ume6 recognition motifs, and arrows denote directional closing (single-headed arrow) or opening (double-headed arrow) of associated NDRs.
While introduction of Chd1-Ume6 to a Δume6 strain was relatively inconsequential for global off-target transcript abundance, genes containing Ume6 recognition motifs in their promoter regions were preferentially repressed by Chd1-Ume6 (hypergeometric test P < 10−15 for more than 1.5-fold repression), suggesting that positioning of nucleosomes near intergenic Ume6 binding sites is generally repressive. However, effects on transcription were context dependent, and even at loci where remodeling by Chd1-Ume6 significantly reduced NDR size, the extent of transcriptional repression was modest compared to that observed in wild-type cells (Fig. 4C). One limitation of the fusion system is that only a fraction of nucleosomes could be positioned at many binding sites, likely due to the inability of Chd1-Ume6 to mobilize a fraction of the “out of reach” nucleosomes at a given site. Our observed decrease in transcription by Chd1-Ume6 is therefore likely an underestimate of the repressive capacity of positioned nucleosomes at Ume6 binding sites. Nevertheless, our transcription results provide direct evidence that nucleosome positioning by Chd1-Ume6 can be repressive, yet they highlight the requirement of both nucleosome positioning and other factors such as histone deacetylation for full transcriptional repression of Ume6-sensitive genes.
Disruption of Isw2-positioned nucleosomes leads to cryptic transcription at Ume6 target loci
Since Chd1-Ume6 displayed distinct placements of nucleosomes adjacent to URS1 motifs compared with Isw2/Ume6, one question was the extent that Chd1-Ume6 might compete with Isw2/Ume6 in the same cell, potentially disrupting normal nucleosome positioning and transcriptional repression found in wild-type yeast. To explore the possibility of remodeler antagonism, we introduced the Chd1-Ume6 expression plasmid into a wild-type background and measured nucleosome positions and changes in transcription at Ume6 target genes (Fig. 5). Similar to the sliding behavior described above for Δume6 and Δisw2 backgrounds, Chd1-Ume6 shifted nucleosomes on top of Ume6 motifs in a wild-type background despite the presence of functional Isw2 (Fig. 5A). Interestingly, the presence of Isw2/Ume6 allowed Chd1-Ume6 to move a larger subset of nucleosomes, suggesting that Isw2/Ume6 effectively extends the range of URS1-proximal nucleosomes ultimately acted upon by Chd1-Ume6 (Fig. 5B,C). On average, nucleosomes were moved ∼50 bp closer to recruitment sequences by Chd1-Ume6 than by Isw2/Ume6, but we did not observe transcriptional induction comparable to loss of Ume6 at genes containing Ume6 binding motifs in their nucleosome-free regions (NFRs), suggesting that these alternate nucleosome positions were still generally repressive (Fig. 5D). These results are consistent with the previous conclusion that loss of nucleosome positioning through deletion of ISW2 does not lead to strong transcriptional derepression from Ume6-repressed promoters (Goldmark et al. 2000; Fazzio et al. 2001), which we have reproduced using RNA-seq (Supplemental Fig. S6).
Figure 5.

Chd1-Ume6 can disrupt Isw2-directed nucleosome positions and result in cryptic transcription. (A) Difference in nucleosome dyad signal between wild-type cells with and without addition of Chd1-Ume6, clustered (as in Figs. 2, 3) by direction of nucleosome repositioning (left) with average cluster signal from individual strains (right). (B) Average signals within cluster 3 in Figure 5A for indicated backgrounds showing cooperation of endogenous remodeling machinery with Chd1-Ume6 based on increased movement of distal nucleosomes to the +50-bp position. Red- and blue-shaded regions correspond to the range of nucleosomes mobilized by Chd1-Ume6 and endogenous Isw2/Ume6, respectively. The difference between the red and orange traces is due to sequential positioning by Isw2 and Chd1-Ume6 at Ume6 binding sites. (C) Example locus (upstream of RPC17) where initial positioning by Isw2/Ume6 is required for the final Chd1-Ume6 remodeled position observed in a wild-type background. In the absence of Isw2/Ume6 (red), nucleosomes are beyond the reach of Chd1-Ume6, while Isw2 action (blue) moves nucleosomes to a favorable position for further Chd1-Ume6 positioning in the wild-type background (orange). Black asterisk denotes the preferred Δume6 dyad position. (D) Changes in global (left; black dots) or motif-proximal (right; red dots) transcript abundance between the wild-type and wild-type +[Chd1-Ume6] strains. (E) Browser shot of the MEI5/VPS30 locus showing changes in nucleosome positions (top) and transcript abundance (bottom) upon addition of Chd1-Ume6 to a wild-type background. Blue and red signals reflect Watson (right-transcribed) and Crick (left-transcribed) strands, respectively, from normalized strand-specific RNA-seq profiles. (F) Same as E, showing differences in nucleosome dyad signal and strand-specific transcript abundance at the MEI4/ACA1 locus.
