

Summary
Human mitochondria produce ATP and metabolites to support development and maintain cellular homeostasis. Mitochondria harbor multiple copies of a maternally-inherited, non-nuclear genome (mtDNA) that encodes for 13 subunit proteins of the respiratory chain. Mutations in mtDNA occur mainly in the 24 non-coding genes, with specific mutations implicated in early death, neuromuscular and neurodegenerative diseases, cancer, and diabetes. A significant barrier to new insights in mitochondrial biology and clinical applications for mtDNA disorders is our general inability to manipulate the mtDNA sequence. Microinjection, cytoplasmic fusion, nucleic acid import strategies, targeted endonucleases, and newer approaches that include the transfer of genomic DNA, somatic cell reprogramming, and a photothermal nanoblade, attempt to change the mtDNA sequence in target cells with varying efficiencies and limitations. Here, we discuss the current state of manipulating mammalian mtDNA and provide an outlook for mitochondrial reverse genetics, which could further enable mitochondrial research and therapies for mtDNA diseases.
Graphical Abstract
INTRODUCTION
Mammalian mitochondria are double-membrane eukaryotic organelles that are thought to have originated by endosymbiosis of α-proteobacteria of the Rickettsiales family (Thrash et al., 2011; Wallin, 1926; Yang et al., 1985). Although isolated mitochondria are similar to bacteria in size, ~2 μm x 1 μm, they appear granular/singular or as an extended fused, and branching network within the cytoplasm. Inherited maternally, mitochondria generate the energy metabolites ATP, NADH, and FADH2. They function in the breakdown of fatty acids via beta-oxidation and in the biosynthesis of iron-sulfur clusters, heme, and steroids. The flow of biomolecules, such as calcium, citrate, acetyl-CoA, and cytochrome c, between the cytosol and mitochondria modulates disparate cellular functions including enzyme activities, epigenome remodeling, and apoptosis (Weinberg and Chandel, 2015). Reactive oxygen species (ROS), generated during oxidative phosphorylation (OXPHOS), can damage DNA, proteins, and lipids at high concentrations and may interfere with cell proliferation at low levels, and have a role in cell differentiation (Shadel and Horvath, 2015; Weinberg and Chandel, 2009). Thus, mitochondria are more than just the ‘powerhouse’ of a cell as they impact intracellular pathways associated with apoptosis, calcium homeostasis, aging, signaling, and many others (McBride et al., 2006).
Greater than 99% of the total proteins required for mitochondria biogenesis and function (~1,500 proteins) are encoded within the nucleus (Wallace, 2005). However, mitochondria also contain an independent genome (mtDNA) that is circular, double-stranded and 16,569 base pairs in humans. Originally known as cytoplasmic rho (ρ)-factor from studies in yeast (Ephrussi et al., 1949a; Ephrussi et al., 1949b), mtDNA contains no introns and encodes for 37 genes, which include 13 electron transport chain (ETC) polypeptides, 22 tRNAs, and 2 rRNAs. Transcription of the mtDNA initiates from three independent strand-specific promoters, resulting in three different pre-processed polycistronic transcripts (Temperley et al., 2010). Whereas nuclear DNA (nDNA) resides in histone-rich chromatin inside the nucleus, mtDNA forms histone-less nucleoprotein structures called nucleoids that are ~100 nm in diameter and attached to the matrix side of the mitochondrial inner membrane (Kukat and Larsson, 2013; Wang et al., 2013).
Mutations in mtDNA (http://www.mitomap.org) can range from being phenotypically silent to causing devastating familial diseases that typically affect tissues with high energy demands, such as the brain, heart, and muscle (Haas et al., 2007; Schon et al., 2012; Taylor and Turnbull, 2005). Most inherited mtDNA disorders appear in young children and progress with age (Westly, 2010). Although mutations in nucleus encoded genes can contribute to mitochondrial diseases, a recent study determined that ~1 in 5,000 adults living in northeast England have pathogenic mtDNA mutations (Gorman et al., 2015; Schaefer et al., 2008), and ~1 in 200 people are carriers for mtDNA alterations (Elliott et al., 2008; Wallace and Chalkia, 2013). This numeric discrepancy is related to the large number of mtDNA molecules present in an individual, with each cell containing either ~2 – 10 or ~100s – 100,000s of mtDNA copies, respectively, depending mainly on tissue energetic demands (Garcia-Rodriguez et al., 2007; Gilkerson et al., 2008; Kukat and Larsson, 2013; Shoubridge and Wai, 2007). The likelihood that an individual with a deleterious mtDNA mutation displays a phenotypic disorder depends on heteroplasmy, or the ratio of mutant to wild-type mtDNAs in cells. For example, a single nucleotide polymorphism, A3243G, in mtDNA alters the mt-tRNA for leucine (mt-tRNALeu) and, consequently, translation of 13 ETC proteins (Sasarman et al., 2008). Individuals with 10 – 30% of their mtDNA containing A3243G may have diabetes and potentially autism, which is controversial (Pons et al., 2004; Vandenouweland et al., 1992), whereas the more severe MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes) syndrome may occur at 50 – 90% A3243G heteroplasmy (Goto et al., 1990; Picard et al., 2014). Importantly, the symptoms and severity of these disorders also depend on the levels of mutant mtDNA as well as their tissue distribution.
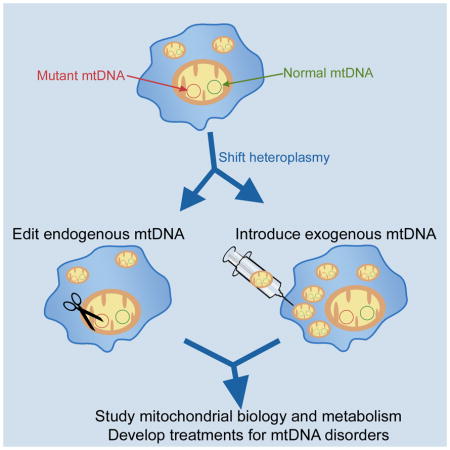
No curative treatments are available for mtDNA disorders. Rather, current approaches aim to maintain general patient health and do not repair, eliminate, or compensate for deleterious mtDNA sequences, except potentially in future offspring as discussed below (Parikh et al., 2009). In contrast to nDNA, it is difficult to alter the mtDNA sequence in mammalian cells. Establishing methods for removing detrimental mtDNA sequences or for transferring mitochondria with specific mtDNA sequences into cells are key initial steps in targeting the cause of mtDNA diseases (Figure 1). Long-term, methods to introduce sequence alterations directly into the mtDNA are desirable for fundamental insights into mitochondrial biology and for potentially enhanced clinical options for those afflicted by a mtDNA disease (Figure 2).
Figure 1.

Transferring Mitochondria for multiple applications. Mitochondria are double-membrane bound organelles that contain their own genome (mtDNA), which is organized into nucleoprotein structures, called nucleoids, attached to the inner membrane facing the mitochondrial matrix. Transfer of exogenous mitochondria into cells that contain or lack (ρ0 cells) mtDNA could improve our understanding of ETC function, metabolism, and the interaction between the mitochondrial and nuclear genomes. Mitochondrial transfer also may hold potential for treating diseases of dysfunctional mitochondria caused by mutations in mtDNA.
Figure 2.

Current methodologies for manipulating mtDNA in mammalian cells. Targeted degradation of endogenous mtDNA to shift heteroplasmy ratios can be performed with mitoTALENs or mitoZFNs. The mtDNA heteroplasmy ratio can also be shifted through a ‘bottleneck’ during the reprogramming of somatic cells to induced pluripotent stem cells (iPSCs). Adeno-associated virus (AAV) transduction can deliver up to ~5 kbp DNA into mitochondria that does not integrate into the mitochondrial genome. RNA and protein import can potentially compensate for dysfunctional mtDNA gene products. Whole mitochondria transfer technologies focus on delivering either isolated mitochondria (co-culture, microinjection, or photothermal nanoblade) or mitochondria from a donor cell (mitocytoplast, cytoplasmic fusion) to a recipient cell. Importantly, none of these methods can generate novel mtDNA sequences. To generate non-native mtDNA sequences, repair cells with mtDNA disorders, or establish cell lines with unique mtDNA mutations for basic studies and disease modeling, new methods are needed to insert, delete, or substitute sequences into existing mtDNA (‘reverse genetics’).
