

Abstract
Objectives: We quantified medical signs and symptoms to construct the Physical Symptom Score (PSS) for use in research to assess somatic disease burden in mucopolysaccharidoses (MPS) to track disease and monitor treatments. We examined scoring reliability, its concurrent validity with other measures, and relationship to age in MPS type I.
Methods: Fifty-four patients with MPS I (36 with Hurler syndrome treated with hematopoietic cell transplant and 18 with attenuated MPS I treated with enzyme replacement therapy), ages 5 to 18 years, were seen longitudinally over 5 years. The summation of frequency and severity of signs of specific organ involvement, surgeries, and hydrocephalus drawn from medical histories comprise the PSS. We examined relationship to age and to daily living skills (DLS) from the Vineland Adaptive Behavior Scale and physical quality of life from the Child Health Questionnaire (CHQ) for each group.
Results: The PSS was associated with age in both groups, indicating increase in disease burden over time. The PSS was significantly negatively associated with DLS (r = −0.48) and CHQ (r = −0.55) in the attenuated MPS I but not in the Hurler group.
Conclusions: The association of somatic disease burden with physical quality of life and ability to carry out daily living skills suggests that the PSS will be useful in the measurement of disease and treatment effects in the attenuated MPS I group. Earlier treatment with transplant and differing parental expectations are possible explanations for its lack of association with other outcomes necessitating an adaptation for Hurler syndrome in the future.
Introduction
Patients with mucopolysaccharidoses type I (MPS I) have medical problems in many organ systems (Neufeld and Muenzer 2001; Clarke and Heppner 2002; Muenzer et al. 2009). They are often severe and likely contribute significantly to their health-related quality of life (QOL) and adaptive status. However, there is no single score that can summarize their disease burden for clinical research purposes. Quantification of these problems will assist in understanding how medical problems contribute to other outcome measures. We have developed a Physical Symptom Score (PSS) to measure disease burden based on the severity and frequency of symptoms in relevant organ systems; we have assessed its scoring reliability and validated it with quality of life and daily living skills measures.
MPS I is one of the lysosomal storage disorders which is an autosomal recessive multi-organ system disease caused by a deficiency of the enzyme, alpha-l-iduronidase (Neufeld and Muenzer 2001; Hopwood and Morris 1990), with an incidence of 1:100,000 live births (Neufeld and Muenzer 2001; Meikle et al. 1999; Moore et al. 2008). The phenotypes of MPS I disorder are historically classified into three syndromes: severe Hurler, milder disorder Scheie, and the intermediate disorder Hurler–Scheie. Hurler–Scheie and Scheie syndromes also have been referred collectively as attenuated MPS I and cannot be easily differentiated diagnostically using clinical, biochemical, or molecular criteria (Neufeld and Muenzer 2001; Clarke and Heppner 2002; Pastores et al. 2007; D’Aco et al. 2012). Hearing difficulties, corneal clouding, organomegaly, skeletal or orthopedic abnormalities, cardiorespiratory problems, central nervous system problems like hydrocephalus with or without shunt placement, other neurological problems such as carpal tunnel syndrome, headache, cognitive impairment, and sometimes cervical cord compression impede functionality on a day-to-day basis and contribute to disease burden. These physical signs and symptoms together with increased urinary excretion of GAG (glycosaminoglycan), and absent or deficient alpha-l-iduronidase enzyme activity are observed in all forms of MPS I (Terlato and Cox 2003; Scott et al. 1995; Beesley et al. 2001), but no method exists to accurately summarize phenotypic disease burden in retrospective or prospective research studies. Genotype–phenotype correlations have been established to a limited degree in Hurler syndrome and even more limited in the attenuated forms (Pastores et al. 2007; Terlato and Cox 2003; Scott et al. 1995; Beesley et al. 2001; Bertola et al. 2011; Ahmed et al. 2014a). Standard of care treatments such as hematopoietic cell transplant (HCT) (Peters et al. 1996; Peters et al. 1998; Souillet et al. 2003; Staba et al. 2004) in Hurler syndrome and enzyme replacement therapy (ERT) (Kakkis et al. 2001; Wraith et al. 2004) for the attenuated syndromes ameliorate some but not all symptoms. This scale will provide a summary score that could be used to assess the overall impact on disease burden of treatments over time.