While we generally did not observe large defects in repression when nucleosomes were specifically perturbed by Chd1-Ume6, there was a small number of notable exceptions that resulted from exposure of normal and cryptic promoters. For example, positioning of nucleosomes by Chd1-Ume6 led to generation of an extensive NDR at the MEI5-VPS30 locus, corresponding to increased bidirectional transcription of MEI5 and VPS30 (Fig. 5E). This increase in NDR size was likely due to the presence of a Ume6 binding motif upstream of the NDR (as opposed to within the NDR for a majority of other motif instances). Similarly, cryptic transcription of a truncated MEI4 transcript and antisense transcription of the adjacent ACA1 gene were observed coincident with reorganization of nucleosomes surrounding a URS1 motif in the MEI4 promoter (Fig. 5F). Interestingly, the de novo exposure of a cryptic antisense transcript in the ACA1 gene did not significantly reduce ACA1 coding expression, which supports a view that antisense transcription in budding yeast does not intrinsically have repressive consequences for the associated sense transcript. Chd1-Ume6 also induced nucleosome positioning at the Ume6 binding motif located in the 3′ NFR of ALP1 (Supplemental Fig. S7A). This corresponded to previously unannotated bidirectional transcripts from within the ALP1 coding region, likely through exposure of a cryptic promoter element. A similar disruptive result was observed at the HED1 promoter, where Chd1-Ume6 moved a strongly positioned nucleosome that likely promoted bidirectional transcription of both the HED1 gene and a cryptic noncoding transcript (Supplemental Fig. S7B). These exceptions demonstrate that precise nucleosome positions can indeed be critical for faithful transcriptional regulation. However, the genome-wide data suggest that many promoters can accommodate significant nucleosome perturbations with little or no effects on transcription, even in the absence of a functional exosome (Supplemental Fig. S8), indicating that the regulation of transcription through precise nucleosome positions is likely complex and context dependent.
Sequence-directed chromatin remodeling creates phased nucleosome arrays
Previous studies have reported that chromatin remodeling factors are responsible for the well positioned +1 and −1 nucleosomes at gene promoters and also for packing nucleosomes into organized arrays against these founding nucleosomes (Mavrich et al. 2008; Gkikopoulos et al. 2011; Zhang et al. 2011; Ganguli et al. 2014). However, the nature of the barriers and mechanism(s) through which these arrays are phased remain unclear (Hughes and Rando 2014). To address whether targeted positioning of one nucleosome could affect packing of adjacent nucleosomes, we characterized downstream nucleosome shifts at genomic Ume6 binding motifs that have Chd1-Ume6–induced changes in nucleosome positions (Fig. 6). Although Chd1-Ume6 only had a limited range in nucleosomes that could be directly mobilized (Fig. 2), we observed more distant nucleosomes also changing positions (Fig. 6A,B). On average, recruitment of Chd1-Ume6 to genomic Ume6 binding sites resulted in approximately five nucleosomes shifting toward the target sequence (Fig. 6C). Because of the strict sequence dependence of the Chd1-Ume6 fusion remodeler, these downstream shifts cannot be attributed to direct Chd1-Ume6 remodeling activity and are likely due to endogenous chromatin remodeling proteins like Chd1 and Isw1 (Gkikopoulos et al. 2011). Thus, the nucleosomes adjacent to URS1 sites that were positioned by Chd1-Ume6 served as “founding” nucleosomes against which others were packed.
Figure 6.

