

Abstract
Background:
Recent studies have implicated specific assembly subtypes of β-amyloid (Aβ) peptide, specifically soluble oligomers (soAβ) as disease-relevant structures that may underlie memory loss in Alzheimer disease. Removing existing soluble and insoluble Aβ assemblies is thought to be essential for any attempt at stabilizing brain function and slowing cognitive decline in Alzheimer disease. IV immunoglobulin (IVIg) therapies have been shown to contain naturally occurring polyclonal antibodies that recognize conformational neoepitopes of soluble or insoluble Aβ assemblies including soAβ. These naturally occurring polyclonal antibodies have been suggested to underlie the apparent clinical benefits of IVIg. However, direct evidence linking anti-Aβ antibodies to the clinical bioactivity of IVIg has been lacking.
Methods:
Five-month-old female Dutch APP E693Q mice were treated for 3 months with neat IVIg or with IVIg that had been affinity-depleted over immobilized Aβ conformers in 1 of 2 assembly states. Memory was assessed in a battery of tests followed by quantification of brain soAβ levels using standard anti-soAβ antibodies.
Results:
We provide evidence that NU4-type soAβ (NU4-soAβ) assemblies accumulate in the brains of Dutch APP E693Q mice and are associated with defects in memory, even in the absence of insoluble Aβ plaques. Memory benefits were associated with depletion from APP E693Q mouse brain of NU4-soAβ and A11-soAβ but not OC-type fibrillar Aβ oligomers.
Conclusions:
We propose that targeting of specific soAβ assembly subtypes may be an important consideration in the therapeutic and/or prophylactic benefit of anti-Aβ antibody drugs.
Alzheimer disease (AD), the most common form of dementia among the elderly, is attended by decades of accumulation of the neurotoxic β-amyloid (Aβ) peptide.1 Removing existing soluble and insoluble Aβ assemblies is thought to be essential for stabilizing brain function and slowing cognitive decline. While prior active or passive immunotherapies have been successful in AD mouse models, success in clinical trials has been elusive.2
IV immunoglobulin (IVIg) consists of purified plasma Ig pooled from thousands of healthy donors, is associated with reduced risk of developing AD,3 and was shown to contain naturally occurring antibodies against Aβ (nAbs-Aβ).4,5 Such nAbs-Aβ appear to be decreased in patients with AD, suggesting that some component(s) of IVIg may be useful for the treatment of early sporadic or familial forms of AD,4,6 and an independent phase 3 trial of IVIg yielded benefit in patients with moderate-stage AD who carried an APOE ε4 allele.7
Immune Globulin (IG), an IVIg therapy developed by Baxter Pharmaceuticals, has shown benefit in AD models8,9 and produced cognitive benefit in early trials.10,11 IG contains nAbs that recognize conformational neoepitopes on detergent-soluble and -insoluble Aβ aggregates. However, direct evidence linking anti-Aβ antibodies to the clinical bioactivity of IG has been lacking. The aim of this study was to test the effects of neat or Aβ-affinity-depleted forms of IG on learning behaviors and pathology in Dutch APP E693Q12 transgenic mice, and to determine whether improved learning behavior(s) might be associated with the depletion of specific soluble oligomeric Aβ (soAβ) immunosubtypes.
METHODS
Experimental animals.
Animal procedures were conducted in accordance with the NIH Guidelines for the Care and Use of Experimental Animals and were approved by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. All mice were given ad libitum access to food and water, and housed in micro-isolator cages under a 12-hour light/dark cycle. Generation of Dutch (APP E693Q) and PS1ΔE9 transgenic mouse lines have been described previously.12 For baseline cued and contextual fear conditioning (FC) behavior, experimentally naive, 6-month-old male and female mice were used: nontransgenic (nTg; n = 8), Dutch (n = 9), and Dutch/PS1ΔE9 (n = 13).
For IG drug-treatment experiments, 5-month-old female Dutch APP E693Q transgenic mice were given weekly subcutaneous injections of either saline (n = 11) or 2 g/kg neat Baxter IG (n = 12), 2 g/kg IG depleted of anti-fibril Aβ antibodies (fibril Aβ-affinity-depleted IG; n = 11), 2 g/kg IG depleted of anti-oligomer Aβ antibodies (oligomer Aβ-affinity-depleted IG; n = 11), or 2 g/kg IG depleted of both anti-oligomer and anti-fibril Aβ antibodies (Aβ-affinity-depleted IG; n = 11) for 3 months.
Preparation of Aβ monomers, oligomers, and fibrils for affinity depletion.
Resin bearing Aβ42 monomers coupled at either the N or C terminus was obtained from Alpha Diagnostic International (San Antonio, TX). Aβ globular oligomers were prepared as previously described.13 The resultant oligomers were characterized by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and stored at 4°C for up to 2 weeks. Stabilized oligomers, for coupling to affinity columns, were cross-linked by treatment with 1 mM glutaraldehyde for 15 minutes at 20°C and terminated in 5 mM ethanolamine at 20°C. The ethanolamine was removed by ultrafiltration using a Vivaspin centrifugal concentrator with a 10-kDa cutoff centrifugal concentrator (Sartorius Stedim, Bohemia, NY). Aβ fibrils were prepared as described.14 HFIP-treated Aβ40 monomer was dissolved in 2 mM NaOH, then centrifuged at 10,000g for 60 minutes to remove amyloid clumps. The supernatant was adjusted to 1× phosphate-buffered saline (PBS) 0.05% NaAz and incubated with agitation at 37°C for 14 days. Fibril structure was confirmed by electron microscopy. Before coupling onto the affinity resin, the fibrils were sonicated as described.15
Depletion of anti-Aβ antibodies from Baxter IG by affinity chromatography.