Our goal is to establish a quantified measure of disease burden by creating a summative scale of the history of medical problems in multiple organ systems. We hypothesize that it will be associated with age, with the ability to carry out daily living skills (DLS), and with quality of life (QOL) outcomes in MPS I. The latter measures will provide information about concurrent validity.
Methods
Subjects
Subjects with MPS I were drawn from four centers contributing to a natural history study “Longitudinal Studies of Brain Structure and Function in MPS Disorders,” NCT01870375 of the Lysosomal Disease Network (Rare Disease Clinical Research Network-RDCRN). Inclusion criteria were (1) age range of 5 to 18 years, (2) availability of medical data to create PSS score at each visit, and (3) completion of either Vineland Adaptive Behavioral Scales (VABS), Second Edition, or Child Health Questionnaire Parent Form 50 (CHQ-PF50), for at least one visit. Each patient had between one and five visits over 5 years.
Procedures
Development of the PSS Scale
Medical and treatment histories, as gathered from interviews with patients or parents of patients and review of medical records, were recorded on case report forms developed for the longitudinal study. When available all clinical files and medical records of patients were reviewed. A scoring system was devised to quantify abnormalities in four medical domains: skeletal/orthopedic, vision, hearing, and cardiorespiratory. These data were scored by a system of frequency and severity of the organ system involvement (Ahmed et al. 2014b). The scoring system also quantified the patient’s number of surgical procedures using general anesthesia, and the absence or presence of hydrocephalus with or without shunt placement. Medical records were examined together with historical information for each domain. For example, to score the skeletal/orthopedic domain, we would collect appropriate information from the physician medical encounters and comments on symptoms such as limited range of motion, hip dysplasia, kyphosis, etc., and determine the frequency of these symptoms. Because we could not always determine the severity, each symptom was categorized as present or absent. Each of the 6 domains was scored 0 (absent) to 3 (severe). The specific criteria for coding each domain are provided in Table 1. Total summary scores were calculated by totaling each domain score. The range of total summary scores could be from 0 to 18.
Table 1.
MPS-specific physical symptom scale
| Feature | Score | Description |
|---|---|---|
| Skeletal/orthopedic | 0 | No orthopedic symptom |
| 1 | 1–2 symptoms | |
| 2 | 3–4 symptoms | |
| 3 | 5–6 symptoms or cord compression | |
| Skeletal/orthopedic symptoms include limited range of motion, kyphosis, scoliosis, hip dysplasia, knock knee, and high arch foot | ||
| Vision | 0 | No visual impairment or symptom |
| 1 | Mild corneal clouding or glaucoma or cataract | |
| 2 | Moderate corneal clouding or both glaucoma and cataract | |
| 3 | Severe corneal clouding or retinal degeneration | |
| Hearing | 0 | None |
| 1 | Mild hearing loss | |
| 2 | Moderate hearing loss | |
| 3 | Severe hearing loss | |
| Cardiorespiratory | 0 | None |
| 1 | 1–2 cardiac or respiratory symptoms | |
| 2 | 3–4 symptoms or presence of sleep apnea | |
| 3 | 5–6 symptoms or history of cardiac surgery | |
| Cardiac and respiratory symptoms include murmur, hypertension, valve disease, cardiac surgery, chronic nasal discharge/obstruction, tonsils/adenoids, respiratory infection/reactive airway disease, and sleep apnea | ||
| Hydrocephalus | 0 | Absent |
| 1 | Hydrocephalus without shunt | |
| 2 | Hydrocephalus with shunt | |
| 3 | Revision of shunt | |
| Number of surgeries | 0 | No surgery |
| 1 | Less than 4 surgeries | |
| 2 | 4 to 8 surgeries | |
| 3 | More than 8 surgeries | |
| Total score = 0–18 | ||
Reliability
To investigate the inter-rater scoring reproducibility, 30 MPS I patients were chosen in a random order and independently scored by an expert who is involved in MPS research and previously was a health professional (E.R.) and compared with the PSS scoring of the author (A.A.). Inter-rater reliability is reported on the total score based on history and medical record review. All the items were scored in a similar manner according to the MPS-specific Physical Symptom Scale by both raters (Table 1).
Validation Measures
We compared the PSS with physical summary (PhS) measure from the Child Health Questionnaires Parent Form 50 (CHQ-PF50) and the daily living skills (DLS) domain from Vineland Adaptive Behavioral Scales (VABS), Second Edition. These two scales were selected because they have embedded measures of functional outcomes of somatic burden of disease (Physical Summary Measure of Quality of Life and Daily Living Skills), allowing for concurrent validation. Ideally, a measure such as the HAQ (Health Assessment Questionnaire) (Pastores et al. 2007) might be used to validate the PSS; however, normative data is not available for children, and the HAQ was not selected as a measure for the longitudinal study for the same reason.
