

Abstract
Transaldolase (TALDO) deficiency has various clinical manifestations including liver dysfunction, hepatosplenomegaly, anemia, thrombocytopenia, and dysmorphic features. We report a case presenting prenatally with hyperechogenic bowel and intrauterine growth restriction. The infant was born small for gestational age, with cutis laxa and hypertrichosis. Postnatally, meconium plug was identified, complicated with intestinal obstruction necessitating laparotomy, partial resection of the intestine, and ileostomy. Liver biopsy revealed cholangiolar proliferation and portal fibrosis. He also suffered from persistent congenital thrombocytopenia requiring platelet transfusions and severe hypothyroidism with normal anatomical and structural gland responding only to the combination of T3 and T4 treatment. Neurologically, severe hypotonia and anisocoria were noted at the age of 2 months. Brain MRI was normal. Shortly after the abdominal surgery, a rapid liver failure ensued, which eventually led to his death. Specific metabolic tests ruled out glycosylation disorders, yet urine analysis using 1H NMR showed accumulation of sedoheptulose which was previously described in patients with transaldolase deficiency. Sequencing of the gene-encoding transaldolase (TALDO1) revealed a homozygous stop mutation c.669C>G; p.Tyr223*. In conclusion, we present an infant with a novel homozygous mutation in TALDO1, causing TALDO deficiency, and extend the clinical characteristics of this rare syndrome.
Keywords: Cutis laxa, Echogenic bowel, Hypothyroidism, Microarray analysis, Transaldolase
Introduction
Transaldolase (TALDO, EC 2.2.1.2) is a key enzyme in the pentose phosphate pathway (PPP). It is crucial in the nonoxidative part, immediately after transketolase (EC 2.2.1.1). These two enzymes enable an important connection between the PPP and glycolysis (Perl 2007). TALDO interconverts a three-carbon moiety between different sugars; thus, it enables the production of fructose-6-phosphate and erythrose-4-phosphate from glyceraldehyde-3-phosphate and sedoheptulose-7-phosphate. In the absence of transaldolase intermediate products such as ribitol, d-arabitol and erythritol may accumulate, and their abnormal elevated concentrations can be measured directly (Wamelink et al. 2007). Although the diagnosis is rare, the description of more cases is needed for further delineation of the enzyme’s deficiency. TALDO deficiency was previously described in several patients, causing a severe disease of the liver, skin, and blood (Eyaid et al. 2013; Verhoeven et al. 2001, 2005). Fetal abnormal findings and maternal disease during pregnancy are infrequent, but were documented previously, presented by IUGR (Verhoeven et al. 2001), maternal HELLP syndrome (Verhoeven et al. 2005), oligohydramnios, fetal splenomegaly, and fetal distress (Wamelink et al. 2008a). Here, we present a case of fetal hyperechogenic bowel, which after birth developed a multisystemic disease, eventually leading to his death. He was found to harbor a novel homozygous mutation in TALDO1.
Clinical Report
A 35-year-old pregnant woman was referred to the genetic clinic at 21 weeks and 6 days of pregnancy, due to grade 2 hyperechogenic bowel detected in the fetus. The estimated hyperechogenic bowel size was 39 × 21 mm. Fetal echocardiogram was interpreted as normal. Maternal history included delivery of two normal children born at term and five miscarriages. The mother was heterozygous for Factor V Leiden and was treated with enoxaparin (Clexane) up to 35 weeks’ gestation. Parental karyotyping was normal. The parents are first-degree cousins, of Arabic Muslim origin. Follow-up during pregnancy revealed fetal dilated bowel loops and intrauterine growth retardation. Genetic evaluation of both parents for 24 common known mutations of cystic fibrosis was negative as was the infectious workup – for cytomegalovirus (CMV) and toxoplasmosis. First-trimester screening was normal, without added risk for any aneuploidy.