Resins for the depletion of anti-Aβ oligomer and anti-Aβ fibril antibodies were prepared as described.16 Baxter IG at 10 mg/mL in PBS was passed at least 5 times over columns at 1 to 2 mL/min to deplete either anti-oligomer or anti-fibril antibodies. To produce IG deficient in antibodies against all types of amyloid, the columns, including monomer amyloid columns, were run in series. Unbound IG from the various columns was concentrated to approximately 500 mg/mL by ultrafiltration through 30k molecular weight cutoff filters (Sartorius Stedim) and dialyzed against PBS before use in the animal studies.
Behavioral testing.
Mice were placed in the testing room 1 hour before testing to acclimatize to the room. All equipment was cleaned between animals. Memory was assessed in the novel object recognition (NOR), Y-maze spontaneous alternation (SA), and contextual/cued FC tests and anxiety-like behavior assessed in the elevated plus maze (EPM) as previously described.17,18
Tissue preparation.
Animals were killed, perfused with ice-cold 1× PBS, and brains were hemisected. One hemisphere was postfixed in 4% paraformaldehyde, stored at 4°C, and prepared for immunohistologic analyses. The remaining hemisphere was snap frozen and stored at −80°C before biochemical analysis. Brain samples were prepared for biochemical analysis as described previously.19
Antibody-based assays.
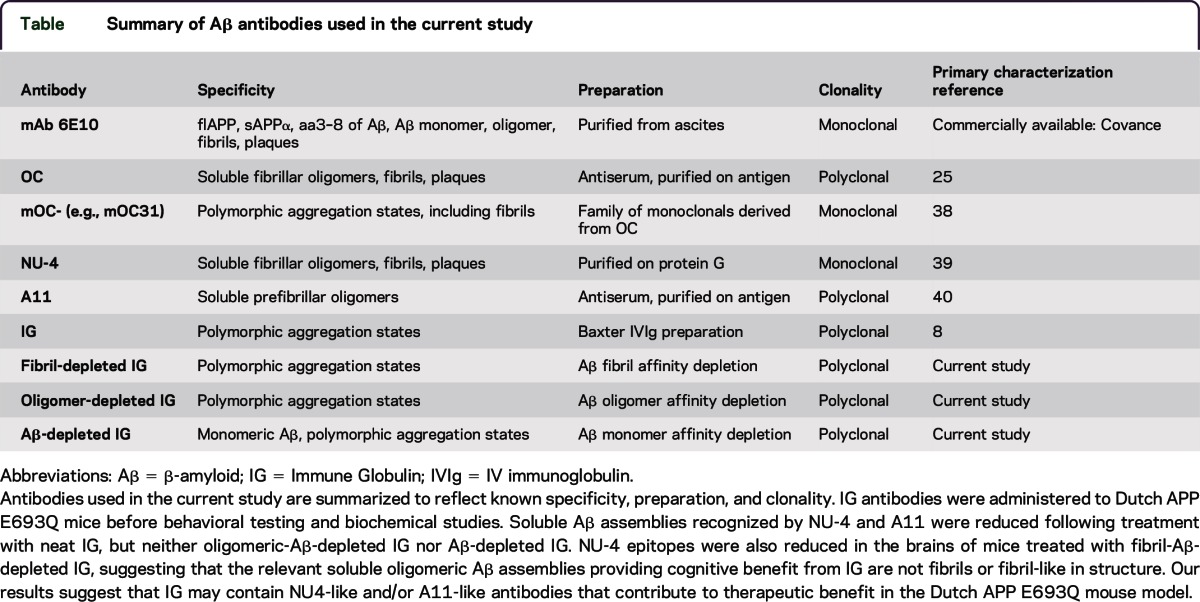
For a list of antibodies used in the current study, or for immunofluorescence and immunohistology, Western blot, dot blot, and ELISA assays described here, see e-Methods at Neurology.org/nn.
Statistical analysis.
Independent-samples t tests were used to determine significant differences between 2 groups. One-way analyses of variance with Bonferroni post hoc analyses were used to compare 3 or more groups. Significance for t tests and analyses of variance are reported with a p ≤ 0.05 using 2-tailed tests with an α level of 0.05. All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA).
RESULTS
Impairment of hippocampus-dependent memory is evident at 6 months of age in Dutch and Dutch APP/PS1ΔE9 mice.
Six-month-old nTg, Dutch APP, and Dutch APP/PS1ΔE9 mice were trained and tested on the cued and contextual FC paradigm. By comparison to their nTg littermates, both Dutch APP and Dutch APP/PS1ΔE9 mice displayed significant impairment of contextual fear memory, whereas no differences were observed among the 3 groups for cued fear memory (figure 1, A and B). No significant differences were observed between male and female mice for performance on cued or contextual FC tasks (data not shown), consistent with our prior observations using the Morris water maze task12; therefore, males and females were combined for analysis. Because both contextual and cued fear memory rely on an intact amygdala,20 but only contextual memory relies additionally on an intact hippocampus, our data suggest that impaired contextual memory reflects a defect in hippocampus-dependent memory. This finding is consistent with prior studies of AD mouse models12,19,20 and patients with AD.21 No difference was observed between Dutch APP and Dutch/PS1ΔE9 mice for contextual memory, suggesting that the cause of cognitive defect may be common (i.e., related to APP E693Q transgene product) among the 2 mouse lines and not related to the accumulation of amyloid plaques that occurs only in Dutch/PS1ΔE9 mice.