The Child Health Questionnaire Parent Form 50 (CHQ-PF50) is a 50-item, parent-completed questionnaire designed to measure children’s general functional health and/or health-related quality of life from parent’s perspective (Landgraf et al. 1996). The CHQ-PF50 has been validated in multiple settings and various childhood disease groups and normed in a large national study of children from 5 to 18 years of age. We used the T-scores (where M = 50 and SD = 15) for the physical summary (PhS) measure. Lower scores on the CHQ indicate greater impairment in functioning. This measure was chosen because it reflects the impact of disease burden on the perception of how it affects physical quality of life.
Vineland Adaptive Behavioral Scales (VABS), Second Edition, is an observer-rated measure of personal and social skills needed for everyday living (Sparrow et al. 2005). This adaptive level of functioning measures along four broad domains (communication, daily living skills (DLS), socialization, and motor skills) and is designed for assessing individuals between ages of 0 and 90 years. The daily living skills domain is a measure of practical skill of self-care, care of home, and community participation. We used the standard scores (where M = 100 and SD = 15) of parent-reported DLS for comparison. Lower scores on the DLS indicate greater impairment in functioning. This measure was chosen because it reflects the impact of disease burden on the day-to-day functioning of the patient.
Statistical Analysis
Descriptive statistics were tabulated separately for attenuated and Hurler forms of MPS I. These included the mean and standard deviation for continuous variables and frequency for categorical variables. Correlations were estimated with generalized estimating equations using normalized covariates with a working independence correlation structure, which corresponds to Pearson’s product moment correlation coefficient (Diggle et al. 2002). Confidence intervals and P-values were based on robust variance estimation to account for the correlated nature of longitudinal measurements. All analyses were conducted using R v3.1.1 (R Core Team 2014).
Results
The attenuated group was slightly older than the Hurler group (Table 2). The age at treatment and years from treatment were also different for the two groups due to differences in standards of care for the age of diagnosis and timing at each treatment. All of the Hurler patients underwent hematopoietic cell transplantation, and all of the attenuated patients were on enzyme placement therapy only.
Table 2.
Patient descriptives; values presented are mean (SD) or N (%) where indicated
| Covariate | Hurler | Attenuated |
|---|---|---|
| (N = 36) | (N = 18) | |
| Male | 18 (50.0%) | 9 (50.0%) |
| Age at first visit (years) | 9.53 (3.64) | 11.70 (4.49) |
| Age at treatment (years) | 1.42 (0.77) | 7.57 (5.02) |
| Time on treatment (years) | 8.11 (3.83) | 4.13 (2.74) |
| PSS score at first visit | 9.39 (2.05) | 7.89 (2.76) |
Reliability: Inter-rater reliability of the PSS scoring was good (intra-class correlation [ICC] = 0.99).
In Table 3 both groups showed a significant correlation with age such that older patients showed more disease burden in both groups (Fig. 1). Of those with multiple visits, no patient showed a decrease and several showed an increase in PSS: 6/22 (27%) for the Hurler patients and 3/11 (27%) for the attenuated patients.
Table 3.
Correlation of PSS with Hurler and attenuated groups
| Variable | Group | Correlation (95% CI) | P-value |
|---|---|---|---|
| Age | Hurler | 0.39 (0.20, 0.55) | <0.001 |
| Age | Attenuated | 0.58 (0.31, 0.76) | 0.001 |
| CHQ-PhS | Hurler | −0.14 (−0.36, 0.09) | 0.235 |
| CHQ-PhS | Attenuated | −0.56 (−0.76, −0.25) | 0.001 |
| Vineland DLS | Hurler | −0.11 (−0.31, 0.11) | 0.328 |
| Vineland DLS | Attenuated | −0.48 (−0.70, −0.18) | 0.003 |
Fig. 1.