Positioning of motif-proximal nucleosomes sets the phase for nucleosome arrays. (A) Nucleosome positioning by Isw2/Ume6 (blue) and Chd1-Ume6 (red) at the HOP2 locus compared with lack of Ume6-directed positioning (black). Solid arrows indicate the direction of movement for motif-proximal nucleosomes, while faded arrows denote secondary shifts in nucleosome positions. Gray dashed lines indicate preferred nucleosome dyad positions in the Δume6 strain. (B) Same as in A for a Ume6 motif at the 3′ end of the ALP1 gene. (C) Difference in nucleosome dyad signal for Δume6 strains with and without Chd1-Ume6 for motif-proximal and distal nucleosomes within 1000 bp of the Ume6 binding motif. Signal is shown for motifs corresponding to cluster 3 (right-to-left repositioning), though cluster 1 displayed similar results. (Top) Average difference in nucleosome dyad signal within the cluster; (bottom) difference in signal at all individual motif instances within the cluster. Direction of nucleosome positioning relative to associated TSS (if applicable) is indicated on right. (D–F) Same as in C, but for differences between (D) the wild type and Δume6, (E) the wild type and Δisw2, or (F) Δisw1/Δchd1 and Δisw1/Δchd1/Δisw2 (Gkikopoulos et al. 2011). (G) Example of nucleosome boundary formation within a coding region, annotated as in A and B. (H) Average difference in nucleosome dyad signal as in C for 77 instances of coding region nucleosome positioning by Chd1-Ume6.
Interestingly, in addition to precise local positioning of URS1-proximal nucleosomes, similar shifts of nucleosome arrays were also induced by Isw2. Like Chd1-Ume6, positioning by Isw2/Ume6 resulted in shifts of up to five flanking nucleosomes toward the recruitment site (Fig. 6A,B,D,E). In comparing nucleosome positions at Ume6 motifs in Δisw2/Δchd1/Δisw1 cells to those in Δchd1/Δisw1 cells (Gkikopoulos et al. 2011), which should reflect the isolated nucleosome positioning activity of Isw2, we observed strong reorganization of single adjacent nucleosomes with weak organization of only one flanking nucleosome (Fig. 6F). The creation of phased arrays does not appear to be dependent on the location of a TSS, as it is achieved near TSSs, TTSs, and intergenic regions without discernible features (Fig. 6A–D). We therefore conclude that in wild-type cells, the shifting of the adjacent nucleosome at Ume6 target sequences requires Isw2, while the shifts in flanking nucleosomes are carried out by Chd1 and/or Isw1. These results are consistent with a mechanism in which Isw2 cooperates with Ume6 to precisely position a single founding nucleosome, while wild-type Chd1 and Isw1 pack adjacent nucleosomes against this established boundary (Fig. 7). Finally, we observed the creation of phased nucleosome arrays when Chd1-Ume6 positioned nucleosomes within coding regions (Fig. 6G,H). In coding regions, packing against the motif-proximal, positioned nucleosome was seen on both sides, likely reflecting the enrichment of nucleosome packing factors like Isw1 and Chd1 within gene bodies. We therefore conclude that the establishment of a single positioned nucleosome through sequence-specific chromatin remodeling is sufficient to create a boundary for phased chromatin array formation in vivo.
Figure 7.
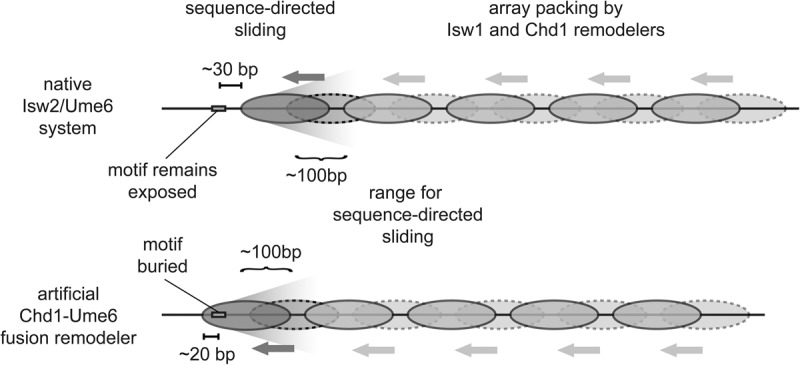
Model for transcription factor-mediated chromatin remodeling at sequence-targeted sites. (Top) In the endogenous system, Isw2 is recruited to specific genomic loci through interaction with Ume6 at target motifs (rectangle). Motif-proximal nucleosomes (dark gray) within a limited range are shifted by Isw2 from an unremodeled position (dashed lines) to a final position such that 30 bp of DNA are exposed between the motif center and the nucleosome edge. (Bottom) The artificial Chd1-Ume6 system works through a similar mechanism where the motif proximal nucleosome is directly moved by the chromatin remodeling protein, but with this remodeler, the recruitment motif is buried by 20 bp of nucleosomal DNA. For both cases of sequence-directed nucleosome sliding, the motif-proximal nucleosome determines the phasing for evenly spaced nucleosome arrays that are packed against these founding nucleosomes by endogenous remodelers.