Figure 1. Impaired contextual memory is associated with levels of soluble fibrillary oligomeric Aβ assemblies.

Six-month-old mice were trained and tested for contextual (A) and cued (B) fear memory. Brain lysates were analyzed for levels of soAβ (C), soluble Aβ42/40 (D), and insoluble aggregated Aβ42 (E). Using conformation-specific antibodies, levels of soluble prefibrillar (F) or fibrillar (G) oAβ were analyzed from brain lysates. (H) Brain sections were stained for soluble prefibrillar oAβ assemblies (A11) or nuclear oligomeric assemblies (M78); scale bars = 40 µm; ×20 magnification, counterstain is DAPI. Data are representative littermates; histograms represent mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001. Aβ = β-amyloid; Agg. = aggregated; AU = arbitrary units; DAPI = 4',6-diamidino-2-phenylindole; nTg = nontransgenic; oAβ = oligomeric β-amyloid; soAβ = soluble oligomeric β-amyloid.
Accumulation of soluble, but not insoluble, oligomeric Aβ assemblies is a common feature among Dutch APP and Dutch APP/PS1ΔE9 mice.
Using a 7n22/7n22 duplicate-epitope sandwich immunoassay, we measured levels of total soAβ assemblies from both mouse lines. No differences were observed between Dutch APP and Dutch APP/PS1ΔE9 littermates for levels of soAβ assemblies (figure 1C). Next, we measured levels of soluble Aβ40 and Aβ42, revealing a significant difference between Dutch APP and Dutch APP/PS1ΔE9 mice for Aβ42/Aβ40 ratio (figure 1D), which is consistent with expression of the PS1ΔE9 transgene.12,22 We also observed a nearly 10-fold-higher accumulation of detergent-insoluble aggregated Aβ42 in the brains of Dutch APP/PS1ΔE9 mice as compared to their Dutch APP littermates (figure 1E).
Based on the observation that both mouse lines accumulate the same amount of soAβ assemblies (figure 1C), we utilized conformation-specific antibodies to assess the native conformation of the soAβ assemblies. We noted a significant decrease in levels of A11-immunoreactive soluble prefibrillar oAβ assemblies among Dutch APP/PS1ΔE9 mice when compared to their Dutch APP littermates (figure 1, F and H), whereas no difference was observed between the 2 genotypes for levels of OC-immunoreactive soluble fibrillar oAβ assemblies (figure 1G).
Previously, we reported marked intraneuronal accumulation of APP/Aβ in the brains of both Dutch APP and Dutch APP/PS1ΔE9 mice.12 We further characterized this pathology, revealing subtle differences in intra- and extraneuronal pathology between the genotypes (figure 2). Whereas Dutch APP mice primarily exhibited perinuclear intracellular 4G8-immunoreactivity without plaque-like M31-immunoreactivity, Dutch APP/PS1ΔE9 mice exhibited punctate intracellular 4G8-immunoreactivity and plaque-like M31-immunoreactivity (figure 2).
Figure 2. Subtle differences in intra- and extraneuronal accumulation of Aβ between Dutch and Dutch/PS1ΔE9 mice.
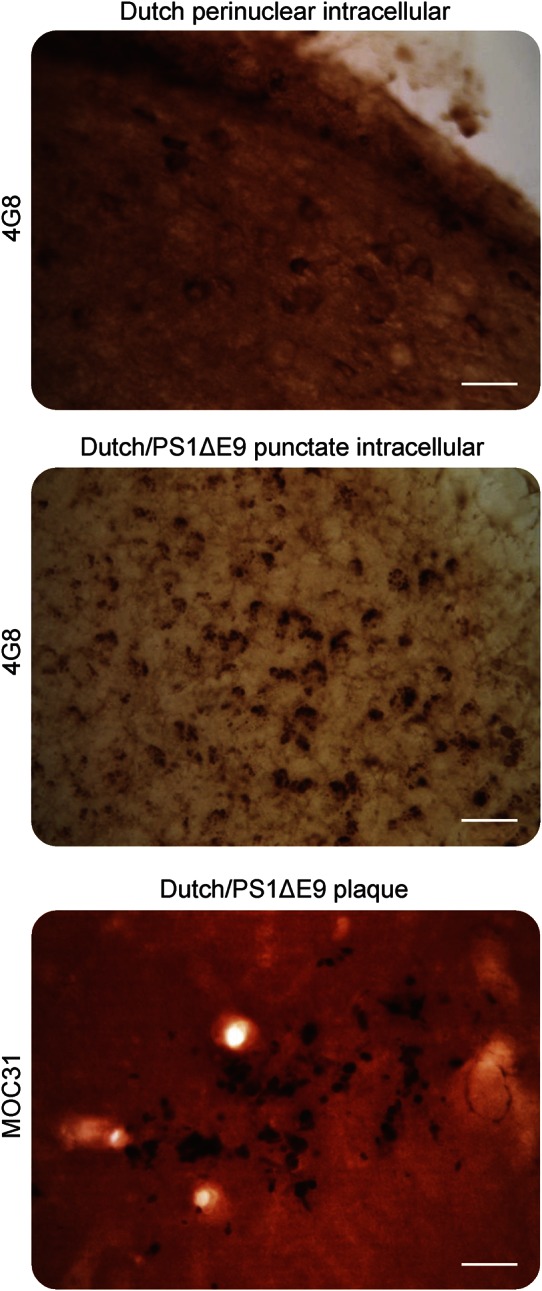
Brains were stained to analyze intracellular accumulation of APP/Aβ (4G8) or extracellular accumulation of Aβ (mOC31). Punctate immunoreactivity to 4G8 was typically observed in Dutch APP/PS1ΔE9 mice, whereas Dutch APP mouse brains typically exhibited perinuclear immunoreactivity. Only Dutch APP/PS1ΔE9 mice accumulated insoluble mOC31-immunoreactive extracellular plaques. Micrographs are representative littermates; scale bars = 10 µm; ×20 magnification. Aβ = β-amyloid.