The couple refused amniocentesis, and further fetal ultrasonographic scan demonstrated bowel echogenicity without dilated bowel loops. A male infant was born at 37 weeks of gestation by vaginal delivery. Birth weight was 2,050 g, the length was 40 cm, and head circumference was 31 cm. He had cutis laxa and hypertrichosis. At 2 days of age, meconium plug was diagnosed, necessitating laparotomy, resection of 15 cm of occluded small intestine, and ileostomy. Surgical closure of the ileostomy at 2 months of age was complicated by Pseudomonas aeruginosa peritonitis. During this operation, a liver biopsy was performed due to the impression of an abnormal liver color. Histopathology demonstrated cholangiolar proliferation and portal fibrosis. Following this operation, the infant deteriorated clinically with rapid liver failure which eventually led to his death. The liver malfunction presented mainly with increased direct bilirubin levels (up to 35 mg/dl), increased ammonia levels, low clotting factor levels (Factor V-12%, Factor VII-8%), however, without considerable elevation in hepatocellular or cholestatic liver enzymes. Congenital thrombocytopenia was observed requiring perioperative platelet transfusions, yet neither anemia nor leucopenia was found. Blood smear showed a low number of small platelets. The thyroid axis hormones were examined routinely and revealed severe hypothyroidism with minimal change under levothyroxine sodium (T4) treatment. Euthyroidism was achieved only by using a combination of triiodothyronine (T3) and T4 treatment. A thyroid scan demonstrated a normal anatomical and structural gland, raising the possibility of enzymatic or receptor-related endocrine disorder. Neurologically, the infant had severe hypotonia since birth. At 3 days of age, after recovering from the first abdominal surgery, he had a comatose event lasting 3 days, from which he recovered spontaneously. Infectious, metabolic, and toxic evaluations were normal. Brain magnetic resonance imaging (MRI) was normal. At the age of 2 months, he developed acute anisocoria. Brain computerized tomography (CT) demonstrated dilated brain ventricles with no focal finding, edema, or midline deviation and normal structures of gray and white matter. Assays of transferrin electrophoresis for glycosylation disorders were normal.
Urine test for metabolites of the pentose cycle using proton nuclear magnetic resonance (1H-NMR) demonstrated a high level of polyols, including sedoheptulose, which characterizes transaldolase deficiency. Erythronic acid level was below detection (normal), as shown in Fig. 1b. The concentration of sedoheptulose found here was 1,350-μmol/mmol creatinine, similar to a patient described by Engelke et al. (2010) which showed a sedoheptulose level of 1,700-μmol/mmol creatinine at the same age (1 month). The normal range for sedoheptulose at ages 0–3 months was previously reported to be lower than 40-μmol/mmol creatinine (Engelke et al. 2010). The concentration of polyols cannot be provided quantitatively since the specific molecules underlying this heavily overlapping signal region are not known. Nevertheless, the integrated intensity ratio of the polyol region (3.60–4.15 ppm) to that of creatinine (signal at 3.13 ppm) was 6–13-fold higher in the spectrum obtained from the patient’s urine compared to spectra obtained from four healthy children aged 3 to 7 years. The limit of detection of erythronic acid under the conditions used here is 60-μmol/mmol creatinine or 130 μM absolute concentration. Therefore, the level of erythronic acid in our case, if any, is lower than this limit. This detection limit is based on (1) the identification and quantification of erythronic acid using the doublet signal of the proton in position 2, at 4.326 ppm (Engelke et al. 2010), and (2) the specific concentration of creatinine in the current sample which was independently measured. Array comparative genomic hybridization (CGH) analysis showed male karyotype with no copy-number changes. Based on the known parental consanguinity, an identity by descent approach utilized CGH for assessment of runs of homozygosity (ROH), a combined homozygous region of 234 Mbp was identified. By focusing on metabolic recessive genetic causes for cutis laxa (Valayannopoulos et al. 2006; Mohamed et al. 2011), within these areas of homozygosity, we could exclude known congenital disorders of glycosylation, Menkes disease, and other previously published forms of autosomal recessive cutis laxa syndromes, and we were left with transaldolase deficiency encoded by TALDO1, localized at chromosomal region 11p15.5, amidst a homozygous region of 7.4 Mbp, out of a total of 234-Mbp homozygous regions.