Discussion
Consequences of engineered chromatin remodeling
By fusing the Chd1 chromatin remodeler to an exogenous DNA binding domain, we have developed a new tool for targeting sequence-specific chromatin remodeling activity in vivo. This technology has allowed us to deliberately and predictably move select nucleosomes through the native Ume6/URS1 targeting system. This work also shows that the strong positioning of single nucleosomes is sufficient for establishing boundaries against which downstream nucleosomes can be packed by other chromatin remodeling factors. We note that this study is not exhaustive, since our chromatin analysis only accounts for changes in nucleosome positioning but not for dynamics or changes in histone density. In addition, by using Ume6 as the recruitment module, the targeted genes affected are primarily involved in meiosis and are thus tightly regulated transcriptionally (Steber and Esposito 1995). With this system, nucleosome positioning is neither indicative of nor predictive for changes in transcription, which underscores the importance of other factors such as histone modifications, activator binding, and nucleosome dynamics for transcriptional regulation. Although the Chd1-Ume6 fusion protein is not a naturally occurring enzyme, it was recently reported that in humans, the incidental fusion of RUNX1, a transcription factor involved in hematopoiesis, to the C terminus of human CHD1 can contribute to leukemogenesis (Yao et al. 2015). Thus, the controlled targeting of the Chd1 remodeler described here is a first step in understanding the potential in vivo consequences of fusions between chromatin remodelers and transcription factors that lead to human diseases.
Implications for global regulation of chromatin structure
Similar to the Chd1-Ume6 fusion remodeler, the endogenous Isw2/Ume6 system moved nucleosomes adjacent to URS1 motifs to defined locations, yet the preferred positions were distinct for these two remodeler systems (Fig. 3C). Positioning by Isw2 maintained nucleosome edges ∼30 bp from nearby URS1 motifs. Leaving room between the nucleosome edge and Ume6 binding site may be important for uninterrupted binding of Ume6 to continuously recruit corepressors such as Rpd3. The precise spacing between URS1 sequence and proximal nucleosome could potentially be important for facilitating additional enzymatic activity on the target nucleosomes since chromatin modifying enzymes like Rpd3 are sensitive to the distance and orientation of histone substrates on chromatin (Lee et al. 2013). In addition, at a subset of promoters, precise positioning by Isw2/Ume6 appeared to prevent cryptic transcription, consistent with previously defined roles of chromatin remodelers in protecting against spurious transcription (Whitehouse et al. 2007; Yadon et al. 2010; Alcid and Tsukiyama 2014). We have demonstrated that by positioning motif-proximal nucleosomes at Ume6-dependent loci, Isw2 establishes boundaries for arrays generated by Isw1 and Chd1 remodelers. Therefore, the activity of Isw2 appears to be largely limited to single nucleosomes at targeted genomic loci, but its effects indirectly propagate into coding regions via packing by other factors. While we have only formally shown that this boundary-forming activity is associated with recruitment by Ume6, Isw2 can be targeted to many NFRs through different recruitment factors (Rizzo et al. 2011; Yadon et al. 2013), and previous studies have shown precise binding architecture of Isw2 with respect to Reb1, Abf1, Mbp1, and Cbf1 binding sites (Yen et al. 2012; Zentner et al. 2013). This single-nucleosome boundary establishment by Isw2 and subsequent packing by Chd1 and/or Isw1 may therefore be instrumental in phasing nucleosome arrays throughout the yeast genome. Such a mechanism implies that global nucleosome positioning can be determined by DNA sequence preferences of transcription factors that recruit chromatin remodeling factors. This variation of a sequence-dictated mechanism for genome-wide nucleosome positioning relies on a combination of targeted nucleosome sliding at a limited number of recruitment sites and sequence nonspecific ATP-dependent nucleosome packing against founding nucleosomes (Fig. 7).