We analyzed brain lysates from nTg, Dutch APP, and Dutch APP/PS1ΔE9 for markers of autophagic/lysosomal stasis including LC3, p62, α-synuclein, and APP in order to determine whether autophagic/lysosomal failure was evident and/or associated with accumulation of soAβ assemblies or insoluble Aβ42. No accumulation of soluble LC3, p62, or α-synuclein was observed by comparison to nTg controls among either Dutch APP or Dutch APP/PS1ΔE9 mice, suggesting that autophagic/lysosomal clearance may be intact at this stage of pathology (figure 3). However, we noted a significant accumulation of insoluble p62 among only Dutch APP/PS1ΔE9 mice by comparison to their nTg and Dutch APP littermates (figure 3, A and C) and this effect was also observed to precede onset of autophagic/lysosomal failure in TgCRND8 mice.19 Based on this result, it is tempting to speculate that the insoluble accumulation of Aβ42 and p62 may represent an early marker of toxicity to the autophagic/lysosomal pathway; however, further aging studies with these mouse lines will be necessary to determine whether autophagic/lysosomal failure occurs at any age.
Figure 3. Only Dutch APP/PS1ΔE9 mice accumulate insoluble p62.

Brain lysates were analyzed for levels of soluble (LC3, p62, α-synuclein) and insoluble (p62) autophagic/lysosomal substrates and APP metabolites (A–E). No impairment of autophagic/lysosomal clearance was observed at this age; however, only Dutch/PS1ΔE9 mice accumulated insoluble p62, which we previously reported to occur before autophagic/lysosomal failure in the brains of TgCRND8 mice19. Western blots are representative littermates, histograms represent mean ± SEM; *p < 0.05; 1-way analysis of variance with Bonferroni post hoc analyses. nTg = nontransgenic.
Treatment of Dutch mice with IG rescued learning behavior.
Next, we sought to determine whether antibodies against various forms of soluble or insoluble Aβ assemblies may be responsible for the benefit provided by IG therapy. We previously reported deficits on hippocampus-dependent memory in the Morris water maze task and FC in the Dutch APP mouse line.12,23 Here, we chose a battery of tasks that target both hippocampus-dependent memory as well as the entorhinal cortex and amygdala to assess complete memory function; these include FC,17,23 NOR,17,23 and Y-maze SA (previously untested), or anxiety-like behaviors using the EPM (previously reported17,23). Three-month treatment of Dutch APP mice by once-weekly subcutaneous injection of neat IG prevented onset of NOR (figure 4A), SA (figure 4B), and FC (figure 4C) deficits, without affecting anxiety-like behavior in the EPM (figure 4D) in female Dutch APP mice. This behavioral protection was completely abrogated by treatment with either saline or fibrillar Aβ-affinity-depleted or total Aβ-affinity-depleted IG. However, a statistically significant benefit in learning behavior tasks (e.g., NOR) was observed among Dutch APP mice treated with oligomer Aβ-affinity-depleted IG (figure 4A).
Figure 4. Prevention of onset of behavioral deficits following 3-month treatment of Baxter IG in Dutch APP E693Q transgenic mice.

Five-month-old female Dutch APP E693Q transgenic mice were given weekly subcutaneous injections of either saline (n = 11) or 2 g/kg neat Baxter IG (n = 12), 2 g/kg IG depleted of anti-fibril Aβ antibodies (fibril Aβ-affinity-depleted IG; n = 11), 2 g/kg IG depleted of anti-oligomer Aβ antibodies (oligomer Aβ-affinity-depleted IG; n = 11), or 2 g/kg IG depleted of both anti-oligomer and anti-fibril Aβ antibodies (Aβ-affinity-depleted IG; n = 11) for 3 months. Unlike treatment of saline, fibril Aβ-affinity-depleted IG and Aβ-affinity-depleted IG, treatment of IG prevented onset of behavioral deficits in novel object recognition (A), Y-maze spontaneous alternation (B), and contextual/cued fear conditioning (C) without affecting anxiety-like behavior in the elevated plus maze (C) in Dutch APP E693Q transgenic mice. Treatment of oligomer Aβ-affinity-depleted IG prevented onset of behavioral deficit in novel object recognition (A) in Dutch APP E693Q transgenic mice. *p < 0.05; **p < 0.01; 1-way analysis of variance with Bonferroni post hoc analyses. Data expressed as mean ± SEM. Aβ = β-amyloid; IG = Immune Globulin.