Nuclear Genome Transfer
To block the inheritance of mutant mtDNA in women at risk for a familial mtDNA disease, mitochondrial replacement procedures are being developed. These methods have in common the transfer of nDNA in various forms from an ovum containing mutant maternal mtDNA into an enucleated donor oocyte or ovum with wild-type mtDNA. Techniques include the transfer of an intact nucleus (Ma et al., 2015a), pronucleus (Craven et al., 2010), a polar body (Wang et al., 2014), or the meiotic spindle-chromosomal complex (Tachibana et al., 2013). Although successfully tested in mice and Rhesus macaques (Shoubridge, 2009; Tachibana et al., 2013; Tachibana et al., 2009; Wang et al., 2014; Wolf and Mitalipov, 2014), reproductive applications in humans warrant consideration of potential adverse effects from the reagents and methods used in the isolation and transfer of nDNA (Morrow et al., 2015). Currently, these techniques target unborn future generations and are not clinically useful in somatic cells or for individuals with a mtDNA disease. Furthermore, the unintended consequences of pairing nuclear and mitochondrial genomes from different sources are unknown, although cellular respiration appears intact despite a potential nDNA-mtDNA mismatch in human cells (Ma et al., 2015a). However, it still remains unclear whether individuals born with a non-selected nDNA-mtDNA mismatch could be affected (Hamilton, 2015). For example, crossed in-bred mice with wild-type C57BL/6J nDNA and wild-type 129S6 or NZB mtDNA show reduced respiration, activity, food consumption, and stress responses compared to matched controls (Sharpley et al., 2012). These nDNA transfer techniques also pose a potential ethical challenge because genetic material from three different individuals are combined to generate the embryos. In 2015, the UK House of Commons approved the use of IVF-based gene therapy approaches to treat mtDNA diseases. The possibility for these embryos generated by nuclear genome transfer has created a debate over an individual’s identity and the dissemination of third-party donor mtDNA to subsequent generations (Herbert and Turnbull, 2015; Richardson et al., 2015; Vogel, 2014).
Heteroplasmy Reduction through a ‘Bottleneck’
A shift in the heteroplasmy ratio toward wild-type or mutant mtDNAs occurs by a reduction in mtDNA content caused by a ‘genetic bottleneck’ during early embryonic differentiation (Brown et al., 2001; Taylor and Turnbull, 2005). A similar reduction ‘bottleneck’ may also occur during the reprogramming of heteroplasmic somatic cells to induced pluripotent stem cells (iPSCs), resulting in iPSCs with a range of wild-type to mutant mtDNA ratios (Cherry et al., 2013; Folmes et al., 2013; Fujikura et al., 2012; Teslaa and Teitell, 2015). It is unknown whether all starting heteroplasmic cell types and all mtDNA mutations experience a bottleneck during reprogramming to provide enriched or homoplasmic mutant iPSCs for disease modeling or fundamental studies. Of interest, a recent study showed different ratios of wild-type versus mutant mtDNA in single fibroblasts within a population before reprogramming, suggesting selection as a potential alternative mechanism for generating iPSCs with elevated wild-type or mutant mtDNA ratios (Ma et al., 2015b). This result further suggests that the segregation of mutant mtDNAs may occur more broadly in non-germ line cell types. These data could provide a potential alternative strategy to embryos generated by nuclear genome transfer involving somatic cell reprogramming from a female to obtain iPSCs lacking mutant mtDNA, followed by differentiation into functional oocytes (as possible) for various forms of nDNA transfer (Hayashi and Saitou, 2013; Park et al., 2009). In theory, the development of such an approach would require genetic material only from two individuals and wild-type homoplasmic iPSCs could find use in regenerative medicine applications.
DNA Nucleases and mtDNA Editing Enzymes
The nuclear expression of engineered constructs encoding mitochondria-targeted and site-specific DNA nucleases has been used to selectively eliminate mutant (or wild-type) mtDNA sequences in heteroplasmic cells and shift mtDNA ratios. In rodent and human cytoplasmic hybrid (‘cybrid’) cell lines, mitochondria-targeted PstI or SmaI restriction enzymes shifted the mtDNA heteroplasmy ratios from mutant to wild-type, resulting in increased cell respiration (Srivastava and Moraes, 2001; Tanaka et al., 2002). Cultured mouse hepatocytes transfected with a mitochondria-targeted ApaLI restriction enzyme showed a rapid shift to mtDNA homoplasmy within 6h (Bayona-Bafaluy et al., 2005). Expression of mitochondria-targeted EcoRI in the tibialis anterior muscle of hamsters decreased the activity of mitochondria-encoded cytochrome c oxidase without targeting similar nuclear pseudogenes (Tanaka et al., 2002). Adeno-associated virus (AAV) transfection of NZB × BALB/c mice with mitochondria-targeted endonucleases shifted whole animal mtDNA heteroplasmy ratios (Bayona-Bafaluy et al., 2005) and successfully targeted mtDNAs exclusively in liver, skeletal muscle, heart, and germ line (Bacman et al., 2012; Bacman et al., 2010; Bacman et al., 2007; Reddy et al., 2015). Despite the success of mitochondria-targeted endonucleases, it is difficult to identify target sites present in only the wild-type or mutant mtDNAs in a cell and there are a limited number of endonucleases with known cleavage sites. In fact, of ~200 different mtDNA mutations associated with human mtDNA disorders, only two have a restriction enzyme site that can be selectively targeted by an existing endonuclease (Reddy et al., 2015).
To circumvent the limitations of restriction enzymes, sequence non-specific nucleases have been fused to DNA recognition domains of proteins to target and cleave a broader range of mtDNA sequences. mtDNA cleavage produces a double-stranded DNA break that results in its degradation (Bayona-Bafaluy et al., 2005). For example, certain zinc finger proteins can bind to three nucleotides that comprise a codon. Zinc finger DNA binding modules have been engineered for almost all of the 64 nucleotide codon combinations. The addition of the human DNMT3a methyltransferase to a specific zinc finger construct resulted in the methylation of mtDNA at a predetermined nucleotide (Minczuk et al., 2006). By pairing specific zinc finger modules, a mitochondria-targeting sequence, and a DNA nuclease, expression constructs encoding for mitochondrial Zinc Finger Nucleases (mitoZFNs) have been generated that can target, cleave, and eliminate specific mtDNA sequences (Gaj et al., 2013; Minczuk et al., 2006). mitoZFNs containing the non-specific FokI nuclease have been engineered to recognize a 12 base pair sequence and cleave mutant mtDNA that differs from wild-type mtDNA at only one base pair (Minczuk et al., 2008). mitoZFNs in human cell lines that target either a T8993G mtDNA mutation associated with Leigh Syndrome (LS) and Neuropathy, Ataxia, Retinitis Pigmentosa (NARP) or a several kbp-sized deletion linked to Kearns-Sayre Syndrome (KSS) resulted in a reduction of mutant mtDNAs and a shift toward wild-type mtDNAs (Gammage et al., 2014).
Mitochondria-targeted Transcription Activator-Like Effector Nucleases (mitoTALENs) provide another type of protein-based gene cleaving enzyme that relies on the 34 amino acid repeat-containing TAL effector proteins originally discovered in the proteobacteria Xanthomonas. TALE nucleases can recognize single nucleotides to enable the binding to and cleavage of almost any sequence as long as the sequence starts with a thymidine. To date, mitoTALENs have successfully altered the mtDNA heteroplasmy ratio in NZB × BALB/c mouse oocytes (Reddy et al., 2015) and have eliminated mtDNAs with specific point mutations or large deletions in patient-derived cells (Bacman et al., 2013; Hashimoto et al., 2015).
Despite the promise of mitoZFNs and mitoTALENs, these gene cleaving enzymes have limitations. For example, a zinc finger module does not exist for every nucleotide codon which excludes certain sequences from mitoZFN targeting. TALENs are immune from this limitation because each TALE nuclease recognizes a specific nucleotide. However, both ZFNs and TALENs are challenging to generate because of the substantial protein engineering required to recognize specific DNA sequences. Furthermore, the delivery of large amounts of correctly engineered mitoZFNs or mitoTALENs to mitochondria is a challenge (Moraes, 2014). For mitoTALENs, a vector encoding all of the components can exceed the ~5 kbp DNA size limit that AAVs can accommodate to infect cells (Hashimoto et al., 2015). The recently described CRISPR-Cas9 genome editing approach may solve this issue, as it relies on only one protein (Cas9 nuclease) and one guide RNA to selectively cleave DNA sequences. However, it is unclear whether CRISPR-Cas9 could cleave or edit mtDNAs because it is challenging to import the guide RNA component into mitochondria (Doudna and Charpentier, 2014; Hashimoto et al., 2015; Liang et al., 2015). Finally, targeting mtDNAs that comprise a high percentage of the total mtDNAs in a cell with ZFN, TALEN, and CRISPR-Cas9 gene cleaving tools may inadvertently reduce the total mtDNA content below a functional threshold, causing additional problems (Moraes, 2014).
Targeted RNA and Protein Import into Mitochondria
Many organisms import tRNAs encoded in the nucleus into the mitochondrion. In vitro studies showed that tRNAs from the budding yeast Saccharomyces cerevisiae could be imported into isolated human mitochondria (Kolesnikova et al., 2000). Subsequent experiments in which yeast tRNAs were expressed in the nucleus of patient-derived fibroblasts containing a Myoclonic Epilepsy with Ragged Red Fibers (MERRF) mutation in a mitochondrial-encoded tRNA showed that tRNA import partially restored respiration (Kolesnikova et al., 2004). To try to improve import efficiency, the RNA Import Complex (RIC) of the kinetoplastid protozoa Leishmania reportedly augmented the import of human mt-tRNALys into isolated mitoplasts and helped to restore mtRNA translation in isolated mitochondria from MERRF and KSS cells expressing RIC (Mahata et al., 2005). It was also reported that expressing RIC in human cells with mtDNA mutations in tRNA genes enabled the import of all tRNAs, except glycine, into mitochondria, although studies with RIC have been difficult to independently replicate (Mahata et al., 2006). Recently, polynucleotide phosphorylase (PNPase), an enzyme with 3’–5’ exoribonuclease and poly-A-polymerase biochemical activities, was shown to augment the import of small, nucleus-encoded noncoding RNAs into the mitochondrial matrix (Wang et al., 2010). The addition of a 20-ribonucleotide stem-loop sequence from RNase P or MRP RNAs to tRNAs resulted in augmented tRNA import into the mitochondrial matrix (Wang et al., 2012). However, augmented RNA import mediated by PNPase remains inefficient, especially in vivo, and the mechanism augmenting import is not well understood.