Potential interplay among nucleosome positions, chromatin remodeling, and transcription factor binding
Our characterization of sequence-targeted chromatin remodeling has revealed that certain nucleosome positions are refractory to targeted chromatin remodeling. While more distant nucleosomes can be shifted by Isw2 such that the nucleosome edge is brought up to ∼30 bp away from Ume6 binding motifs, nucleosomes that are closer than this 30-bp limit are not repositioned by Isw2. Accordingly, the closer preferred positioning exhibited by Chd1-Ume6 appears to give this fusion remodeler dominance over nucleosome positioning by Isw2. While it is not known if similar antagonism occurs in vivo, it has recently been shown that the ISW1a and SWI/SNF chromatin remodelers appear to shift nucleosomes in opposite directions in vitro relative to the same transcription factor binding site (Li et al. 2015). Furthermore, SWI/SNF remodelers can displace bound transcription factors (Nagaich et al. 2004; Li et al. 2015). It is thus possible that remodeler-intrinsic responses to transcription factor targeting may underlie global establishment of and condition-specific transitions in nucleosome positioning in vivo.
Conclusions
The method of fusing the Chd1 chromatin remodeler to exogenous DNA binding domains provides a versatile strategy for inducing specific and sequence-targeted nucleosome positioning in vivo. Chd1 has been functionally fused to several DNA binding domains of different classes and can even be specifically targeted to nucleosomes via biotinylation (McKnight et al. 2011; Patel et al. 2013). A key advantage of targeting a chromatin remodeler via a sequence-specific DNA binding factor is that by burying recruitment sites with neighboring nucleosomes, accessibility for endogenous factors should be reduced genome-wide. In addition to targeting through DNA binding preferences of endogenous factors, combining Chd1 with designed targeting systems used previously with chromatin modifiers—such as TALE, engineered zinc fingers, and CRISPR/Cas9 (Mendenhall et al. 2013; Keung et al. 2014; Hilton et al. 2015; Konermann et al. 2015)—opens up the possibility of burying any genomic locus by neighboring nucleosomes. The ability to precisely control nucleosome positions with sequence-targeted fusion remodelers in vivo will be a useful tool for investigating other natural systems that establish and regulate chromatin structure, and offers the potential to manipulate DNA accessibility at desired loci for therapeutic purposes.
Methods
Strains and growth conditions
Wild-type, Δisw2, Δume6, and Δrpd3 backgrounds are from a W303 parent strain and are identical to those used previously (Goldmark et al. 2000). The Δisw2/Δrpd3 strain was a RAD5+ prototroph derived from W303. For all analyses, cells were grown to mid-log phase (OD600 = 0.4–0.6) in YPD. The Chd1-Ume6 expression plasmid was created by directly fusing residues 118–1014 of S. cerevisiae Chd1 to residues 764–836 of the S. cerevisiae Ume6 protein via seamless PCR insertion mutagenesis. The fusion construct was then amplified with an N-terminal NLS (KKKRK), a 5′ BamHI site, and a 3′ HindIII site and cloned into p416-ADH1 (Mumberg et al. 1995). The Chd1-Ume6 expression plasmid was introduced into indicated backgrounds through standard yeast transformation methods. For in vitro analysis, Chd1118-1014(ΔDBD) or Chd1-Ume6 was cloned into pDEST17, transformed into BL21 Star (DE3) cells with trigger factor and RIL plasmid (Stratagene), and purified via nickel affinity, ion exchange, and size-exclusion chromatography as described previously for other Chd1 constructs (Hauk et al. 2010; McKnight et al. 2011).
Nucleosome sliding assay
Recombinant yeast histones were purified as previously described (Tsukiyama Lab online protocol http://research.fhcrc.org/tsukiyama/en/protocols.html) (Luger et al. 1999) and dialyzed by gradient salt dialysis onto the Widom 601 positioning sequence (Lowary and Widom 1998) with 80 bp of linker DNA and, where indicated, a URS1 site (TGGCGGCT) located +8 to +15 with respect to the nucleosome edge and a 5′ FAM label. Purified end-positioned mononucleosomes (50 nM) were incubated with 10 nM Chd1 protein constructs in nucleosome sliding buffer (10 mM HEPES at pH 7.8, 10 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 5 mM ATP, 0.2 mg/mL BSA, and 100 mM KCl). Nucleosome sliding reactions were quenched by adding 1 µg of unlabeled competitor plasmid and placed on ice. To resolve nucleosome positions, native gels (6% acrylamide, 60:1 acrylamide:bisacrylamide ratio in 1× TBE) were run for 2 h at 150 volts at 4°C in prechilled 0.25× TBE running buffer and visualized using a Typhoon 9410 variable imager (GE Healthcare).