Treatment of mice with neat IG did not affect brain levels of Aβ40 (figure 5, A–D) or Aβ42 (figure 5, E–H), or its ratio (figure 5, I–L), but rather reduced brain levels of both toxic prefibrillar A11-immunoreactive soAβ (figure 5M) and NU-4-immunoreactive assemblies (figure 5N) in Dutch APP mice. Levels of OC-immunoreactive soluble fibrillar oligomers were not affected following treatment with neat IG in Dutch mice (figure 5O). Treatment of mice with fibrillar Aβ-affinity-depleted IG apparently reduced brain levels of NU-4-immunoreactive (figure 5N) but not A11-immunoreactive (figure 5M) soAβ assemblies in Dutch APP mice. Thus, neat IG treatment was associated with clinical benefit and reduction of 2 immunosubtypes of soAβ assemblies (i.e., both A11-type and NU-4-type oligomers). Among 3 immunoaffinity-depleted IG preparations, only one was clinically effective, and, unexpectedly, that bioactive fraction was the fibrillar Aβ-affinity-depleted IG that was associated with reduction in the content of NU-4-type soAβ assemblies. We cannot determine whether reduction in the content of A11-type soAβ assemblies might also be associated with clinical benefit. The relevance of this point is discussed in detail below.
Figure 5. Reduction of oligomeric Aβ following 3-month treatment of Baxter IG in Dutch APP E693Q transgenic mice.

Five-month-old female Dutch APP E693Q transgenic mice were given weekly subcutaneous injections of either saline (n = 11) or 2 g/kg neat Baxter IG (n = 12), 2 g/kg IG depleted of anti-23 fibril Aβ antibodies (fibril Aβ-affinity-depleted IG; n = 11), 2 g/kg IG depleted of anti-oligomer Aβ antibodies (oligomer Aβ-affinity-depleted IG; n = 11), or 2 g/kg IG depleted of both anti-oligomer and anti-fibril Aβ antibodies (Aβ-affinity-depleted IG; n = 11) for 3 months. Aβ40 (A–D), Aβ42 (E–H), or Aβ42/40 ratio (I–L) were unaffected with all treatments. Treatment with IG reduced brain levels of prefibrillar oligomers (M) and oligomers (N) but not fibrillar oligomers (O) in Dutch APP E693Q transgenic mice. Treatment of fibril Aβ-affinity-depleted IG reduced brain levels of oligomers in Dutch APP E693Q transgenic mice (N). (A, E, I) Total Aβ. (B, F, J) TBS fraction. (C, G, H) Triton-X fraction. (D, H, I) Formic acid fraction. (M, N, O) TBS fraction (n = 5/group). *p < 0.0; **p < 0.01; 1-way analysis of variance with Bonferroni post hoc analyses. Data expressed as mean ± SEM. Aβ = β-amyloid; IG = Immune Globulin.
DISCUSSION
The current study indicates the utility of rodent models to study multiple stages of AD-related pathology, the relationship of Aβ pathology to cognitive status, and the ability to separate canonical Aβ plaque pathology from underlying toxic mechanisms. Here, we describe a hippocampus-dependent contextual fear memory deficit at 6 months of age in Dutch APP and Dutch APP/PS1ΔE9 mice, and the accumulation of cognitive deficit–associated soAβ assemblies that are common to both lines. Of note, prior studies indicate that A11-immunoreactive prefibrillar soAβ assemblies are associated with conversion from soluble low-n multimers to insoluble fibrillar assemblies, whereas OC-immunoreactive soluble fibrillar soAβ assemblies do not form fibrils and, rather, seed formation of more soluble fibrillar soAβ assemblies.24 Moreover, levels of soAβ assemblies were recently found to be predictive of cognitive status at death among patients with AD,25 validating our findings (as discussed above) and the utility of these mouse models of AD. Here, we noted a significant decrease in A11-immunoreactive soAβ assemblies among Dutch APP/PS1ΔE9 mice as compared to their Dutch APP littermates (figure 1, F–H; table). Our conclusions are supported by recent work by Liu et al.26 who analyzed contributions of A11- and OC-immunoreactive soAβ assemblies to cognitive failure in Tg2576 mice and concluded that A11-immunoreactive, but not OC-immunoreactive, soAβ assemblies were associated with impaired memory. Based on these reports and our current results, we propose that A11- and/or NU-4-immunoreactive soAβ assemblies constitute key soAβ assemblies related to early hippocampal and entorhinal cortex-related memory failures in AD. Moreover, we propose that the Dutch APP E693Q mutation can support stabilization of these structures in a soluble state, perpetuating memory failure despite a failure to polymerize into insoluble fibrillar assemblies. A more dramatic example of this situation is exemplified by the Arctic APP E693G mutation wherein typical clinical AD develops despite the inability of the mutant peptide to form plaques, a property evidenced by the failure of [11C] Pittsburgh compound B to detect cerebral amyloidosis at any stage of the Arctic APP E693G–related familial AD.27
Table.
Summary of Aβ antibodies used in the current study
Our group recently performed an integrative genomic analysis of the transcriptomes from human AD brain tissue or 2 independent mouse lines (including the Dutch APP mouse line) to identify altered molecules and pathways between wild-type and transgenic mice, and human patients with AD.28 These studies revealed consistent alterations in APP/Aβ metabolism, epigenetic control of neurogenesis, cytoskeletal organization, and extracellular matrix regulation. Comparison of these transcriptomic results with human AD postmortem gene expression data indicated significant similarities in pathway alterations between mouse models and patients with AD. In addition, it has been reported that changes in neuroinflammation-related gene regulation at early stages of AD was not related to neurofibrillary tangles, Aβ plaque burden, total Aβ40 or Aβ42, or membrane-bound fibrillar Aβ, but rather to increased levels of soAβ.29 Taken together, these studies suggest that soAβ assemblies represent a critical driver of early-stage disease processes, leading to cognitive and cellular dysfunction observed at later stages of disease.