Allotopic nucleus expression and cytosolic translation of mitochondria-encoded ETC genes was originally shown in S. cerevisiae (Law et al., 1988). In human cybrid cells containing a T8993G ATPase 6 mtDNA mutation that causes LS, a nucleus-expressed MTATP6 gene fused with a mitochondrial targeting sequence generated a fusion protein that was successfully imported and incorporated into complex V of the respiratory chain, resulting in improved ATP synthesis and cell growth (Manfredi et al., 2002). Nucleus-expressed mitochondria-targeted tRNAs have also been used to improve the translation and respiration of cells with a MELAS mtDNA mutation (Karicheva et al., 2011). Despite these encouraging results, developing a safe allotopic gene delivery method for therapy and the possibility for unintended side effects on cell function from recoded proteins transiting from the nucleus to mitochondria needs further study (Manfredi et al., 2002).
An alternative to expressing mitochondrial genes in the nucleus is to directly deliver them to mitochondria using AAV. Engineered AAV vectors are useful for nuclear gene therapy because of their ability to transduce a wide variety of cells and their lack of human pathology (Vasileva and Jessberger, 2005). The incorporation of a mitochondria targeting sequence into the AAV outer capsid protein enabled the vitreous injection of a mutant NADH dehydrogenase subunit 4 (ND4) gene to mitochondria in the eyes of wild-type mice, resulting in a phenotype similar to Leber’s Hereditary Optic Neuropathy (LHON) (Yu et al., 2012b). Conversely, when the AAV vector targeted wild-type ND4 to the mitochondria in the eyes of LHON-like mice, the mutant phenotype was rescued (Yu et al., 2012a). The AAV vector can accommodate DNA sequences up to ~5 kbp in size, enabling the delivery of all 37 mitochondrial genes into a human cell. However, mtDNA deletions > ~5 kbp, as may occur in KSS, may be a challenge to incorporate into AAVs and the mechanism of DNA entry into mitochondria is unknown, as is whether AAV delivery could interfere with additional, ETC-independent mitochondrial functions (Yu et al., 2012a). Interestingly, AAV delivered DNA sequences into mitochondria do not integrate into mtDNA and remain episomal for an unknown length of time because integration requires non-homologous end joining (NHEJ) or homologous recombination (HR), DNA repair processes that are infrequent in mammalian mitochondria (Alexeyev et al., 2013). Low level NHEJ and HR processes may also limit the use of CRISPR-Cas9 mtDNA editing.
Direct Uptake by Co-Culture with Isolated Mitochondria
The uptake of isolated mitochondria by passive co-culture with cells in vitro has been reported with some conflicting results. One report showed that isolated mitochondria could not be taken up by mammalian cell lines in co-culture media (Spees et al., 2006). However, chloramphenicol and efrapeptin antibiotic sensitive cells became resistant when co-incubated with isolated mitochondria derived from resistant cells (Clark and Shay, 1982). Consistent with the latter results, cellular uptake of mitochondria from the media was discovered to be temperature dependent, could occur between species, and was shown for MDA-MB-435, COLO 205, and MCF-7 cell lines (Katrangi et al., 2007). The incorporation of isolated mitochondria into endometrial gland-derived mesenchymal cells was also shown to be dose-dependent (Kitani et al., 2014a; Kitani et al., 2014b). Rho-zero (ρ0) cells can be generated that lack mtDNA and, therefore, cannot respire or generate ATP by OXPHOS and become uridine auxotrophs due to the inactivation of ETC-dependent dihydroorotate dehydrogenase (King and Attardi, 1989). The uptake of isolated mitochondria into ρ0 cells increased cell viability and respiration (Kitani et al., 2014b). The uptake mechanism did not involve clathrin-mediated endocytosis, but data obtained using inhibitors against microtubule assembly, actin polymerization, and Na+/H+ exchange suggested that non-selective macropinocytosis was a key component (Kitani et al., 2014b).
Results showing direct mitochondrial transfer by co-culture in vitro naturally led to in vivo studies. Autologous mitochondria were injected into a rabbit model of regional ischemia and improved recovery from the injury (Masuzawa et al., 2013). Data showed that most of the injected mitochondria remained extracellular with some mitochondria taken up by cells, although it was unclear how mitochondria were taken up from the extracellular fluid (Masuzawa et al., 2013).
Cell-to-Cell Mitochondria Transfer
The most common approach for transferring mitochondria from one cell to another is cytoplasmic fusion, which generates transmitochondrial cytoplasmic hybrid cells known as cybrids. In this procedure, enucleated mitochondria donor cells with specific mtDNA haplotypes are fused with ρ0 cells to yield cybrid cell lines that are isolated by growth in medium lacking uridine and containing selection drugs (King and Attadi, 1996; Moraes et al., 2001; Vithayathil et al., 2012). Cybrids have been used to study diseases arising from mtDNA mutations and other mitochondriopathies, such as Parkinson and Alzheimer diseases (Wilkins et al., 2014). However, results from cybrid studies can be challenging to interpret because mitochondria, along with miRNAs, lncRNAs, signaling proteins, and other organelles among other biomolecules are also transferred to ρ0 recipient cells.
Increasing evidence suggests mitochondria can be transferred from cell-to-cell by other mechanisms. One mechanism involves tunneling nanotubes (TNTs), cellular structures 50–200 nm in diameter and spanning several cell widths in length (Rustom et al., 2004). Studies examining contact interactions between endothelial progenitor cells and cardiomyocytes discovered labeled mitochondria in TNTs (Koyanagi et al., 2005). Further work showed that cell-to-cell transfer of mitochondria through TNTs depends on many factors, including p53, F-actin, Cx43, and stress (Domhan et al., 2011; Islam et al., 2012; Wang et al., 2011). It was recently shown that mitochondrial transfer from mesenchymal stem cells (MSCs) to epithelial cells through TNTs is dependent on the mitochondrial Rho GTPase 1 (Miro1), with overexpression and knockdown of this enzyme resulting in enhanced or decreased mitochondria transfer, respectively (Ahmad et al., 2014).
TNTs transfer mitochondria between stem cells, (Acquistapace et al., 2011; Li et al., 2014), stromal endothelial cells (Pasquier et al., 2013), human retinal pigment epithelial cells (Wittig et al., 2012), renal epithelial cells (Domhan et al., 2011), vascular smooth muscle cells (VSMCs) (Vallabhaneni et al., 2012), human mesothelioma cells (Lou et al., 2012), macrophages (Onfelt et al., 2006), and astrocytes (Wang et al., 2011). Although bidirectional mitochondria transfer has been observed in some cell types (Domhan et al., 2011; Lou et al., 2012; Onfelt et al., 2006), mitochondria transfer is mainly unidirectional. For example, labeled mitochondria from cardiomyocytes transit TNTs to endothelial progenitor cells, but not vice-versa (Koyanagi et al., 2005). Also, mitochondria transfer occurred from MSCs to endothelial cells, but not the reverse (Otsu et al., 2009). Finally, although cytoplasmic transfer was bidirectional between cardiomyocytes and MSCs, mitochondria were only observed migrating to cardiomyocytes (Plotnikov et al., 2008). Although it is suggested that stressed cells develop TNTs that target unstressed cells (Wang et al., 2011), it is unclear what biological processes determine the direction mitochondria transit in TNTs.
Naturally, the transfer of mitochondria from one cell to another can alter the recipient cell’s function. For example, mitochondria transfer from MSCs to ρ0 cells increases OXPHOS, ATP generation, and mitochondrial membrane potential (Cho et al., 2012; Spees et al., 2006). Mitochondria transfer may also degrade recipient cell functions and have been suggested to result in capillary degradation potentially from increased mitochondrial ROS (Otsu et al., 2009). Cell-to-cell transfer of mitochondria can also effect non-mitochondria cellular changes. For example, transfer of human multipotent adipose-derived stem cell mitochondria to mouse cardiomyocytes is suggested to be important for somatic cell reprogramming (Acquistapace et al., 2011). Also, the transfer of mitochondria from VSMCs to MSCs regulated MSC proliferation (Vallabhaneni et al., 2012). Finally, the transfer of mitochondria from endothelial cells to cancer cells could impart chemoresistance in the cancer cells (Pasquier et al., 2013).
Accumulating evidence suggests an in vivo role for mitochondria transfer via TNTs in cases of lung injury. In lipopolysaccharide-treated mouse lungs, bone marrow stromal cells (BMSCs) produced TNTs and microvesicles that transferred mitochondria to alveolar epithelium, resulting in the recipient cells having increased ATP levels and surviving acute lung injury (Islam et al., 2012). In a rat model of chronic obstructive pulmonary disease (COPD), mitochondria transferred from intravenously provided iPSC-MSCs and BMSCs lessened alveolar destruction (Li et al., 2014). Furthermore, mitochondria transfer was correlated with the ability of stem cells to repair damaged epithelial cells associated with lung injury and asthma in mouse models (Ahmad et al., 2014). Finally, mouse cancer models with tumor cells lacking mtDNA (ρ0 tumor cells) showed reduced mitochondrial function and tumor growth, but somehow could obtain mtDNA from the tumor microenvironment to restore respiration and increase tumorigenicity (Tan et al., 2015). Although TNTs may play a role in this mtDNA acquisition, it remains unclear how these mitochondria are transferred.