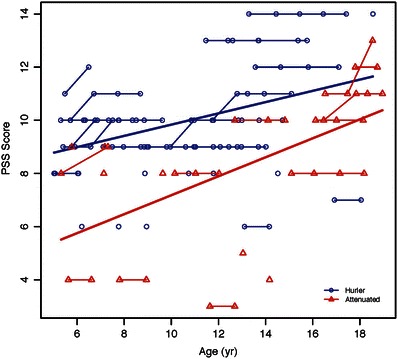
Association of PSS with age among Hurler and attenuated MPS I groups
In the attenuated group, PSS was negatively associated with the PhS from the CHQ (Fig. 2) and DLS from the Vineland (Fig. 3) such that lower scores for quality of life and daily living skills are associated with higher PSS, indicating more functional impairment. PSS was not significantly associated with PhS from CHQ and DLS from Vineland in the Hurler group.
Fig. 2.
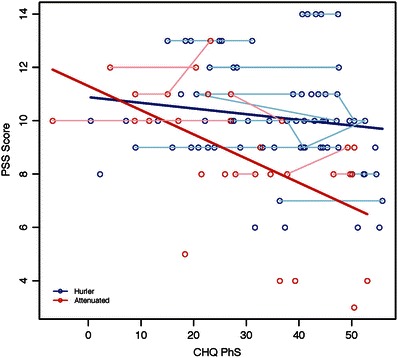
Association of PSS with CHQ-PhS among Hurler and attenuated MPS I groups
Fig. 3.
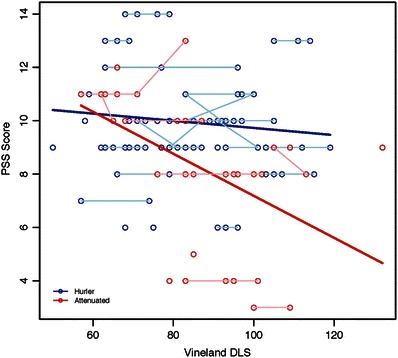
Association of PSS with Vineland DLS among Hurler and attenuated MPS I groups
Discussion
We have constructed a somatic disease severity score to reflect the degree of disease burden based on a multicenter longitudinal cohort of Hurler and attenuated (Hurler–Scheie and Scheie) forms of MPS I patients. Our goal was to design an MPS-specific physical symptom scale to quantify somatic disease burden for comparison with other outcome measures and for use in clinical trials.
Our analysis supports the scoring reliability of this measure for both MPS I groups and the validity of the PSS for attenuated MPS I patients. We have presented evidence for the association of PSS with age in both MPS I groups. In both groups, the percent increase of PSS was the same.
We know that despite ERT treatment, patients with attenuated forms of MPS I continue to undergo surgeries for orthopedic problems, cervical cord compression, and shunt placement for hydrocephalus. However, a good treatment response would be avoiding further decline. To be most useful, treatments would need to start early, before substantial progression (i.e., high PSS).
The PSS provides a shorthand measure of these accumulated problems. We found that these problems are associated with difficulties in carrying out activities of daily living and physical quality of life in the attenuated MPS I patients. While other measures such as the HAQ, a direct measure of disability, might also provide concurrent validation, the advantage of the VABS and the CHQ is that they both have normative data for children. The association of physical handicap with activities of daily living is consistent with previous reports that on a direct measure of physical performance, MPS I patients have difficulties in conducting daily functional activities (Haley et al. 2006). Correlation with such a measure in future studies might provide additional validity for the PSS.
In contrast to the attenuated group, while showing an increase with age, the PSS was not associated with decreased daily living skills or quality of life among Hurler patients. There are several possible factors. As the majority of problems occurred in these patients at the time of HCT, the elapsed time since the occurrence of these symptoms is long. Because they occurred earlier, they may not be as handicapping, the symptoms are more stable, or they may have learned to live with their disease burden. Further work is necessary to adapt the PSS for children with Hurler syndrome for it to be a useful measure of late effects.
For the attenuated MPS I group, this measure of disease burden can be both a marker of long-term treatment efficacy and tracking individual domains of somatic involvement in disease progression. In MPS II, effects of PSS were documented in a previous publication where it was found that for every PSS point, a measure of variability in attention is decreased by 12 points (p value <0.001) in mild mucopolysaccharidosis type II (Yund et al. 2015).
It should be noted that this scale is not meant to be a clinical tool. It is a summary of the patient’s history of medical problems. Thus, it does not define specific approaches to the severity or method of obtaining medical data. Because this scale is based on medical records, it relies on many physician assessments, which may not use the same criteria to judge a sign of MPS. Thus, while the scoring may be reliable, we are basing this scoring on the judgment of physicians who may differ in the criteria they use or their data collection methods. This is similar to the problem encountered in registry data. Pastores et al. note that registries suffer from a lack of standardization of assessment and data collection methods (Pastores et al. 2007). It should be noted that multiple sites enter data to a registry, but in this case, single well-trained personnel are compiling data for the PSS, eliminating one source of variation.
