

Abstract
Movement disorders such as ataxia are a recognized complication of classical galactosaemia, even in diet-compliant patients. Here, we report the coexistence of classical galactosaemia and Friedreich ataxia (FRDA) in nine children from seven Irish Traveller families. These two autosomal recessive disorders, the loci for which are located on either side of the centromere of chromosome 9, appear to be in linkage disequilibrium in this subgroup. Both conditions are known to occur with increased frequency amongst the Irish Traveller population.
Each member of our cohort had been diagnosed with galactosaemia in the neonatal period, and all are homozygous for the common Q188R mutation in the GALT gene. Eight of the nine patients later presented with progressive ataxia, between the ages of 5–13 years. Another child presented in cardiac failure secondary to dilated cardiomyopathy at 7 years of age. He was not ataxic at presentation and, one year from diagnosis, his neurological examination remains normal. The diagnosis of FRDA was confirmed by detecting the common pathogenic GAA expansion in both alleles of the frataxin gene (FXN) in each patient.
Neurological symptoms are easily attributed to an underlying diagnosis of galactosaemia. It is important to consider a diagnosis of Friedreich ataxia in a child from the Irish Traveller population with galactosaemia who presents with ataxia or cardiomyopathy.
Introduction
Classical galactosaemia is an autosomal recessively inherited disorder of galactose metabolism. Mutations in the GALT gene result in reduced activity of the galactose-1-phosphate uridyltransferase (GALT) enzyme leading to toxic accumulation of galactose and its metabolites (Murphy et al. 1999). Presentation is typically in the neonatal period, with feeding problems, hepatic failure and coagulopathy soon after the introduction of galactose-containing feeds (Rubio-Agusti et al. 2013). Without urgent treatment by dietary restriction of galactose, these infants are at risk of E. coli sepsis, multi-organ failure and death.
Classical galactosaemia is relatively common in the Irish population, with an overall birth incidence of 1 in 16,476 (Coss et al. 2013). This is in part due to a high incidence in the endogamous Irish Traveller population, with approximately 1 in 430 live births being affected in this group.
While dietary restriction of lactose and galactose is life saving in the neonatal period, long-term complications of galactosaemia occur even in diet-compliant patients (Coss et al. 2013). These include neurological complications such as ataxia and tremor. Although movement disorders are well described in galactosaemia, with reported rates varying from 6 to 45% (Coss et al. 2013; Ridel et al. 2005; Rubio-Agusti et al. 2013; Waggoner et al. 1990), their pathogenesis is, as yet, poorly understood.
Friedreich ataxia (FRDA) is a neurodegenerative disorder, which also demonstrates autosomal recessive inheritance. It is the commonest inherited ataxia, with a prevalence of up to 1 in 30,000 in parts of Western Europe, including Ireland (Collins 2013; Schulz et al. 2009). The prevalence in the Irish Traveller population has not been ascertained, although anecdotal evidence suggests it is common in this population (D. Barton and S.A. Lynch, personal communication). FRDA occurs due to mutations in the frataxin (FXN) gene, resulting in a marked reduction in production of this mitochondrial protein. Over 95% of cases are due to homozygous GAA triplet repeat expansions in the first intron of the FXN gene (Schulz et al. 2009). This is the mutational mechanism found amongst Irish Travellers.
FRDA usually presents in the first two decades of life with gait instability, ataxia, dysarthria and impaired sensation. Non-neurological manifestations include scoliosis, hypertrophic cardiomyopathy and diabetes mellitus. The disease is progressive, with an average of 10 years passing between onset of ataxia and the patient becoming wheelchair dependent (Collins 2013; Delatycki 2012). Current treatment of FRDA is supportive only, although novel agents are under investigation.
Here, we report the coexistence of genetically proven FRDA and classical galactosaemia in a cohort of paediatric patients from the Irish Traveller population.
Methods
Two centres were involved in this study: the National Centre for Inherited Metabolic Disorders at Children’s University Hospital, Temple Street in Dublin, Ireland, and the Department of Metabolic Paediatrics at the Royal Hospital for Sick Children in Belfast, Northern Ireland. Between these two centres, care is provided for all paediatric galactosaemia patients on the island of Ireland.
A review of the 114 Irish and Northern Irish paediatric galactosaemia patients identified nine children who have also been diagnosed with FRDA. Following approval from the local Research and Ethics Committee, a detailed, retrospective chart review was performed for each of these nine cases. This included collecting basic demographic data, patient history and physical examination findings, as well as accessing imaging, test results and educational psychology reports.