Microinjection for Mitochondria Transfer
The transfer of micron-sized cargo into cells by microinjection is usually associated with the delivery of nuclei into eggs in IVF procedures, although microinjection has also been used to transfer isolated mitochondria into cells. Using a 3 micron internal diameter needle, erythromycin-resistant mitochondria were transferred into large and antibiotic-sensitive Paramecium cells, resulting in erythromycin resistant cells (Knowles, 1974). Chloramphenicol resistant mitochondria were also transferred by microinjection into sensitive 143BTK– and HT1080-6TG human cells using a smaller and more clog-prone 1 micron needle that was necessary because of the smaller size of human cells compared to Paramecium (King and Attardi, 1988). Although the procedure was inefficient and transferred only 1 mitochondrion per cell, the delivered mitochondria completely replaced the endogenous mitochondria under chloramphenicol selection within 35 generations. Finally, microinjection of ρ0 cells with mitochondria containing wild-type mtDNA resulted in cells that grew on selection media lacking uridine (King and Attardi, 1989).
Although the microinjection of mitochondria into cells is well-documented, the persistence of transferred mitochondria in developing cells and organisms has shown mixed results. Mouse testes or liver mitochondria injected into fertilized mouse eggs resulted in zygotes that survived, developed, and reproduced, but the transferred mitochondria were undetectable in tissues, potentially due to the limited number of mitochondria transferred (Ebert et al., 1989). By contrast, the injection of mitochondria from Mus spretus into fertilized Mus musculus ova yielded detectable levels of transferred mitochondria in some blastocysts (Pinkert et al., 1997). Microinjection to generate transmitochondrial mice resulted in <10% of animals being heteroplasmic, suggesting that some of the injected cells did not have replicating mitochondria (Irwin et al., 1999). Additional issues that could have limited efficiency include the possibility that mitochondria were damaged by the small bore needles during injection or that certain mtDNA haplotypes are energetically unfavorable and therefore out-competed. For example, the microinjection of spermatid mitochondria into mouse embryos resulted in their elimination from cells prior to birth, whereas transferred liver mitochondria from a different host were retained in the offspring (Shitara et al., 2000). Finally, the microinjection of mitochondria derived from different mouse cells differentially altered the course of parthenogenetic development of murine oocytes (Takeda et al., 2005).
Despite promising results, microinjection has not been widely adopted for mitochondria transfer into mammalian cells mainly because it is labor intensive and inefficient. Microinjection transfers material by insertion of a needle into the cytoplasm which can damage cells, particularly non-egg cells, by excess mechanical stress. To minimize damage, microinjection needles for human somatic cells have an internal diameter of ~1 micron or less. This small orifice leads to frequent clogging during delivery of large cargo, such as mitochondria. To work around this problem, a 6 micron needle was used to inject mitochondria into rodent eggs that are larger and more mechanically forgiving, perhaps because of a thick zona pellucida, than somatic cells (Yang and Koob, 2012). Subsequently, a 20 micron needle removed a portion of the cytoplasm containing the injected mitochondria, and these mitocytoplasts were fused to mouse ρ0 somatic cells using a viral-based membrane fusion technique (Yang and Koob, 2012). This interspecies transfer method restored mtDNA and respiratory function in the ρ0 cells. Although contaminating rodent mitochondria was eliminated in mouse recipient cells and homoplasmy obtained, other rodent egg contaminants were transferred and this technique exhibited low throughput and was relatively inefficient.
Photothermal Nanoblade
A recent technological development provides a controlled, higher-throughput method for transferring mitochondria with desired mtDNA haploytpes into mammalian cells (Wu et al., 2010). A microcapillary pipette is coated at its ~3 micron diameter tip with light-absorbing titanium and positioned to lightly touch a cell membrane where it is exposed to a 532 nm wavelength non-damaging laser pulse (Wu et al., 2011; Wu et al., 2010). The laser pulse causes a rapidly expanding and collapsing vapor bubble to form at the pipette tip and generates a transient membrane incision by shear stress to enable the pressure driven delivery of large cargo into cells. Successfully delivered materials include DNA (Chiou et al., 2012), conjugated quantum dots (Xu et al., 2012), and live intracellular bacterial pathogens (French et al., 2011; Teh et al., 2014), with high efficiency and retained cell viability. The nanoblade approach has also been modified to deliver isolated MDA-MB-453 breast carcinoma cell line mitochondria into 143BTK–ρ0 cells. Analysis of the resulting clonal lines showed these mitochondria delivered cells were stable for mtDNA rescue, grew on uridine-free selection media, respired, and reset the metabolite profile to that of the parental 143BTK– cells (Wu et al., manuscript under review).
An important limitation of the single pipette photothermal nanoblade is that it can only deliver cargo to one cell at a time. Consequently, this approach not only remains a low throughput technique with ~100 cell deliveries/ hour, but the successful cell line generation is also operator dependent. Therefore, a massively parallel, high-throughput cargo transfer platform that is less operator dependent has been developed based on the principles of the nanoblade, to enable the large cargo delivery of up to ~100,000 cells/min (Wu et al., 2015). Similar to the nanoblade, this Biophotonic Laser Assisted Surgery Tool (BLAST) platform can deliver nanoparticles, proteins, and intracellular bacteria into mammalian cells with high efficiency and retained cell viability (Wu et al., 2015). Compared to microinjection techniques, the nanoblade tip and BLAST platform delivery portals are larger and never enter into the cell, reducing mechanical trauma and thereby increasing efficiency. The cavitation bubble generated by each device locally cuts a contacting membrane and this damage is repaired and resealed rapidly. The BLAST platform is currently being modified and tested for efficient mitochondrial transfer into mammalian somatic cells.
Is Mitochondrial Reverse Genetics in the Cards?
Reverse genetics is an approach in which the functions of specific genes and their encoded proteins are deciphered based on nucleotide sequence substitutions, deletions, and insertions. The application of reverse genetics to the nuclear DNA has uncovered the functions of a huge number of genes and proteins independently and as part of larger genome and proteome networks. Mitochondria transfer techniques such as co-culture, cell-to-cell transfer, cytoplasmic fusion, microinjection, and the photothermal nanoblade can only exchange pre-existing mtDNA sequences. Embryos generated by nuclear genome transfer use nDNA exchange techniques to obtain viable zygotes with functional, but only pre-existing, mtDNAs. Reprogramming bottlenecks and various forms of targeted endonucleases can shift heteroplasmy ratios by removing specific mtDNA sequences, but these approaches cannot yet be used to generate non-native mtDNA sequences. This is because the DNA repair machinery required to generate nucleotide substitutions, deletions, or insertions appears to be lacking inside mitochondria. Even if the proteins required for HR or NHEJ could be imported into mitochondria, it is unknown whether this machinery would function similarly on mitochondrial nucleoids as on histone-rich chromatin or what kind of havoc these proteins could generate in mtDNA. However, to gain further insight into the functional relationships amongst mitochondrial genes and with nucleus encoded genes, or to potentially develop gene replacement therapies for mtDNA diseases, continued technical advances toward mitochondrial reverse genetics are required. Another key difference compared with gene manipulations in the nucleus is that mitochondrial genes are subject to the rules of population genetics whereas nuclear genes are under Mendelian genetic rules. Given the significant roadblocks to reverse mitochondrial genetics in vivo, it is at least worth considering the possibility for manipulating mtDNA ex vivo, followed by the reintroduction into mitochondria and subsequent transfer into cells using one or more of the aforementioned approaches.
Ex-vivo approaches are varied and include cloning mtDNA into a vector enabling genetic manipulations and purification by standard techniques. The 16.3 kbp mouse mtDNA was inserted into a centromeric yeast/bacteria shuttle vector by HR, resulting in a ~22 kbp hybrid plasmid (Wheeler et al., 1997). However, only one bacterial clone was obtained when a point mutation was introduced into the mitochondrial 16S rRNA gene in order to convey chloramphenicol resistance (Bigger et al., 2000). Independently, mouse mtDNA was cloned into an E. coli plasmid producing a 17.8 kbp vector (Yoon and Koob, 2003). By randomly inserting an E. coli γ-ori origin of DNA replication and the chloramphenicol resistance marker by in vitro transposition reactions, this vector was shown to stably replicate in E. coli at low copy numbers. However, transposon insertion can randomly disrupt mitochondrial genes and the vectors are prone to degradation in E. coli when expressed at high copy numbers (Yoon and Koob, 2003). To get around size-related problems, mouse mtDNA was cloned into a Bacillus subtilis vector that can stably maintain a million base pairs (Yonemura et al., 2007) and the mtDNA was cut into 4 – 5 pieces and individually cloned into a vector that stably replicates at ~10 – 15 copies per E. coli (Yoon et al., 2009). Although mouse mtDNA has been modified in E. coli, B. subtilis, and S. cerevisiae vectors, these vectors and their exogenous sequences may be problematic in mammalian cells. Attempts to place human mtDNA into plasmids showed that regions including mt-tRNAThr and the D-loop may be unstable (Bigger et al., 2000; Drouin, 1980; Kearsey et al., 1980). Also, human mtDNA is unstable in multi-copy vectors or single-copy phage artificial chromosome vectors in E. coli and cannot be cloned into multi-copy yeast vectors (Bigger et al., 2011). The human mtDNA was cloned into a single-copy yeast plasmid by HR but could not be shuttle cloned into E. coli, indicating a conflict between human mtDNA and bacteria. Further work is required to generate a suitable system for modifying human mtDNA.