MNase digestion and sequencing of nucleosomal DNA
Nucleosome mapping by MNase-seq was performed as previously described (Rodriguez et al. 2014). Briefly, 200 mL of cells was grown to OD600 = 0.4–0.6, crosslinked with 1% formaldehyde, quenched with glycine, spheroplasted with zymolyase (100T), pelleted, split into three separate reactions, and digested with 10, 20, or 40 units of MNase (Worthington) in the presence of 30 units of exonuclease III (NEB) for 10 min at 37°C. MNase digestion was quenched with EDTA/SDS, and samples were treated with 200 µg of proteinase K overnight at 65°C. DNA was extracted by phenol/chloroform extraction, then ethanol precipitated, and treated with RNase A. Appropriately digested samples (resulting in ∼80% mononucleosomes) were treated with 10 units of alkaline phosphatase (NEB), and the mononucleosomal band was purified by gel extraction from low-melt agarose (GeneMate). Sequencing libraries were created from purified phosphatase-treated mononucleosomal DNA using the Illumina TruSeq sample prep kit. Paired-end sequencing was performed on Illumina HiSeq 2500 on high-output mode (Fred Hutchinson Cancer Research Center shared resources).
MNase-seq data processing
Paired-end reads were trimmed of adapter sequences with a custom Python script using HTSeq (Anders et al. 2015) and aligned to the S. cerevisiae reference genome (Saccharomyces_cerevisiae.ER4.65.dna.toplevel.fa) (Cunningham et al. 2015) with Bowtie 2 (Langmead and Salzberg 2012). Aligned reads were filtered computationally to select for mononucleosome-sized fragments (100 bp < fragment < 200 bp). MNase-seq data for Δisw2/Δchd1/Δisw1 and Δisw1/Δchd1 strains were obtained from the GEO database with accession number GSE31015 (Gkikopoulos et al. 2011). For mononucleosome-sized fragments, the dyad position was calculated by taking the midpoint of paired-end reads, and the per-base dyad coverage was calculated across the S. cerevisiae genome. Dyad counts were normalized such that the average count at a genomic position (excluding the rDNA locus) was equal to 1.0 (i.e., data were scaled so reads/bp = 1.0). Predicted nucleosome footprint was inferred by expanding signal from each nucleosome dyad 72 bp in both directions. Nucleosome alignment to variations of the URS1 motif and at other transcription factor binding sites was performed as previously described (McKnight et al. 2015). Briefly, average dyad signal at each position relative to a motif center was calculated from all intergenic instances of the respective motif. Instances of individual motifs were identified using the Saccharomyces Genome Database pattern matching tool (http://www.yeastgenome.org/cgi-bin/PATMATCH/nph-patmatch), and transcription factor motifs were obtained from the JASPAR database (Mathelier et al. 2014).
Strand-specific RNA sequencing and processing
RNA was isolated by hot acid phenol extraction followed by purification and DNase I treatment using the RNeasy kit (Qiagen). Depletion of rRNA was performed using the Ribo-Zero kit (Epicenter) per the manufacturer's protocol. Strand-specific sequencing libraries were constructed using the dUTP method combined with TruSeq (Illumina) (Parkhomchuk et al. 2009; Sultan et al. 2012). Paired-end sequencing was performed by the Fred Hutchinson Cancer Research Center shared resources facility with Illumina HiSeq 2500 on high-output mode. Individual sequencing reads were trimmed of adapter sequences using HTSeq (Anders et al. 2015) and aligned to the S. cerevisiae genome (Saccharomyces_cerevisiae.EF4.65.dna.toplevel.fa) (Cunningham et al. 2015) with TopHat2 (Kim et al. 2013). Aligned reads were filtered using SAMtools (-f 3 -F 256) (Li et al. 2009), and differential expression analysis was performed using Cufflinks (Trapnell et al. 2012) with a maskfile for tRNA and rRNA genes. Strand-specific RNA transcripts were separated using SAMtools, with flags -f 83 and -f 163 for Watson strands or -f 99 and -f 147 for Crick strands, and then visualized using Integrated Genome Browser.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE72572.
Acknowledgments
We thank Laura Lee, Srinivas Ramachandran, and Joseph Boerma for critiquing the manuscript and Kyle Horvath for help with preliminary experiments. This work was supported by a Leukemia and Lymphoma Society Fellow Award CDP-5078-14 for J.N.M., NIGMS R01GM058465 for T.T., and NIGMS R01-084192 for G.D.B.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.199919.115.