IVIg formulations have been previously assessed in other mouse models of AD, demonstrating that peripheral administration of IVIg crossed the blood–brain barrier and binding of Aβ deposits in APP/PS-1 mice. In ex vivo APP/PS-1 brain sections, IVIg enhanced microglial-mediated Aβ clearance and depletion of nAbs-Aβ in IVIg significantly reduced clearance.8 Treatment of TgCRND8 mice with nAbs-Aβs isolated from IVIg reduced plaque load in mice treated at 3 months of age but not those treated later during disease progression, at 12 months of age.30 When memory was assessed in Tg2576 mice, nAbs-Aβ treatment improved object recognition.30 In contrast, when APP/PS1ΔE9 mice were treated with IVIg for 3 or 8 months, no evidence of reduced Aβ was reported.9 Instead, increased monomeric Aβ40 and Aβ42 levels, enhancement of neurogenesis, suppression of proinflammatory cytokine gene expression, and evidence of modulation of microglial activation was observed in mice treated for 8 months.9 More recently, another study in Tg2576 mice saw an improvement in memory and synaptic plasticity following IVIg treatment that was not linked to altered monomeric Aβ levels, but rather by the activation of complement components.31
IVIg contains antibodies against both monomeric and soAβ.32 In vitro studies in N2a neuroblastoma cells have revealed that IVIg can disaggregate Aβ fibrils and promote Aβ removal33 while preventing neurotoxicity-associated soAβ assemblies.5 Similarly, in cultured primary mouse hippocampal neurons, treatment with IVIg has been shown to reduce Aβ fibril formation and reduce Aβ neurotoxicity.8 Many studies, including ours, revealed no detectable reduction in monomeric Aβ. When IVIg was assessed in the 3xTgAD model of AD, an improvement in NOR, reduced immunologic CD4/CD8 blood ratio, reduced interleukin (IL)-5/IL-10 ratio in the cortex together with limited effects on tau but reduced Aβ42/40 ratio and levels of soluble 56-kDA Aβ oligomers were reported.34 Our study is the first to focus on oligomer immunosubtypes post IVIg treatment. In the present study, IG protection was not associated with altered levels of monomeric Aβ40 and Aβ42 levels but rather with decreased levels of some but not all immunosubtypes of soluble prefibrillar oligomeric Aβ assemblies. A11-immunosubtype and/or NU4-immunosubtype soAβ assemblies appear to be the key conformer(s) driving memory failure. This study suggests that IG may contain A11-like and NU-4–like antibodies that confer some protection in the Dutch APP mouse line. An unexpected result of this study is the improvement in NOR in the oligomer Aβ-affinity-depleted IG-treated mice with no change in Aβ biochemistry. One explanation for this result is that the oligomer preparation used for fibrillar and oligomer Aβ affinity depletion of IG may not contain a complete and comprehensive representation of the structurally diverse spectrum of soluble Aβ assemblies. In other words, these 2 affinity-depleted IG fractions designated “oligomeric” or “fibrillar” may represent an oversimplified binary classification. Another possibility is that the nAbs may contain catalytic antibodies that induce changes in the conformations of the soAβ assemblies as part of their therapeutic bioactivity. “Chaperone-like” activity has been reported for other antibodies35,36 such that the antibodies induce changes in the conformation of soAβ assemblies as part of their therapeutic action and/or as a biochemical reaction in IG-treated brain homogenates during assays. We are currently characterizing the chaperone content of our IG fractions in order to assess this possibility properly.
Baxter IG has been proposed as a potential treatment for AD, especially at moderate stage and in carriers of APOE ε4 alleles.37 IG contains natural human polyclonal IgG antibodies that recognize conformational neoepitopes expressed on soluble and insoluble Aβ assemblies. The current study demonstrates for the first time that 3 months of neat IG treatment of Dutch APP transgenic Aβ-oligomer-forming mice is associated with prevention of memory deficits in NOR, SA, and FC that is abrogated by affinity depletion of IG using certain conformers of soluble Aβ assemblies. These principles might well be kept in mind during the continued development and optimization of active and passive anti-Aβ immunotherapies and during the continued early-stage work on creating therapeutic chaperones that have the capacity to stabilize soAβ assemblies in nontoxic conformations.
Supplementary Material
GLOSSARY
- Aβ
β-amyloid
- AD
Alzheimer disease
- EPM
elevated plus maze
- FC
fear conditioning
- IG
Immune Globulin
- IVIg
IV immunoglobulin
- nAb
naturally occurring antibody
- NOR
novel object recognition
- nTg
nontransgenic
- oAβ
oligomeric β-amyloid
- PBS
phosphate-buffered saline
- SA
spontaneous alternation
- soAβ
soluble oligomeric β-amyloid
Footnotes
Funding information and disclosures are provided at the end of the article. Go to Neurology.org/nn for full disclosure forms. The Article Processing Charge was paid by the authors.
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
E.M.K. designed the experiments with M.E., P.S., N.R.R., C.M.A., C.G.G., S.G., and J.W.S. E.M.K., S.H.K., J.C.K., A.H., R.A., A.L., A.S., and P.S. performed the experiments. E.M.K. and J.W.S. analyzed the data. E.M.K., P.S., N.R.R., S.G., and J.W.S. wrote the manuscript.