Another, more significant hurdle to overcome is the re-introduction of manipulated mtDNA back into mitochondria. Although short, linear pieces of DNA conjugated to mitochondrial-targeting peptides can pass through the mitochondrial membranes (Flierl et al., 2003; Vestweber and Schatz, 1989), this method does not enable the import of circular or large DNAs (Yamada et al., 2008). Liposome-based nanoparticles, known as MITO-Porters, have imported macromolecules, such as GFP and DNase I, into the mitochondrial matrix (Yamada et al., 2008; Yamada et al., 2011). Although these mitochondrial-targeted nanoparticles have a diameter of ~200 nm and could potentially fit mtDNA, it is unclear whether the mtDNA would be degraded once inside the mitochondria, as has been observed for other molecules (Yamada et al., 2008). Nanoparticles based on the amphiphile dequalinium have delivered cargo into mitochondria. Dequalinium has a delocalized cationic charge and self-assembles around DNA in a structure termed the DQAsome, which can interact with the cardiolipin rich mitochondrial membrane to import DNA (D’Souza et al., 2003; Weissig et al., 2001; Weissig et al., 2000; Weissig and Torchilin, 2000).
Electroporation has been used to transfer a 7.2 kbp pCMVβ plasmid into isolated mitochondria (Collombet et al., 1997). The mouse mtDNA was cloned into an E. coli vector and electroporated into isolated mouse ρ0 mitochondria, with mtRNA transcripts detected by in organello assays (Yoon and Koob, 2003, 2005). Despite this encouraging ex vivo result, these mitochondria did not function when transferred into mammalian cells, which could have been a result of damage by the intracellular transfer process (Yoon and Koob, 2005).
Bacterial conjugation is a form of sexual reproduction that involves the transfer of plasmid DNA from F-plasmid (the fertility factor that enables bacterial conjugation) positive bacteria to F-plasmid negative bacteria via cell contact. Since mitochondria are thought to originate from bacteria, conjugation was attempted between a F+ bacterium and F- mitochondria and a plasmid containing a T7 promoter was successfully transferred and expressed in T7 RNA polymerase-containing mouse mitochondria (Yoon and Koob, 2005). It is unclear whether the entire mtDNA can also be transferred using conjugation. However, conjugation transfers single-stranded DNA and it is also unclear whether synthesis of a complementary second-strand would occur inside mitochondria. Also, the donor bacterium transfers both a plasmid and the F-plasmid into recipients and it is unclear whether mitochondria in vivo would tolerate the bacterial F-plasmid. Finally, for any ex vivo mtDNA transfer method, a completely unknown potential roadblock exists for inserting exogenous, naked mtDNA into mitochondrial nucleoids.
If the obstacles to editing the mtDNA and delivering it back into mitochondria ex-vivo can be overcome, the next step would be to identify ideal recipient mitochondria and cells. The simplest path is to transfer the engineered mtDNA into isolated ρ0 mitochondria where there would be no competition from endogenous mtDNA. Furthermore, the path of least resistance would be to then place these engineered mitochondria into ρ0 mammalian cells. This path would be potentially incompatible in specific clinical settings of mutant mtDNA and there are a limited number of different ρ0 cell lines available (Hashiguchi and Zhang-Akiyama, 2009). The generation of new ρ0 lines requires DNA intercalating agents, such as ethidium bromide and ditercalinium chloride, which are also potential nDNA mutagens. A potential safer alternative for generating additional ρ0 cell lines is the transient use of targeted endonucleases to deplete the mtDNA (Kukat et al., 2008; Schubert et al., 2015).
CONCLUDING REMARKS
mtDNA mutations often result in devastating disorders for which there are currently no effective treatments or curative therapies. Whereas effective approaches for some monogenic diseases caused by nDNA mutations have and are being developed, the mtDNA stands out as particularly challenging to manipulate and therefore has been left out of the reverse genetics revolution. This discussion has highlighted our present capabilities for manipulating mtDNA, and while remarkable progress has been made in transferring pre-existing mtDNA sequences between cells, it is clear that our toolset is limited and hindered by key roadblocks (Table 1). Success at developing reproducible techniques for genetically altering mtDNA, either in vivo or ex vivo with cellular reintroduction, may create new treatment possibilities for mtDNA disorders and also help to improve our understanding of mitochondrial function and cell metabolism.
Table 1.
Limitations of Current Methods to Alter mtDNA in Cells
| Technique | Limitations & Considerations |
|---|---|
| Mitochondrial targeting | |
|
| |
| mitoTALENs, mitoZFNs | Can be complex protein engineering; Targeted degradation, not reparative; Heteroplasmy shifting; 5 kbp size limitation if AAV vector used |
| CRISPR/Cas9 | Deficient or inefficient guide RNA import; Low NHEJ and HR rates |
| Adeno-associated virus (AAV) transduction | 5 kbp DNA size limit; Mitochondria targeting with unknown consequences; Compensatory and not reparative; Non-integrating; Unclear how mtDNA fragment is maintained and expressed episomally |
| RNA import | Inefficient; Mechanism not understood; Compensatory and not reparative |
|
| |
| Mitochondria transfer | |
|
| |
| Nuclear/spindle/pronuclear/polar body transfer | Efficiency, technical, and ethical considerations; Pre-existing mtDNAs only |
| Co-culture with isolated mitochondria | Sporadic, low frequency |
| Transmitochondrial cytoplasmic fusions (cybrids) | Unwanted miRNAs, lncRNAs, proteins, organelles |
| Cell-to-cell transfer | Specific cell types and conditions; Unclear how to control transfer process; Mechanism(s) not fully understood |
| Microinjection* | Low efficiency; Microinjection needle prone to clogging; Needle imparts mechanical stress and cell trauma |
| Mitocytoplast fusion* | Low efficiency; Interspecies/fusion impurities |
| Photothermal nanoblade technology* | Operator and technique dependent |
|
| |
| Other | |
|
| |
| Bottleneck to shift mtDNA heteroplasmy | Unclear if works with all cell types and mtDNA mutations; Stochastic at reprogramming rates |
Potential approach to transfer mitochondria with modified mtDNA sequences
Acknowledgments
Supported by a Ruth L. Kirschstein National Research Service Award CA009120; an Air Force Office of Scientific Research grant FA9550-15-1-0406; an UC Discovery Biotechnology grant 178517; National Institute of Health grants GM114188, GM073981, CA90571, CA156674, and CA185189; a National Science Foundation grant CBET-1404080; California Institute for Regenerative Medicine grants RB1-01397 and RT3-07678; a Prostate Cancer Foundation Challenge Award; a Broad Stem Cell Research Center Innovator Award; and by NanoCav, LLC. We also acknowledge the Library of Science and Medical Illustrations (http://www.somersault1824.com/).