References
- Alcid EA, Tsukiyama T. 2014. ATP-dependent chromatin remodeling shapes the long noncoding RNA landscape. Genes Dev 28: 2348–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SF, Steber CM, Esposito RE, Coleman JE. 1995. UME6, a negative regulator of meiosis in Saccharomyces cerevisiae, contains a C-terminal Zn2Cys6 binuclear cluster that binds the URS1 DNA sequence in a zinc-dependent manner. Protein Sci 4: 1832–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2015. Ensembl 2015. Nucleic Acids Res 43: D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Kagalwala MN, Bartholomew B. 2006. Regulation of ISW2 by concerted action of histone H4 tail and extranucleosomal DNA. Mol Cell Biol 26: 7388–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Tsukiyama T. 2003. Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol Cell 12: 1333–1340. [DOI] [PubMed] [Google Scholar]
- Fazzio TG, Kooperberg C, Goldmark JP, Neal C, Basom R, Delrow J, Tsukiyama T. 2001. Widespread collaboration of Isw2 and Sin3-Rpd3 chromatin remodeling complexes in transcriptional repression. Mol Cell Biol 21: 6450–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguli D, Chereji RV, Iben JR, Cole HA, Clark DJ. 2014. RSC-dependent constructive and destructive interference between opposing arrays of phased nucleosomes in yeast. Genome Res 24: 1637–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Bachman N, Delrow J, Boeke JD, Tsukiyama T. 2005. Genome-wide identification of Isw2 chromatin-remodeling targets by localization of a catalytically inactive mutant. Genes Dev 19: 942–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen-Hughes T. 2011. A role for Snf2-related nucleosome-spacing enzymes in genome-wide nucleosome organization. Science 333: 1758–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T. 2000. The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103: 423–433. [DOI] [PubMed] [Google Scholar]
- Gordan R, Murphy KF, McCord RP, Zhu C, Vedenko A, Bulyk ML. 2011. Curated collection of yeast transcription factor DNA binding specificity data reveals novel structural and gene regulatory insights. Genome Biol 12: R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon SE, Rizzo JM, Tatomer DC, Lieb JD, Buck MJ. 2011. The stress response factors Yap6, Cin5, Phd1, and Skn7 direct targeting of the conserved co-repressor Tup1-Ssn6 in S. cerevisiae. PLoS One 6: e19060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G, McKnight JN, Nodelman IM, Bowman GD. 2010. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell 39: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton IB, D'Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. 2015. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33: 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Rando OJ. 2014. Mechanisms underlying nucleosome positioning in vivo. Annu Rev Biophys 43: 41–63. [DOI] [PubMed] [Google Scholar]
- Kadosh D, Struhl K. 1997. Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89: 365–371. [DOI] [PubMed] [Google Scholar]
- Kadosh D, Struhl K. 1998a. Histone deacetylase activity of Rpd3 is important for transcriptional repression in vivo. Genes Dev 12: 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadosh D, Struhl K. 1998b. Targeted recruitment of the Sin3-Rpd3 histone deacetylase complex generates a highly localized domain of repressed chromatin in vivo. Mol Cell Biol 18: 5121–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagalwala MN, Glaus BJ, Dang W, Zofall M, Bartholomew B. 2004. Topography of the ISW2–nucleosome complex: insights into nucleosome spacing and chromatin remodeling. EMBO J 23: 2092–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent NA, Karabetsou N, Politis PK, Mellor J. 2001. In vivo chromatin remodeling by yeast ISWI homologs Isw1p and Isw2p. Genes Dev 15: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung AJ, Bashor CJ, Kiriakov S, Collins JJ, Khalil AS. 2014. Using targeted chromatin regulators to engineer combinatorial and spatial transcriptional regulation. Cell 158: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517: 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, Wu J, Li B. 2013. Chromatin remodelers fine-tune H3K36me-directed deacetylation of neighbor nucleosomes by Rpd3S. Mol Cell 52: 255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hada A, Sen P, Olufemi L, Hall MA, Smith BY, Forth S, McKnight JN, Patel A, Bowman GD, et al. 2015. Dynamic regulation of transcription factors by nucleosome remodeling. eLife 4 10.7554/eLife.06249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, Widom J. 1998. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276: 19–42. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ. 1999. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol 119: 1–16. [DOI] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen CY, Chou A, Ienasescu H, et al. 2014. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42: D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, Ioshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, Pugh BF. 2008. A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res 18: 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, Jenkins KR, Nodelman IM, Escobar T, Bowman GD. 2011. Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol Cell Biol 31: 4746–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, Boerma JW, Breeden LL, Tsukiyama T. 2015. Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol Cell 59: 732–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. 2013. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 31: 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. 1995. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Walker DA, Wolford R, Hager GL. 2004. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell 14: 163–174. [DOI] [PubMed] [Google Scholar]
- Nodelman IM, Bowman GD. 2013. Nucleosome sliding by Chd1 does not require rigid coupling between DNA-binding and ATPase domains. EMBO Rep 14: 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HD, Luche RM, Cooper TG. 1992. The yeast UME6 gene product is required for transcriptional repression mediated by the CAR1 URS1 repressor binding site. Nucleic Acids Res 20: 1909–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res 37: e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Chakravarthy S, Morrone S, Nodelman IM, McKnight JN, Bowman GD. 2013. Decoupling nucleosome recognition from DNA binding dramatically alters the properties of the Chd1 chromatin remodeler. Nucleic Acids Res 41: 1637–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo JM, Mieczkowski PA, Buck MJ. 2011. Tup1 stabilizes promoter nucleosome positioning and occupancy at transcriptionally plastic genes. Nucleic Acids Res 39: 8803–8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, McKnight JN, Tsukiyama T. 2014. Genome-wide analysis of nucleosome positions, occupancy, and accessibility in yeast: nucleosome mapping, high-resolution histone ChIP, and NCAM. Curr Protoc Mol Biol 108: 21 28 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundlett SE, Carmen AA, Suka N, Turner BM, Grunstein M. 1998. Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 392: 831–835. [DOI] [PubMed] [Google Scholar]
- Steber CM, Esposito RE. 1995. UME6 is a central component of a developmental regulatory switch controlling meiosis-specific gene expression. Proc Natl Acad Sci 92: 12490–12494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, Owen-Hughes T. 2006. Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem 281: 16279–16288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Dokel S, Amstislavskiy V, Wuttig D, Sultmann H, Lehrach H, Yaspo ML. 2012. A simple strand-specific RNA-Seq library preparation protocol combining the Illumina TruSeq RNA and the dUTP methods. Biochem Biophys Res Commun 422: 643–646. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse I, Tsukiyama T. 2006. Antagonistic forces that position nucleosomes in vivo. Nat Struct Mol Biol 13: 633–640. [DOI] [PubMed] [Google Scholar]
- Whitehouse I, Rando OJ, Delrow J, Tsukiyama T. 2007. Chromatin remodelling at promoters suppresses antisense transcription. Nature 450: 1031–1035. [DOI] [PubMed] [Google Scholar]
- Yadon AN, Van de Mark D, Basom R, Delrow J, Whitehouse I, Tsukiyama T. 2010. Chromatin remodeling around nucleosome-free regions leads to repression of noncoding RNA transcription. Mol Cell Biol 30: 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadon AN, Singh BN, Hampsey M, Tsukiyama T. 2013. DNA looping facilitates targeting of a chromatin remodeling enzyme. Mol Cell 50: 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Pan J, Wu C, Shen H, Xie J, Wang Q, Wen L, Wang Q, Ma L, Wu L, et al. 2015. Transcriptome sequencing reveals CHD1 as a novel fusion partner of RUNX1 in acute myeloid leukemia with t(5;21)(q21;q22). Mol Cancer 14: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen K, Vinayachandran V, Batta K, Koerber RT, Pugh BF. 2012. Genome-wide nucleosome specificity and directionality of chromatin remodelers. Cell 149: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tsukiyama T, Henikoff S. 2013. ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet 9: e1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Reese JC. 2004a. Redundant mechanisms are used by Ssn6-Tup1 in repressing chromosomal gene transcription in Saccharomyces cerevisiae. J Biol Chem 279: 39240–39250. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Reese JC. 2004b. Ssn6–Tup1 requires the ISW2 complex to position nucleosomes in Saccharomyces cerevisiae. EMBO J 23: 2246–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Wippo CJ, Wal M, Ward E, Korber P, Pugh BF. 2011. A packing mechanism for nucleosome organization reconstituted across a eukaryotic genome. Science 332: 977–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Persinger J, Bartholomew B. 2004. Functional role of extranucleosomal DNA and the entry site of the nucleosome in chromatin remodeling by ISW2. Mol Cell Biol 24: 10047–10057. [DOI] [PMC free article] [PubMed] [Google Scholar]