In conclusion, a reliable MPS I-specific Physical Symptom Score (PSS) has been developed which may be useful in future investigations of the effects of treatment and disease progression. The association of somatic disease burden with outcomes such as age, physical quality of life, and ability to carry out daily living skills supports the validity of this measure in attenuated MPS I patients. Research needs to be carried out to adapt the measure so it is more sensitive in children with Hurler syndrome. Future investigations will also focus on other correlates such as emotional and social status, genotypes, biomarkers, and MRI markers of the brain.
Acknowledgments
We are grateful to all of the families at the participating sites, as well as the principal investigators who contributed patient material and information. We are also thankful to Evelyn Redtree for her assistance with this research.
We acknowledge the contribution of data for the development of this measure from the following participating investigators and centers: Dr. Paul Harmatz at Children’s Hospital and Research Center, California, Dr. Julian Raiman at Hospital for Sick Children, Toronto, Canada and Dr. Morton Cowan at University of California, San Francisco.
The Lysosomal Disease Network (U54NS065768) supported this study through a longitudinal study called “Longitudinal Studies of Brain Structure and Function in MPS Disorders.” The Lysosomal Disease Network (U54NS065768) is a part of the National Institute of Health (NIH) Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS (National Center for Advancing Translational Sciences), funded through a collaboration between NCATS and the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This project was co-funded by Genzyme-Sanofi.
This project was supported in part by the National Center for Advancing Translational Sciences, National Institutes of Health, UMN-CTSI (UL1TR000114 Dr. Rudser). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
We are very thankful for the support provided by the Center for Neurobehavioral Development (CNBD).
Synopsis of the Article
A reliable, validated MPS-specific Physical Symptom Score (PSS) has been developed that assesses somatic disease burden and used to evaluate treatment outcomes and disease progression for research purposes in the attenuated group of MPS I.
Compliance with Ethics Guidelines
Contributions of Individual Authors
A. Ahmed wrote this article and contributed pertinent aspects of the planning, conduct, and reporting of the work described in the article, provided scientific expertise, and created the PSS.
K Rudser performed the data and statistical analysis and edited the manuscript.
A. Kunin-Batson scored and advised regarding the CHQ-PF50 and edited the manuscript.
K Delaney managed subject recruitment, scored DLS from Vineland Adaptive Behavioral Scales (VABS), Second Edition, and provided expertise about Vineland.
C. Whitley recruited and managed the LDN (Lysosomal Disease Network) study as a P. I. from which these subjects were selected and ensured their cooperation with study protocol.
E. Shapiro provided scientific expertise and co-wrote the manuscript.
Corresponding and Responsible Author
Alia Ahmed, M.D.
Conflict of Interest
Elsa G. Shapiro declares that she has received funds as a consultant from Genzyme-Sanofi, BioMarin, ArmaGen, REGENXBIO, and Shire.
Kathleen Delaney declares that she has received funds as a consultant from Shire, ArmaGen, BioMarin, and REGENXBIO.
Chester B. Whitley discloses the following financial relationships:
Consultant and research support from Shire, BioMarin, Genzyme-Sanofi, and Synageva BioPharma Corp.
Alia Ahmed declares that she has no conflict of interest.
Kyle Rudser declares that he has no conflict of interest.
Alicia Kunin-Batson declares that she has no conflict of interest.
The authors confirm independence from the funders of this research; the content of the article has not been influenced by the funders.
Ethics board approval: This study was approved by the University of Minnesota Institutional Review Board: Human Subjects Committee.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
This article does not contain any studies with animal subjects performed by any of the authors.