Footnotes
AUTHOR CONTRIBUTIONS
A.N.P. and M.A.T. conceived the review, wrote the manuscript, and generated the figures and table. T-H.W. and P-Y.C. contributed ideas and helped edit the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquistapace A, Bru T, Lesault PF, Figeac F, Coudert AE, le Coz O, Christov C, Baudin X, Auber F, Yiou R, et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells. 2011;29:812–824. doi: 10.1002/stem.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, et al. Miro1 regulates intercellular mitochondrial transport and enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014;33:994–1010. doi: 10.1002/embj.201386030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyev M, Shokolenko I, Wilson G, LeDoux S. The maintenance of mitochondrial DNA integrity--critical analysis and update. CSH Perspect Biol. 2013;5:a012641. doi: 10.1101/cshperspect.a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Duan D, Moraes CT. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012;19:1101–1106. doi: 10.1038/gt.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Garcia S, Moraes CT. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010;17:713–720. doi: 10.1038/gt.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Hernandez D, Moraes CT. Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a ‘differential multiple cleavage-site’ model. Gene Ther. 2007;14:1309–1318. doi: 10.1038/sj.gt.3302981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacman SR, Williams SL, Pinto M, Peralta S, Moraes CT. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med. 2013;19:1111–1113. doi: 10.1038/nm.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc Natl Acad Sci USA. 2005;102:14392–14397. doi: 10.1073/pnas.0502896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigger B, Tolmachov O, Collombet JM, Coutelle C. Introduction of chloramphenicol resistance into the modified mouse mitochondrial genome: cloning of unstable sequences by passage through yeast. Anal Biochem. 2000;277:236–242. doi: 10.1006/abio.1999.4382. [DOI] [PubMed] [Google Scholar]
- Bigger BW, Liao AY, Sergijenko A, Coutelle C. Trial and error: how the unclonable human mitochondrial genome was cloned in yeast. Pharm Res. 2011;28:2863–2870. doi: 10.1007/s11095-011-0527-1. [DOI] [PubMed] [Google Scholar]
- Brown DT, Samuels DC, Michael EM, Turnbull DM, Chinnery PF. Random genetic drift determines the level of mutant mtDNA in human primary oocytes. Am J Hum Genet. 2001;68:533–536. doi: 10.1086/318190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry ABC, Gagne KE, Mcloughlin EM, Baccei A, Gorman B, Hartung O, Miller JD, Zhang J, Zon RL, Ince TA, et al. Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells. 2013;31:1287–1297. doi: 10.1002/stem.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou PY, Wu TH, Teitell MA. Photothermal nanoblade for single cell surgery and cargo delivery. Proc Spie. 2012;8460 [Google Scholar]
- Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS, Kang GH, Lee JB, Park KS, Lee HK. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PloS ONE. 2012;7 doi: 10.1371/journal.pone.0032778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Shay JW. Mitochondrial transformation of mammalian cells. Nature. 1982;295:605–607. doi: 10.1038/295605a0. [DOI] [PubMed] [Google Scholar]
- Collombet JM, Wheeler VC, Vogel F, Coutelle C. Introduction of plasmid DNA into isolated mitochondria by electroporation. J Biol Chem. 1997;272:5342–5347. doi: 10.1074/jbc.272.8.5342. [DOI] [PubMed] [Google Scholar]
- Craven L, Tuppen HA, Greggains GD, Harbottle SJ, Murphy JL, Cree LM, Murdoch AP, Chinnery PF, Taylor RW, Lightowlers RN, et al. Pronuclear transfer in human embryos to prevent transmission of mitochondrial DNA disease. Nature. 2010;465:82–85. doi: 10.1038/nature08958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza GG, Rammohan R, Cheng SM, Torchilin VP, Weissig V. DQAsome-mediated delivery of plasmid DNA toward mitochondria in living cells. J Control Release. 2003;92:189–197. doi: 10.1016/s0168-3659(03)00297-9. [DOI] [PubMed] [Google Scholar]
- Domhan S, Ma LL, Tai A, Anaya Z, Beheshti A, Zeier M, Hlatky L, Abdollahi A. Intercellular communication by exchange of cytoplasmic material via tunneling nano-tube like structures in primary human renal epithelial cells. PloS ONE. 2011;6 doi: 10.1371/journal.pone.0021283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Drouin J. Cloning of human mitochondrial DNA in Escherichia coli. J Mol Biol. 1980;140:15–34. doi: 10.1016/0022-2836(80)90354-x. [DOI] [PubMed] [Google Scholar]
- Ebert KM, Alcivar A, Liem H, Goggins R, Hecht NB. Mouse zygotes injected with mitochondria develop normally but the exogenous mitochondria are not detectable in the progeny. Mol Reprod Dev. 1989;1:156–163. doi: 10.1002/mrd.1080010303. [DOI] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrussi B, Hottinguer H, Chimenes AM. Action de lacriflavine sur les levures. La mutation petite colonie. Ann Inst Pasteur. 1949a;76:351–367. [Google Scholar]
- Ephrussi B, Hottinguer H, Tavlitzki J. Action de lacriflavine sur les levures. Etude genetique du mutant petite colonie. Ann Inst Pasteur. 1949b;76:419–442. [Google Scholar]
- Flierl A, Jackson C, Cottrell B, Murdock D, Seibel P, Wallace DC. Targeted delivery of DNA to the mitochondrial compartment via import sequence-conjugated peptide nucleic acid. Mol Ther. 2003;7:550–557. doi: 10.1016/s1525-0016(03)00037-6. [DOI] [PubMed] [Google Scholar]
- Folmes CDL, Martinez-Fernandez A, Perales-Clemente E, Li X, Mcdonald A, Oglesbee D, Hrstka SC, Perez-Terzic C, Terzic A, Nelson TJ. Disease-causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell Ccones derived from a patient with MELAS. Stem Cells. 2013;31:1298–1308. doi: 10.1002/stem.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French CT, Toesca IJ, Wu TH, Teslaa T, Beaty SM, Wong W, Liu MH, Schroder I, Chiou PY, Teitell MA, et al. Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc Natl Acad Sci USA. 2011;108:12095–12100. doi: 10.1073/pnas.1107183108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikura J, Nakao K, Sone M, Noguchi M, Mori E, Naito M, Taura D, Harada-Shiba M, Kishimoto I, Watanabe A, et al. Induced pluripotent stem cells generated from diabetic patients with mitochondrial DNA A3243G mutation. Diabetologia. 2012;55:1689–1698. doi: 10.1007/s00125-012-2508-2. [DOI] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammage PA, Rorbach J, Vincent AI, Rebar EJ, Minczuk M. Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med. 2014;6:458–466. doi: 10.1002/emmm.201303672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez LJ, Gay AC, Pon LA. Puf3p, a Pumilio family RNA binding protein, localizes to mitochondria and regulates mitochondrial biogenesis and motility in budding yeast. J Cell Biol. 2007;176:197–207. doi: 10.1083/jcb.200606054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilkerson RW, Schon EA, Hernandez E, Davidson MM. Mitochondrial nucleoids maintain genetic autonomy but allow for functional complementation. J Cell Biol. 2008;181:1117–1128. doi: 10.1083/jcb.200712101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348:651–653. doi: 10.1038/348651a0. [DOI] [PubMed] [Google Scholar]
- Haas RH, Parikh S, Falk MJ, Saneto RP, Wolf NI, Darin N, Cohen BH. Mitochondrial disease: A practical approach for primary care physicians. Pediatrics. 2007;120:1326–1333. doi: 10.1542/peds.2007-0391. [DOI] [PubMed] [Google Scholar]
- Hamilton G. The hidden risks for ‘three-person’ babies. Nature. 2015;525:444–446. doi: 10.1038/525444a. [DOI] [PubMed] [Google Scholar]
- Hashiguchi K, Zhang-Akiyama QM. Establishment of human cell Lines lacking mitochondrial DNA. In: Stuart JA, editor. Mitochondrial DNA. Humana Press; 2009. pp. 383–391. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Bacman SR, Peralta S, Falk MJ, Chomyn A, Chan DC, Williams SL, Moraes CT. MitoTALEN: A general approach to reduce mutant mtDNA loads and restore oxidative phosphorylation function in mitochondrial diseases. Mol Ther. 2015 doi: 10.1038/mt.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Saitou M. Generation of eggs from mouse embryonic stem cells and induced pluripotent stem cells. Nat Protoc. 2013;8:1513–1524. doi: 10.1038/nprot.2013.090. [DOI] [PubMed] [Google Scholar]
- Herbert M, Turnbull D. Mitochondrial replacement to prevent the transmission of mitochondrial DNA disease. EMBO Rep. 2015;16:539–540. doi: 10.15252/embr.201540354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MH, Johnson LW, Pinkert CA. Isolation and microinjection of somatic cell-derived mitochondria and germline heteroplasmy in transmitochondrial mice. Transgenic Res. 1999;8:119–123. doi: 10.1023/a:1008925419758. [DOI] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18:759–U153. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karicheva OZ, Kolesnikova OA, Schirtz T, Vysokikh MY, Mager-Heckel AM, Lombes A, Boucheham A, Krasheninnikov IA, Martin RP, Entelis N, et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res. 2011;39:8173–8186. doi: 10.1093/nar/gkr546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katrangi E, D’Souza G, Boddapati SV, Kulawiec M, Singh KK, Bigger B, Weissig V. Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 2007;10:561–570. doi: 10.1089/rej.2007.0575. [DOI] [PubMed] [Google Scholar]
- Kearsey SE, Flanagan JG, Craig IW. Cloning of mouse mitochondrial DNA in Escherichia coli affects bacterial viability. Gene. 1980;12:249–255. doi: 10.1016/0378-1119(80)90107-9. [DOI] [PubMed] [Google Scholar]