STUDY FUNDING
This work was supported in part by a grant from Baxter Pharmaceuticals, Inc. J.W.S. is supported by NIH grant K12 GM068524. This work was additionally supported by NIH grants P50 AG05138 (to Mary Sano; S.G.), U01 AG046170 (S.G., M.E.), R34 AG049649 (S.G.), R01 NS075685 (S.G.), R01 MH065635 (C.M.A.), R21 AT005510 (S.G.), RF1 AG042965 (S.G.), VA MERIT Review Grant I01 RX000684 (S.G.), the Cure Alzheimer's Fund (S.G.), and gifts from the Louis B. Mayer Foundation, the Sarah and Gideon Gartner Trust, the Rudin Foundation, and the Werber Family Foundation (all to S.G.).
DISCLOSURE
E. Knight, S.H. Kim, and J. Kottwitz report no disclosures. A. Hatami is an associate editor for Journal of Alzheimer's Disease, received research support from NIH, Cure Alzheimer's Fund. R. Albay and A. Suzuki report no disclosures. A. Lublin has been employed by Sanofi Genzyme, received research support from University of California, Riverside, holds stock or stock options in Geron and Ionis. C. Alberini is on the editorial board for Neural Plasticity, Neurobiology of Learning and Memory, was on the editorial board for Cell Science, is an associate editor for Frontiers in Neuroscience, received research support from NIH. W.L. Klein served on the scientific advisory board for Acumen Pharmaceuticals, received research support from NIH, Baxter-NU Alliance, NUCATS/Northwestern. P. Szabo reports no disclosures. N. Relkin served on the scientific advisory board for Anavex, Herbal Science Group, Eisai, was an associate editor for Neurology Alert, has patents pending for Multiplexed CSF markers for Alzheimer's disease, Diffusion tensor histogram analysis for diagnosis of hydrocephalus: an analytic method for identifying a DTI pattern associated with normal pressure hydrocephalus, Volumetric MRI for predicting response to IVIg treatment of Alzheimer's disease, Cytokine analysis for predicting response to IVIg treatment of Alzheimer's disease, Systems and methods for automating the retrieval of portioned search results, receives royalties from UpToDate, has consulted for Aisai, Forest, Hydrocephalus Association. M. Ehrlich is on the editorial board for ASN Neuro, received research support from NIH, NYSTEM. C. Glabe served on the editorial board for Journal of Molecular Neurodegeneration, has patents and pending patents for immunotherapy and immunodiagnostics for AD, consults relating to intellectual property, receives license fee and royalty payments for antibodies for research purposes only and patent issues, received research support from NIH, Cure Alzheimer's Fund, was involved in legal proceedings for Sidley LLP. S. Gandy is an associate editor for ADAD, Molecular Neurodegeneration, is a consulting editor for JCI, received research support from Avid/Lilly, Constellation Wines. J. Steele is a scientific advisory board member for OrPhi Therapeutics Inc., was editor-in-chief and consulting editor for Journal of Postdoctoral Research, has a pending patent for Mitoprotection for treatment of lysosomal storage diseases, consulted for Amicus Therapeutics, received research support from CurePSP, DPD Deficiency Foundation, is a cofounder and on the board of directors for OrPhi Therapeutics Inc., holds stock or stock options in Amicus Therapeutics, GlaxoSmithKline. Go to Neurology.org/nn for full disclosure forms.
POTENTIAL CONFLICT OF INTEREST STATEMENT
J.W.S. is a cofounder, shareholder, and member of the board of directors and scientific advisory board of OrPhi Therapeutics Inc. (Carlsbad, CA). Within the past 5 years, S.G. has held research grants from Amicus Therapeutics. S.G. is also a member of the Data and Safety Monitoring Board for the Pfizer-Janssen Alzheimer's Immunotherapy Alliance.
REFERENCES
- 1.Gandy S. Lifelong management of amyloid-beta metabolism to prevent Alzheimer's disease. N Engl J Med 2012;367:864–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gandy S, Sano M. Alzheimer disease: solanezumab—prospects for meaningful interventions in AD? Nat Rev Neurol 2015;11:669–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knight EM, Gandy S. Immunomodulation and AD: down but not out. J Clin Immunol 2014;34(suppl 1):S70–S73. [DOI] [PubMed] [Google Scholar]
- 4.Dodel R, Hampel H, Depboylu C, et al. Human antibodies against amyloid beta peptide: a potential treatment for Alzheimer's disease. Ann Neurol 2002;52:253–256. [DOI] [PubMed] [Google Scholar]
- 5.Szabo P, Relkin N, Weksler ME. Natural human antibodies to amyloid beta peptide. Autoimmun Rev 2008;7:415–420. [DOI] [PubMed] [Google Scholar]
- 6.Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol 2002;37:943–948. [DOI] [PubMed] [Google Scholar]
- 7.Relkin N. Intravenous immunoglobulin for Alzheimer's disease. Clin Exp Immunol 2014;178(suppl 1):27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magga J, Puli L, Pihlaja R, et al. Human intravenous immunoglobulin provides protection against Aβ toxicity by multiple mechanisms in a mouse model of Alzheimer's disease. J Neuroinflammation 2010;7:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puli L, Pomeshchik Y, Olas K, Malm T, Koistinaho J, Tanila H. Effects of human intravenous immunoglobulin on amyloid pathology and neuroinflammation in a mouse model of Alzheimer's disease. J Neuroinflammation 2012;9:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Relkin NR, Szabo P, Adamiak B, et al. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging 2009;30:1728–1736. [DOI] [PubMed] [Google Scholar]
- 11.Tsakanikas D, Shah K, Flores C, Assuras S, Relkin NR. P4-351: effects of uninterrrupted intravenous immunoglobulin treatment of Alzheimer's disease for nine months. Alzheimers Dement 2008;4:T776. [Google Scholar]