Footnotes
Competing interests: None declared
Contributor Information
A. Ahmed, Email: ahmed306@umn.edu
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Ahmed A, Whitley CB, Cooksley R et al (2014a) Neurocognitive and neuropsychiatric phenotypes associated with the mutation L238Q of the alpha-L-iduronidase gene in Hurler-Scheie syndrome. Mol Genet Metab 111(2):123–127 [DOI] [PMC free article] [PubMed]
- Ahmed A, Kunin-Batson A, Redtree E, Whitley C, Shapiro E. MPS (mucopolysaccharidosis) specific physical symptom score – development, reliability and validity. Mol Genet Metab. 2014;111(2):S17–S18. doi: 10.1016/j.ymgme.2013.11.014. [DOI] [Google Scholar]
- Beesley CE, Meaney CA, Greenland G, et al. Mutational analysis of 85 mucopolysaccharidosis type I families: frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum Genet. 2001;109:503–511. doi: 10.1007/s004390100606. [DOI] [PubMed] [Google Scholar]
- Bertola F, Filocamo M, Casati G, et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum Mutat. 2011;32(6):189–210. doi: 10.1002/humu.21479. [DOI] [PubMed] [Google Scholar]
- Clarke LA, Heppner J (2002) Mucopolysaccharidosis Type I, (Updated 2011 Jul 21). In: Pagon RA, Adam MP, Bird TD et al (eds) Gene Reviews™ [Internet]. University of Washington, Seattle. 1993–2013. http://www.ncbi.nlm.nih.gov/books/NBK1162/
- R Core Team (2014) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/
- D’Aco K, Underhill L, Rangachari L, et al. Diagnosis and treatment trends in mucopolysaccharidosis I: findings from the MPS I Registry. Eur J Pediatr. 2012;171:911–919. doi: 10.1007/s00431-011-1644-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle P, Heagerty P, Liang K, Zeger S. Analysis of longitudinal data. New York: Oxford University Press; 2002. [Google Scholar]
- Haley SM, Fragala-Pinkham MA, Dumas HM, Ni P, Skrinar AM, Cox GF. A physical performance measure for individuals with mucopolysaccharidosis type I. Dev Med Child Neurol. 2006;48:576–581. doi: 10.1017/S0012162206001216. [DOI] [PubMed] [Google Scholar]
- Hopwood JJ, Morris CP. The mucopolysaccharidosis: diagnosis, molecular genetics and treatment. Mol Biol Med. 1990;7(5):381–404. [PubMed] [Google Scholar]
- Kakkis ED, Muenzer J, Tiller GE, et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344:182–188. doi: 10.1056/NEJM200101183440304. [DOI] [PubMed] [Google Scholar]
- Landgraf JM, Abetz L, Ware JE. The CHQ user’s manual. 1. Boston: The Health Institute, New England Medical Center; 1996. [Google Scholar]
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- Moore D, Connock MJ, Wraith JE, Lavery C. The prevalence of and survival in mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008;3:24. doi: 10.1186/1750-1172-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzer J, Wraith JE, Clarke LA. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19–29. doi: 10.1542/peds.2008-0416. [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J. The mucopolysaccharidoses. In: Scriver C, Beaudet A, Sly W, Valle D, Childs R, Kinzler K, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- Pastores GM, Arn P, Beck M, et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol Genet Metab. 2007;91:37–47. doi: 10.1016/j.ymgme.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Peters C, Balthazor M, Shapiro E, et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood. 1996;87:4894–4902. [PubMed] [Google Scholar]
- Peters C, Shapiro E, Anderson J, et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The Storage Disease Collaborative Study Group. Blood. 1998;91:2601–2608. [PubMed] [Google Scholar]
- Scott HS, Bunge S, Gal A, Clarke LA, Morris CP, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat. 1995;6:288–302. doi: 10.1002/humu.1380060403. [DOI] [PubMed] [Google Scholar]
- Souillet G, GuVon N, Maire I, et al. Outcome of 27 patients with Hurler’s syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant. 2003;31:1105–1117. doi: 10.1038/sj.bmt.1704105. [DOI] [PubMed] [Google Scholar]
- Sparrow SS, Cicchetti DV, Balla DA (2005) Vineland Adaptive Behavior Scales. 2nd Edition. Psychological Corporation, San Antonio
- Staba SL, Escolar ML, Poe M, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350:1960–1969. doi: 10.1056/NEJMoa032613. [DOI] [PubMed] [Google Scholar]
- Terlato NJ, Cox GF. Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A comprehensive review of the literature. Genet Med. 2003;5:286–294. doi: 10.1097/01.GIM.0000078027.83236.49. [DOI] [PubMed] [Google Scholar]
- Wraith JE, Clarke LA, Beck M, et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase) J Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- Yund B, Rudser K, Ahmed A, et al. Cognitive, medical, and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol Genet Metab. 2015;114(2):170–177. doi: 10.1016/j.ymgme.2014.12.299. [DOI] [PMC free article] [PubMed] [Google Scholar]