An extensive literature review failed to reveal any previously reported incidences of coexistent galactosaemia and FRDA in this, or any other, population.
Results
Nine of a total 114 Irish and Northern Irish paediatric galactosaemia patients have been diagnosed with FRDA. These nine patients (four male, five female) include two sets of siblings. While the seven families are not known to be related, all are members of the Irish Traveller population. There is known parental consanguinity in seven of the nine cases.
All were diagnosed with galactosaemia in the newborn period (range: day of life 2–8) and all are homozygous for the common Q188R mutation (GALT).
The onset of ataxia ranged from 5 to 13 years of age, with the diagnosis of FRDA being made between 6 and 15 years (see Table 1). This diagnosis was confirmed by detecting the common pathogenic GAA expansion (>66 repeats) in both alleles of the FXN gene in each patient. The index case presented with gradually progressive ataxia and tremor from the age of five. Following a normal MRI brain, he was seen by a Consultant Paediatric Neurologist, (BL) who suspected FRDA based on clinical history and detailed neurological examination. This diagnosis was confirmed by frataxin gene analysis, by which time our patient was 10 years of age.
Table 1.
Diagnosis of FRDA
| Case no. | Current age (years) | Age at symptom onset (years) | Age at diagnosis (years) | Duration pre-diagnosis (years) | Karyotype | MRI brain |
|---|---|---|---|---|---|---|
| 1a | 16 | 5 | 10 | 5 | N | Y |
| 2 | 17 | 10 | 11.5 | 1.5 | N | Y |
| 3 | 17.5 | 13 | 15 | 2 | Y | N |
| 4 | 16 | 13 | 14 | 1 | N | Y |
| 5 | 11 | 5 | 9 | 4 | N | N |
| 6 | 8.5 | 6 | 7.5 | 1.5 | N | N |
| 7 | 8 | 5 | 6 | 1 | N | N |
| 8 | 8 | 7.25 | 7.5 | 0.25 | N | N |
| 9 | 8.5 | 7.5 | 8.5 | 1 | N | Y |
| Median (range) | 11 (8–17.5) | 7.25 (5–13) | 9 (6–15) | 1.5 (0.25–5) | [1/9] | [4/9] |
aIndex case
Y test conducted, N test not conducted
Subsequent to Case 1’s diagnosis, his older sister was reviewed in clinic and found to have similar, albeit milder, ataxia, tremor and areflexia. Her MRI brain was also normal, and diagnosis of FRDA was confirmed on FXN gene analysis shortly after. Since that time, seven further patients have been diagnosed in Dublin and Belfast. The majority of these children had significantly shorter lag periods between symptom onset and diagnosis than our initial case, owing to a raised index of suspicion for the disease in this population. Two of these patients had brain imaging prior to diagnosis, which were again normal.
Case 8 had failed to attend his galactosaemia outpatient clinics for some years prior to presenting to another centre, aged 7 years, with decompensated cardiac failure secondary to dilated cardiomyopathy. A full cardiomyopathy screen was performed, which yielded the same frataxin mutation seen in our other cases. One year on from diagnosis, his neurological exam remains normal.
The characteristics of FRDA seen in each of our cases are outlined in Table 2. At present, seven of our patients remain independently mobile; Case 1 is fully wheelchair dependent and Case 2 requires a manual wheelchair when mobilizing outside the home. Five patients have a mild degree of left ventricular hypertrophy on echocardiogram, while three of our most recently diagnosed patients are still awaiting cardiology review. To date, no patients have developed diabetes mellitus.
Table 2.
Features of FRDA
| Case no. | Ataxia | Areflexia | Tremor | Dysarthria | Pes cavus | Scoliosis | Cardiomyopathy | Hearing loss |
|---|---|---|---|---|---|---|---|---|
| 1a | +++ | + | + | + | + | +++ | + | − |
| 2 | ++ | + | + | + | + | + | + | − |
| 3 | + | + | + | + | + | + | + | + |
| 4 | + | + | + | − | − | + | + | − |
| 5 | + | + | + | + | + | + | U | − |
| 6 | + | + | + | − | + | − | U | − |
| 7 | + | + | + | + | + | + | + | − |
| 8 | − | − | − | − | − | − | +++ | − |
| 9 | + | + | − | − | U | − | U | − |
| Total (n = 9) | 8 | 8 | 7 | 5 | 6 | 6 | 6 | 1 |
aIndex case
+ feature present, +++ feature present and severe, − feature not present, U unknown as yet
Discussion
The GALT and FXN genes are located on either side of the centromere of chromosome 9, at positions 9p13.3 and 9q21.11, respectively. Karyotyping was performed in one patient in order to rule out a common pericentric inversion of chromosome 9, which may have accounted for the co-segregation of these disorders. This, however, was normal. In the absence of such a structural chromosomal abnormality, the co-segregation of the two disorders in these families suggests that crossover during meiosis is occurring telomeric to both genes and that the two genes are in linkage disequilibrium (see Fig. 1).