- 12.Gandy S, Simon AJ, Steele JW, et al. Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers. Ann Neurol 2010;68:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barghorn S, Nimmrich V, Striebinger A, et al. Globular amyloid beta-peptide oligomer: a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem 2005;95:834–847. [DOI] [PubMed] [Google Scholar]
- 14.O'Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA 2002;99:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 2004;279:17490–17499. [DOI] [PubMed] [Google Scholar]
- 16.Szabo P, Mujalli DM, Rotondi ML, et al. Measurement of anti-beta amyloid antibodies in human blood. J Neuroimmunol 2010;227:167–174. [DOI] [PubMed] [Google Scholar]
- 17.Knight EM, Williams HN, Stevens AC, et al. Evidence that small molecule enhancement of beta-hexosaminidase activity corrects the behavioral phenotype in Dutch APP(E693Q) mice through reduction of ganglioside-bound Abeta. Mol Psychiatry 2015;20:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight EM, Martins IV, Gumusgoz S, Allan SM, Lawrence CB. High-fat diet-induced memory impairment in triple-transgenic Alzheimer's disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging 2014;35:1821–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steele JW, Lachenmayer ML, Ju S, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer's mouse model. Mol Psychiatry 2013;18:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobsen JS, Wu CC, Redwine JM, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 2006;103:5161–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laczo J, Vlcek K, Vyhnalek M, et al. Spatial navigation testing discriminates two types of amnestic mild cognitive impairment. Behav Brain Res 2009;202:252–259. [DOI] [PubMed] [Google Scholar]
- 22.Gandy S, Zhang YW, Ikin A, et al. Alzheimer's presenilin 1 modulates sorting of APP and its carboxyl-terminal fragments in cerebral neurons in vivo. J Neurochem 2007;102:619–626. [DOI] [PubMed] [Google Scholar]
- 23.Kim SH, Steele JW, Lee SW, et al. Proneurogenic group II mGluR antagonist improves learning and reduces anxiety in Alzheimer Abeta oligomer mouse. Mol Psychiatry 2014;19:1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu JW, Breydo L, Isas JM, et al. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J Biol Chem 2010;285:6071–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomic JL, Pensalfini A, Head E, Glabe CG. Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol Dis 2009;35:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P, Reed MN, Kotilinek LA, et al. Quaternary structure defines a large class of amyloid-beta oligomers neutralized by sequestration. Cell Rep 2015;11:1760–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scholl M, Wall A, Thordardottir S, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology 2012;79:229–236. [DOI] [PubMed] [Google Scholar]
- 28.Readhead B, Haure-Mirande JV, Zhang B, et al. Molecular systems evaluation of oligomerogenic APP and fibrillogenic APP/PSEN1 mouse models identifies shared features with human Alzheimer's brain molecular pathology. Mol Psychiatry Epub 2016 Jan 19.
- 29.Lopez-Gonzalez I, Schluter A, Aso E, et al. Neuroinflammatory signals in Alzheimer disease and APP/PS1 transgenic mice: correlations with plaques, tangles, and oligomeric species. J Neuropathol Exp Neurol 2015;74:319–344. [DOI] [PubMed] [Google Scholar]
- 30.Dodel R, Balakrishnan K, Keyvani K, et al. Naturally occurring autoantibodies against beta-amyloid: investigating their role in transgenic animal and in vitro models of Alzheimer's disease. J Neurosci 2011;31:5847–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gong B, Pan Y, Zhao W, et al. IVIG immunotherapy protects against synaptic dysfunction in Alzheimer's disease through complement anaphylatoxin C5a-mediated AMPA-CREB-C/EBP signaling pathway. Mol Immunol 2013;56:619–629. [DOI] [PubMed] [Google Scholar]
- 32.Klaver AC, Finke JM, Digambaranath J, Balasubramaniam M, Loeffler DA. Antibody concentrations to Abeta1-42 monomer and soluble oligomers in untreated and antibody-antigen-dissociated intravenous immunoglobulin preparations. Int Immunopharmacol 2010;10:115–119. [DOI] [PubMed] [Google Scholar]
- 33.Istrin G, Bosis E, Solomon B. Intravenous immunoglobulin enhances the clearance of fibrillar amyloid-beta peptide. J Neurosci Res 2006;84:434–443. [DOI] [PubMed] [Google Scholar]
- 34.St-Amour I, Pare I, Tremblay C, Coulombe K, Bazin R, Calon F. IVIg protects the 3xTg-AD mouse model of Alzheimer's disease from memory deficit and Abeta pathology. J Neuroinflammation 2014;11:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu YH, Bu XL, Liang CR, et al. An N-terminal antibody promotes the transformation of amyloid fibrils into oligomers and enhances the neurotoxicity of amyloid-beta: the dust-raising effect. J Neuroinflammation 2015;12:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA 1997;94:4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Relkin N. Clinical trials of intravenous immunoglobulin for Alzheimer's disease. J Clin Immunol 2014;34(suppl 1):S74–S79. [DOI] [PubMed] [Google Scholar]
- 38.Hatami A, Albay R, III, Monjazeb S, Milton S, Glabe C. Monoclonal antibodies against Abeta42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J Biol Chem 2014;289:32131–32143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert MP, Velasco PT, Chang L, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem 2007;100:23–35. [DOI] [PubMed] [Google Scholar]
- 40.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003;300:486–489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.