- King MP, Attadi G. Mitochondria-mediated transformation of human rho(0) cells. Methods Enzymol. 1996;264:313–334. doi: 10.1016/s0076-6879(96)64030-0. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Injection of mitochondria into human cells leads to a rapid replacement of the endogenous mitochondrial DNA. Cell. 1988;52:811–819. doi: 10.1016/0092-8674(88)90423-0. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kitani T, Kami D, Kawasaki T, Nakata M, Matoba S, Gojo S. Direct human mitochondrial transfer: a novel concept based on the endosymbiotic theory. Transplant Proc. 2014a;46:1233–1236. doi: 10.1016/j.transproceed.2013.11.133. [DOI] [PubMed] [Google Scholar]
- Kitani T, Kami D, Matoba S, Gojo S. Internalization of isolated functional mitochondria: involvement of macropinocytosis. J Cell Mol Med. 2014b;18:1694–1703. doi: 10.1111/jcmm.12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JK. An improved microinjection technique in Paramecium aurelia. Transfer of mitochondria conferring erythromycin-resistance. Exp Cell Res. 1974;88:79–87. doi: 10.1016/0014-4827(74)90620-x. [DOI] [PubMed] [Google Scholar]
- Kolesnikova OA, Entelis NS, Jacquin-Becker C, Goltzene F, Chrzanowska-Lightowlers ZM, Lightowlers RN, Martin RP, Tarassov I. Nuclear DNA-encoded tRNAs targeted into mitochondria can rescue a mitochondrial DNA mutation associated with the MERRF syndrome in cultured human cells. Hum Mol Genet. 2004;13:2519–2534. doi: 10.1093/hmg/ddh267. [DOI] [PubMed] [Google Scholar]
- Kolesnikova OA, Entelis NS, Mireau H, Fox TD, Martin RP, Tarassov IA. Suppression of mutations in mitochondrial DNA by tRNAs imported from the cytoplasm. Science. 2000;289:1931–1933. doi: 10.1126/science.289.5486.1931. [DOI] [PubMed] [Google Scholar]
- Koyanagi M, Brandes RP, Haendeler J, Zeiher AM, Dimmeler S. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes - A novel mechanism for cell fate changes? Circ. Res. 2005;96:1039–1041. doi: 10.1161/01.RES.0000168650.23479.0c. [DOI] [PubMed] [Google Scholar]
- Kukat A, Kukat C, Brocher J, Schafer I, Krohne G, Trounce IA, Villani G, Seibel P. Generation of rho(0) cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukat C, Larsson NG. mtDNA makes a U-turn for the mitochondrial nucleoid. Trends Cell Biol. 2013;23:457–463. doi: 10.1016/j.tcb.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Law RHP, Farrell LB, Nero D, Devenish RJ, Nagley P. Studies on the import into mitochondria of yeast ATP synthase subunit 8 and 9 encoded by artificial nuclear genes. FEBS Lett. 1988;236:501–505. doi: 10.1016/0014-5793(88)80086-3. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang YL, Yeung SC, Liang YM, Liang XT, Ding Y, Ip MSM, Tse HF, Mak JCW, Lian QZ. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Resp Cell Mol. 2014;51:455–465. doi: 10.1165/rcmb.2013-0529OC. [DOI] [PubMed] [Google Scholar]
- Liang PP, Xu YW, Zhang XY, Ding CH, Huang R, Zhang Z, Lv J, Xie XW, Chen YX, Li YJ, et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 2015;6:363–372. doi: 10.1007/s13238-015-0153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K, Moore MAS. Tunneling nanotubes provide a unique vonduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PloS ONE. 2012;7 doi: 10.1371/journal.pone.0033093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Folmes CD, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, et al. Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature. 2015a doi: 10.1038/nature14546. [DOI] [PubMed] [Google Scholar]
- Ma J, Purcell H, Showalter L, Aagaard KM. Mitochondrial DNA sequence variation is largely conserved at birth with rare de novo mutations in neonates. Am J Obstet Gynecol. 2015b;212 doi: 10.1016/j.ajog.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahata B, Bhattacharyya SN, Mukherjee S, Adhya S. Correction of translational defects in patient-derived mutant mitochondria by complex-mediated import of a cytoplasmic tRNA. J Biol Chem. 2005;280:5141–5144. doi: 10.1074/jbc.C400572200. [DOI] [PubMed] [Google Scholar]
- Mahata B, Mukherjee S, Mishra S, Bandyopadhyay A, Adhya S. Functional delivery of a cytosolic tRNA into mutant mitochondria of human cells. Science. 2006;314:471–474. doi: 10.1126/science.1129754. [DOI] [PubMed] [Google Scholar]
- Manfredi G, Fu J, Ojaimi J, Sadlock JE, Kwong JQ, Guy J, Schon EA. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–399. doi: 10.1038/ng851. [DOI] [PubMed] [Google Scholar]
- Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C, Seth P, Bloch DB, Levitsky S, Cowan DB, et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2013;304:H966–H982. doi: 10.1152/ajpheart.00883.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–R560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Minczuk M, Papworth MA, Kolasinska P, Murphy MP, Klug A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc Natl Acad Sci USA. 2006;103:19689–19694. doi: 10.1073/pnas.0609502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minczuk M, Papworth MA, Miller JC, Murphy MP, Klug A. Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA. Nucleic Acids Res. 2008;36:3926–3938. doi: 10.1093/nar/gkn313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes CT. A magic bullet to specifically eliminate mutated mitochondrial genomes from patients’ cells. EMBO Mol Med. 2014;6:434–435. doi: 10.1002/emmm.201303769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes CT, Dey R, Barrientos A. Transmitochondrial technology in animal cells. Method Cell Biol. 2001;65:397–412. doi: 10.1016/s0091-679x(01)65023-4. [DOI] [PubMed] [Google Scholar]
- Morrow EH, Reinhardt K, Wolff JN, Dowling DK. Risks inherent to mitochondrial replacement. EMBO Rep. 2015;16:541–544. doi: 10.15252/embr.201439110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onfelt B, Nedvetzki S, Benninger RKP, Purbhoo MA, Sowinski S, Hume AN, Seabra MC, Neil MAA, French PMW, Davis DM. Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol. 2006;177:8476–8483. doi: 10.4049/jimmunol.177.12.8476. [DOI] [PubMed] [Google Scholar]
- Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009;113:4197–4205. doi: 10.1182/blood-2008-09-176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh S, Saneto R, Falk MJ, Anselm I, Cohen BH, Haas R, Soc MM. A modern approach to the treatment of mitochondrial disease. Curr Treat Option Neurol. 2009;11:414–430. doi: 10.1007/s11940-009-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TS, Galic Z, Conway AE, Lindgren A, van Handel BJ, Magnusson M, Richter L, Teitell MA, Mikkola HK, Lowry WE, et al. Derivation of primordial germ cells from human embryonic and induced pluripotent stem cells is significantly improved by coculture with human fetal gonadal cells. Stem Cells. 2009;27:783–795. doi: 10.1002/stem.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. 2013;11 doi: 10.1186/1479-5876-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Zhang JW, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, et al. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci USA. 2014;111:E4033–E4042. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkert CA, Irwin MH, Johnson LW, Moffatt RJ. Mitochondria transfer into mouse ova by microinjection. Transgenic Res. 1997;6:379–383. doi: 10.1023/a:1018431316831. [DOI] [PubMed] [Google Scholar]
- Plotnikov EY, Khryapenkova TG, Vasileva AK, Marey MV, Galkina SI, Isaev NK, Sheval EV, Polyakov VY, Sukhikh GT, Zorov DB. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J Cell Mol Med. 2008;12:1622–1631. doi: 10.1111/j.1582-4934.2007.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons R, Andreu AL, Checcarelli N, Vila MR, Engelstad K, Sue CM, Shungu D, Haggerty R, De Vivo DC, DiMauro S. Mitochondrial DNA abnormalities and autistic spectrum disorders. J Pediatr. 2004;144:81–85. doi: 10.1016/j.jpeds.2003.10.023. [DOI] [PubMed] [Google Scholar]
- Reddy P, Ocampo A, Suzuki K, Luo J, Bacman SR, Williams SL, Sugawara A, Okamura D, Tsunekawa Y, Wu J, et al. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell. 2015;161 doi: 10.1016/j.cell.2015.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson J, Irving L, Hyslop LA, Choudhary M, Murdoch A, Turnbull DM, Herbert M. Concise reviews: assisted reproductive technologies to prevent transmission of mitochondrial DNA disease. Stem Cells. 2015;33:639–645. doi: 10.1002/stem.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- Sasarman F, Antonicka H, Shoubridge EA. The A3243G tRNA(Leu(UUR)) MELAS mutation causes amino acid misincorporation and a combined respiratory chain assembly defect partially suppressed by overexpression of EFTu and EFG2. Hum Mol Genet. 2008;17:3697–3707. doi: 10.1093/hmg/ddn265. [DOI] [PubMed] [Google Scholar]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, Chinnery PF, Turnbull DM. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–39. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert S, Heller S, Loffler B, Schafer I, Seibel M, Villani G, Seibel P. Generation of rho zero cells: visualization and quantification of the mtDNA depletion process. Int J Mol Sci. 2015;16:9850–9865. doi: 10.3390/ijms16059850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K, Lin CS, Masubuchi S, Friend N, Koike M, et al. Heteroplasmy of mouse mtDNA Is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333–343. doi: 10.1016/j.cell.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitara H, Kaneda H, Sato A, Inoue K, Ogura A, Yonekawa H, Hayashi JI. Selective and continuous elimination of mitochondria microinjected into mouse eggs from spermatids, but not from liver cells, occurs throughout embryogenesis. Genetics. 2000;156:1277–1284. doi: 10.1093/genetics/156.3.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoubridge EA. Developmental biology: asexual healing. Nature. 2009;461:354–355. doi: 10.1038/461354a. [DOI] [PubMed] [Google Scholar]