Fig. 1.
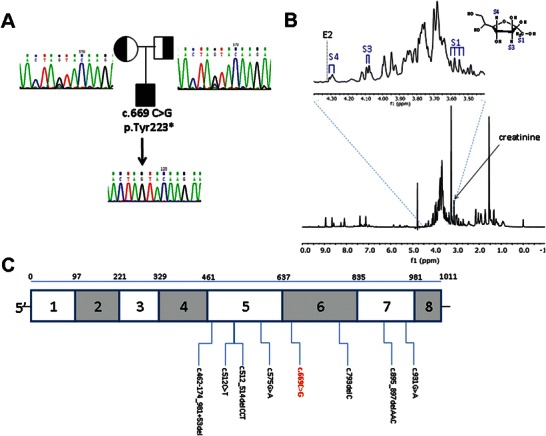
TALDO deficiency in the patient. (a) TALDO1 homozygous pathogenic mutation c.669C>G; p.Tyr223* in the patient and his parents. Sanger sequencing of the patient and his parents: both parents are heterozygous for the mutation and a wild-type allele. TALDO1 mutation is not found in controls of the same background (not included). (b) A 1H-NMR spectrum of the patient urine sample showing abnormally high levels of sugars at about 3.3–4.5 ppm. Sedoheptulose (a ketoheptose) was identified using the S1, S3, and S4 signals of sedoheptulose which were previously characterized in a urine sample from a patient with TALDO deficiency (Engelke et al. 2010). Erythronic acid was not detected in the patient’s urine, as depicted by the lack of the characteristic doublet signal at 4.325 (position marked E2) (Engelke et al 2010). (c) TALDO1 gene cDNA schematic structure. Mutations are presented on the diagram. The following are the mutations previously described: c.512_514delCCT, p.Ser171del; c.512C>T, p.Ser171Phe; c.574C>T, p.Arg192Cys; c.575G>A, p.Arg192His; c.793delC, p.Gln265ArgfsX56; c.895_897delAAC, p.Asn299del; c.931G>A, p.Gly311Arg; c.462-174_981+53del. The mutation in the patient described in this article is colored in red
Sequencing TALDO1 identified a homozygous nonsense point mutations c.669C>G; p.Tyr223* [NM_006755.1] (Fig. 1a). The presence of the above mutation is predicted to target the TALDO1 mRNA for nonsense-mediated decay (NMD).
Materials and Methods
Array CGH
Ten milliliter of peripheral blood was centrifuged at 2,500 g for 10 min and 200-μl buffy coat was taken for isolation of genomic DNA using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA (0.1 μg) was labeled using the Affymetrix Cytogenetics Reagent Kit, and labeled DNA was applied to an Affymetrix Cytogenetics Array (2.7 million probes, Affymetrix Inc., Santa Clara, CA) according to the manufacturer’s instructions. The array was scanned, and the data were analyzed using the Affymetrix Chromosome Analysis Suite.
Urine Sample Preparation
The urine samples were centrifuged before analysis. A volume of 70 μl of a 20-mmol/l trimethylsilyl-2,2,3,3-tetradeuteriumpropionic acid (TSP, sodium salt; Sigma-Aldrich) D2O solution was added to 700 μl of urine as a chemical shift reference (δ = 0.00) and as a lock signal. The pH of the urine was adjusted to 2.51 ± 0.05 with HCl. The sample of 770 μl was then transferred to a 5-mm NMR tube.