Fig. 1.
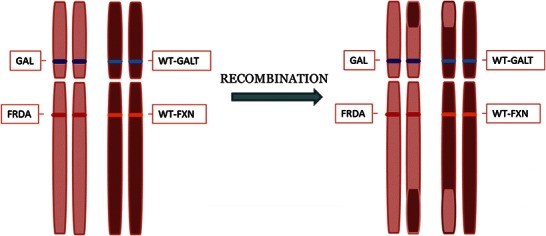
Crossover of genetic material between homologous chromosomes during meiosis I. WT-GALT wild-type galactose-1-phosphate uridyltransferase enzyme gene (normal function), WT-FXN wild type frataxin gene (normal function), GAL classical galactosaemia-causing mutation in GALT, FRDA Friedreich ataxia-causing mutation in FXN
The coexistence of these conditions occurs only in a subset of the Traveller population, with other families known to be affected by either classical galactosaemia or FRDA alone. However, genetic recombination in our cohort of families may have resulted in some patients being homozygous for only one of the two diseases. To date, three siblings (from three different families) of our nine index cases have been diagnosed with galactosaemia alone. At four and seven years old, two of these children could be in the pre-symptomatic phase of FRDA; however, the third sibling is in his mid-teens and as yet is showing no evidence of ataxia, dysarthria or tremor. Pre-symptomatic testing for FRDA has not been performed in these three patients. In two cases (Republic of Ireland), testing has not been offered, as pre-symptomatic testing of a minor for a condition which has no treatment is considered unethical. Another case (Northern Ireland) has been offered pre-symptomatic testing; however, this has been declined by the parents. No family members are known to be affected with FRDA alone.
The diagnosis of galactosaemia can have a significant impact on the quality of life of both affected children and their carers (Bosch et al. 2009; Lambert and Boneh 2004). No literature describes the impact of a second chronic, life-limiting diagnosis in such children. The Traveller population in Ireland is known to have poorer quality engagement with health services (McGorrian et al. 2012). One patient in our cohort had been lost to galactosaemia follow-up for 3 years prior to presenting with severe decompensated cardiac failure, secondary to dilated cardiomyopathy. It was at this point that a diagnosis of FRDA was suspected and confirmed. He has no neurological symptoms of FRDA to date. This particular case highlights the possible need to reconsider the need for pre-symptomatic testing for FRDA in siblings of patients affected by both conditions, in particular those siblings with a diagnosis of galactosaemia.
Conclusion
Movement disorders are well described in classical galactosaemia, irrespective of dietary compliance (Rubio-Agusti et al. 2013; Coss et al. 2013). No previous reports of the coexistence of FRDA and galactosaemia exist. As a result, neurological symptoms may be easily attributed to an underlying diagnosis of galactosaemia, and a coexistent diagnosis could be missed. Clinicians should therefore have a high index of suspicion for a diagnosis of Friedreich ataxia in Irish Traveller patients with galactosaemia, who present with unusual symptoms.
Acknowledgments
Thank you to Prof. David Barton at the National Centre for Medical Genetics for helpful discussion regarding genetic mechanisms and for insight into the prevalence of FRDA in the Traveller population.
Take-Home Message
Clinicians should have a high index of suspicion for Friedreich ataxia in Irish Traveller patients with classical galactosaemia who present with ataxia or cardiomyopathy.
Compliance with Ethics Guidelines
Conflicts of Interest
Siobhán Neville declares that she has no conflict of interest.
Siobhan O’Sullivan declares that she has no conflict of interest.
Bronagh Sweeney declares that she has no conflict of interest.
Bryan Lynch declares that he has no conflict of interest.
Donncha Hanrahan declares that he has no conflict of interest.