- Shoubridge EA, Wai T. Mitochondrial DNA and the mammalian oocyte. Curr Top Dev Biol. 2007;77:87–111. doi: 10.1016/S0070-2153(06)77004-1. [DOI] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA. 2006;103:1283–1288. doi: 10.1073/pnas.0510511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum Mol Genet. 2001;10:3093–3099. doi: 10.1093/hmg/10.26.3093. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Woodward J, Sanchis DM, Ma H, Gutierrez NM, Tippner-Hedges R, Kang E, Lee HS, et al. Towards germline gene therapy of inherited mitochondrial diseases. Nature. 2013;493:627–631. doi: 10.1038/nature11647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J, Li Y, Ramsey C, Kolotushkina O, Mitalipov S. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature. 2009;461:367–372. doi: 10.1038/nature08368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Tasai M, Iwamoto M, Onishi A, Tagami T, Nirasawa K, Hanada H, Pinkert CA. Microinjection of cytoplasm or mitochondria derived from somatic cells affects parthenogenetic development of murine oocytes. Biol Reprod. 2005;72:1397–1404. doi: 10.1095/biolreprod.104.036129. [DOI] [PubMed] [Google Scholar]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015;21:81–94. doi: 10.1016/j.cmet.2014.12.003. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Borgeld HJ, Zhang J, Muramatsu S, Gong JS, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci. 2002;9:534–541. doi: 10.1159/000064726. [DOI] [PubMed] [Google Scholar]
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh BE, French CT, Chen YH, Chen IGJ, Wu TH, Sagullo E, Chiou PY, Teitell MA, Miller JF, Gan YH. Type three secretion system-mediated escape of Burkholderia pseudomallei into the host cytosol is critical for the activation of NF kappa B. BMC Microbiol. 2014;14 doi: 10.1186/1471-2180-14-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial mRNAs-like members of all families, similar but different. Biochim Biophys Acta. 2010;1797:1081–1085. doi: 10.1016/j.bbabio.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslaa T, Teitell MA. Pluripotent stem cell energy metabolism: an update. EMBO J. 2015;34:138–153. doi: 10.15252/embj.201490446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappe MS, Giovannoni SJ. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci Rep. 2011;1 doi: 10.1038/srep00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhaneni KC, Haller H, Dumler I. Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev. 2012;21:3104–3113. doi: 10.1089/scd.2011.0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenouweland JMW, Lemkes HHPJ, Ruitenbeek W, Sandkuijl LA, Devijlder MF, Struyvenberg PAA, Vandekamp JJP, Maassen JA. Mutation in mitochondrial tRNA(Leu(UUR)) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat Genet. 1992;1:368–371. doi: 10.1038/ng0892-368. [DOI] [PubMed] [Google Scholar]
- Vasileva A, Jessberger R. Precise hit: adeno-associated virus in gene targeting. Nat Rev Microbiol. 2005;3:837–847. doi: 10.1038/nrmicro1266. [DOI] [PubMed] [Google Scholar]
- Vestweber D, Schatz G. DNA-protein conjugates can enter mitochondria via the protein import pathway. Nature. 1989;338:170–172. doi: 10.1038/338170a0. [DOI] [PubMed] [Google Scholar]
- Vithayathil SA, Ma YW, Kaipparettu BA. Transmitochondrial cybrids: tools for functional studies of mutant mitochondria. In: Wong L-JC, editor. Mitochondrial Disorders. Humana Press; 2012. pp. 219–230. [DOI] [PubMed] [Google Scholar]
- Vogel G. Assisted Reproduction. FDA considers trials of three-parent embryos’. Science. 2014;343:827–828. doi: 10.1126/science.343.6173.827. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. CSH Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin IE. Bacteria and the origin of species. Science. 1926;64:173–175. doi: 10.1126/science.64.1651.173. [DOI] [PubMed] [Google Scholar]
- Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, Fan KC, Hong JS, French SW, McCaffery JM, et al. PNPASE Regulates RNA Import into Mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Shimada E, Zhang J, Hong JS, Smith GM, Teitell MA, Koehler CM. Correcting human mitochondrial mutations with targeted RNA import. Proc Natl Acad Sci USA. 2012;109:4840–4845. doi: 10.1073/pnas.1116792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Sha HY, Ji DM, Zhang HL, Chen DW, Cao YX, Zhu JH. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell. 2014;157:1591–1604. doi: 10.1016/j.cell.2014.04.042. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cui J, Sun X, Zhang Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011;18:732–742. doi: 10.1038/cdd.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YE, Marinov GK, Wold BJ, Chan DC. Genome-wide analysis reveals coating of the mitochondrial genome by TFAM. PloS ONE. 2013;8 doi: 10.1371/journal.pone.0074513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Chandel NS. Reactive oxygen species-dependent signaling regulates cancer. Cell Mol Life Sci. 2009;66:3663–3673. doi: 10.1007/s00018-009-0099-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nature Chem Biol. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissig V, D’Souza GGM, Torchilin VP. DQAsome/DNA complexes release DNA upon contact with isolated mouse liver mitochondria. J Control Release. 2001;75:401–408. doi: 10.1016/s0168-3659(01)00392-3. [DOI] [PubMed] [Google Scholar]
- Weissig V, Lizano C, Torchilin VP. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000;7:1–5. doi: 10.1080/107175400266722. [DOI] [PubMed] [Google Scholar]
- Weissig V, Torchilin VP. Mitochondriotropic cationic vesicles: a strategy towards mitochondrial gene therapy. Curr Pharm Biotechnol. 2000;1:325–346. doi: 10.2174/1389201003378870. [DOI] [PubMed] [Google Scholar]
- Westly E. When powerhouses fail. Nat Med. 2010;16:625–627. doi: 10.1038/nm0610-625. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Aitken M, Coutelle C. Modification of the mouse mitochondrial genome by insertion of an exogenous gene. Gene. 1997;198:203–209. doi: 10.1016/s0378-1119(97)00315-6. [DOI] [PubMed] [Google Scholar]
- Wilkins HM, Carl SM, Swerdlow RH. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox Biol. 2014;2:619–631. doi: 10.1016/j.redox.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittig D, Wang X, Walter C, Gerdes HH, Funk RHW, Roehlecke C. Multi-level communication of human retinal pigment epithelial cells via tunneling nanotubes. PloS ONE. 2012;7 doi: 10.1371/journal.pone.0033195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf DP, Mitalipov S. Mitochondrial replacement therapies can circumvent mtDNA-based disease transmission. Cell Metab. 2014;20:6–8. doi: 10.1016/j.cmet.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TH, Teslaa T, Kalim S, French CT, Moghadam S, Wall R, Miller JF, Witte ON, Teitell MA, Chiou PY. Photothermal nanoblade for large cargo delivery into mammalian cells. Anal Chem. 2011;83:1321–1327. doi: 10.1021/ac102532w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TH, Teslaa T, Teitell MA, Chiou PY. Photothermal nanoblade for patterned cell membrane cutting. Opt Express. 2010;18:23153–23160. doi: 10.1364/OE.18.023153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Wu TH, Clemens DL, Lee BY, Wen XM, Horwitz MA, Teitell MA, Chiou PY. Massively parallel delivery of large cargo into mammalian cells with light pulses. Nat Methods. 2015;12:439-+. doi: 10.1038/nmeth.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu JM, Teslaa T, Wu TH, Chiou PY, Teitell MA, Weiss S. Nanoblade delivery and incorporation of quantum dot conjugates into tubulin networks in live cells. Nano Lett. 2012;12:5669–5672. doi: 10.1021/nl302821g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Akita H, Kamiya H, Kogure K, Yamamoto T, Shinohara Y, Yamashita K, Kobayashi H, Kikuchi H, Harashima H. MITO-Porter: a liposome-based carrier system for delivery of macromolecules into mitochondria via membrane fusion. Biochim Biophys Acta. 2008;1778:423–432. doi: 10.1016/j.bbamem.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Furukawa R, Yasuzaki Y, Harashima H. Dual function MITO-Porter, a nano carrier integrating both efficient cytoplasmic delivery and mitochondrial macromolecule delivery. Mol Ther. 2011;19:1449–1456. doi: 10.1038/mt.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial origins. Proc Natl Acad Sci USA. 1985;82:4443–4447. doi: 10.1073/pnas.82.13.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YW, Koob MD. Transferring isolated mitochondria into tissue culture cells. Nucleic Acids Res. 2012;40 doi: 10.1093/nar/gks639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura I, Nakada K, Sato A, Hayashi J, Fujita K, Kaneko S, Itaya M. Direct cloning of full-length mouse mitochondrial DNA using a Bacillus subtilis genome vector. Gene. 2007;391:171–177. doi: 10.1016/j.gene.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Yoon YG, Koob MD. Efficient cloning and engineering of entire mitochondrial genomes in Escherichia coli and transfer into transcriptionally active mitochondria. Nucleic Acids Res. 2003;31:1407–1415. doi: 10.1093/nar/gkg228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YG, Koob MD. Transformation of isolated mammalian mitochondria by bacterial conjugation. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gni140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YG, Yang YW, Koob MD. PCR-based cloning of the complete mouse mitochondrial genome and stable engineering in Escherichia coli. Biotechnol Lett. 2009;31:1671–1676. doi: 10.1007/s10529-009-0063-9. [DOI] [PubMed] [Google Scholar]
- Yu H, Koilkonda RD, Chou TH, Porciatti V, Ozdemir SS, Chiodo V, Boye SL, Boye SE, Hauswirth WW, Lewin AS, et al. Gene delivery to mitochondria by targeting modified adenoassociated virus suppresses Leber’s hereditary optic neuropathy in a mouse model. Proc Natl Acad Sci USA. 2012a;109:E1238–E1247. doi: 10.1073/pnas.1119577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Ozdemir SS, Koilkonda RD, Chou TH, Porciatti V, Chiodo V, Boye SL, Hauswirth WW, Lewin AS, Guy J. Mutant NADH dehydrogenase subunit 4 gene delivery to mitochondria by targeting sequence-modified adeno-associated virus induces visual loss and optic atrophy in mice. Mol Vis. 2012b;18:1668–1683. [PMC free article] [PubMed] [Google Scholar]