Ina Knerr declares that she has no conflict of interest.
Sally Ann Lynch declares that she has no conflict of interest.
Ellen Crushell declares that she has no conflict of interest.
Informed Consent
This is a descriptive report, and all patient information has been anonymized to ensure patients are not identifiable. All patients have provided informed consent to genetic testing.
This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributions of Individual Authors
Siobhán O’Sullivan, Ina Knerr and Ellen Crushell identified the patients relevant to this study.
Siobhán Neville, Siobhán O’Sullivan and Bronagh Sweeney collected the patient information.
Bryan Lynch and Donncha Hanrahan performed the neurological examinations.
Sally Ann Lynch advised on the inheritance pattern of these two conditions.
Siobhán Neville and Ellen Crushell drafted the original manuscript.
Siobhán Neville and Ellen Crushell designed the included figure and tables.
Ina Knerr, Siobhán O’Sullivan and Sally Ann Lynch amended the original manuscript.
All authors have read and approved the submitted manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Siobhán Neville, Email: neville.siobhan@gmail.com.
Siobhan O’Sullivan, Email: siobhan.o'sullivan@belfasttrust.hscni.net.
Bronagh Sweeney, Email: bronagh.sweeney@belfasttrust.hscni.net.
Bryan Lynch, Email: bryan.lynch@cuh.ie.
Donncha Hanrahan, Email: donncha.hanrahan@belfasttrust.hscni.net.
Ina Knerr, Email: ina.knerr@cuh.ie.
Sally Ann Lynch, Email: sallyann.lynch@olchc.ie.
Ellen Crushell, Email: ellen.crushell@cuh.ie.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Bosch AM, Maurice-Stam H, Wijburg FA, Grootenhuis MA. Remarkable differences: the course of life of young adults with galactosaemia and PKU. J Inherit Metab Dis. 2009;32(6):706–712. doi: 10.1007/s10545-009-1253-2. [DOI] [PubMed] [Google Scholar]
- Collins A. Clinical neurogenetics: friedreich ataxia. Neurol Clin. 2013;31(4):1095–1120. doi: 10.1016/j.ncl.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Coss KP, Doran PP, Owoeye C, et al. Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis. 2013;36(1):21–27. doi: 10.1007/s10545-012-9507-9. [DOI] [PubMed] [Google Scholar]
- Delatycki MB, Corben LA. Clinical Features of Friedreich Ataxia. J Child Neurol. 2012;27(9):1133–1137. doi: 10.1177/0883073812448230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert C, Boneh A. The impact of galactosaemia on quality of life – a pilot study. J Inherit Metab Dis. 2004;27(5):601–608. doi: 10.1023/B:BOLI.0000042957.98782.e4. [DOI] [PubMed] [Google Scholar]
- McGorrian C, Frazer K, Daly L, et al. The health care experiences of Travellers compared to the general population: the All-Ireland Traveller Health Study. J Health Serv Res Policy. 2012;17(3):173–180. doi: 10.1258/jhsrp.2011.011079. [DOI] [PubMed] [Google Scholar]
- Murphy M, McHugh B, Tighe O, Mayne P, O'Neill C, et al. Genetic basis of transferase-deficient galactosaemia in Ireland and the population history of the Irish Travellers. Eur J Hum Genet. 1999;7(5):549–554. doi: 10.1038/sj.ejhg.5200327. [DOI] [PubMed] [Google Scholar]
- Ridel KR, Leslie ND, Gilbert DL. An updated review of the long-term neurological effects of galactosemia. Pediatr Neurol. 2005;33(3):153–161. doi: 10.1016/j.pediatrneurol.2005.02.015. [DOI] [PubMed] [Google Scholar]
- Rubio-Agusti I, Carecchio M, Bhatia KP, et al. Movement disorders in adult patients with classical galactosemia. MovDisord. 2013;28(6):804–810. doi: 10.1002/mds.25348. [DOI] [PubMed] [Google Scholar]
- Schulz JB, Boesch S, Bürk K, et al. Diagnosis and treatment of Friedreich ataxia: a European perspective. Nat Rev Neurol. 2009;5(4):222–340. doi: 10.1038/nrneurol.2009.26. [DOI] [PubMed] [Google Scholar]
- Waggoner DD, Buist NR, Donnell GN. Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13(6):802–818. doi: 10.1007/BF01800204. [DOI] [PubMed] [Google Scholar]