1H-NMR Spectroscopy
An 11.8 T spectrometer (Varian, Palo Alto, CA) equipped with a TRX probe was used for direct 1H detection. Water pre-saturation was followed by 45° pulses with a repetition time of 10 s. One hundred and twenty-eight transients were recorded at 25°C. The spectra were collected with 16 K data points and zero filled to 32 K. A line broadening of 0.2 Hz was applied, and phase and baseline were corrected manually.
The chemical shift was referenced to TSP at 0 ppm. Metabolite signal areas were compared with the area of creatinine singlet (3.13 ppm; the N–CH3 protons) to determine metabolite concentrations expressed as μmol/mmol creatinine. Spectral analysis was carried out using MNova (Mestrelab Research, Santiago de Compostela, Spain).
TALDO1 Sequencing
The seven coding exons of the TALDO1 gene were PCR amplified using blood-extracted DNA (primer sequences are available upon request). The PCR products were purified and sequenced on a 3130 Genetic Analyzer using BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA).
Discussion
Patients with transaldolase (TALDO) deficiency have a variable phenotype yet with common features. Table 1 depicts the various clinical manifestations previously reported in TALDO deficiency.
Table 1.
Clinical features of previously described patients with TALDO deficiency and comparison to the present patient
| Feature | Reported cases (percent of cases) | Present patient |
|---|---|---|
| Consanguinity | 25/31 (80.6) | + |
| Dysmorphism | 18/31 (58) | + |
| Liver dysfunction | 26/31 (83.8) | + |
| Hepatosplenomegaly | 30/31 (96) | +/− (only splenomegaly) |
| Anemia | 27/31 (87) | − |
| Thrombocytopenia | 27/31 (87) | + |
| Cardiac | 23/31 (74) | + |
| Neonatal edema | 8/31 (25.8) | − |
| Renal | 11/31 (35.5) | + |
| Respiratory | 5/31 (16) | − |
| Developmental delay | 2/31 (6) | NA |
| Additional features found | Hypertrichosis Hypotonia |
Hypothyroidism Hyperechogenic bowel Hypertrichosis Neonatal hypotonia |
It is evident that most reported patients are offsprings of consanguineous marriages as expected in such a rare disorder. Most patients have liver dysfunction, hepatosplenomegaly, and hepatic fibrosis (83.8%, Table 1). TALDO deficiency is associated with dysmorphic features including cutis laxa, anti-mongoloid slant and low-set ears, dolichocephaly, exophthalmia, and broad nasal bridge. Also reported are hirsutism/hypertrichosis, heart defects, renal problems, and intermittent hypoglycemia. Mental and motor development is usually normal in surviving patients. In rare cases, clitoromegaly, micropenis, sensorineural and conductive deafness, and rickets were noted (Eyaid et al. 2013; Wamelink et al. 2008a). Brain MRI and magnetic resonance spectroscopy (MRS) did not reveal abnormal findings. TALDO protein and function have been reviewed thoroughly by Wamelink et al. (2008b). As known, TALDO is a key enzyme in the pentose phosphate pathway, responsible of exchanging a three-carbon moiety between different carbohydrates, enabling either the production of more nicotinamide adenine dinucleotide phosphate (NADPH) and ribose-5-phosphate, or by being inhibited, enabling production of more nucleic acids. Therefore, TALDO function is essential for cell survival (Wamelink et al. 2008b; Perl 2007; Perl et al. 2011).
The novel TALDO1 mutation found in the present described family is a homozygous STOP mutation. This mutation creates an mRNA predicted to be degraded through nonsense-mediated mRNA decay (NMD) process. This case displays perinatal findings not previously reported in TALDO deficiency, namely, fetal echogenic bowel, meconium ileus, neonatal involvement of CNS, and severe hypothyroidism. Generally, hyperechogenic bowel is encountered in 1% of all pregnancies. Associated conditions are aneuploidy, infections (CMV, toxoplasmosis), and cystic fibrosis (CF) (Al-Kouatly et al. 2001). There are different conditions associated with failure to pass meconium – including Hirschsprung’s disease, meconium plug syndrome, meconium ileus, and anorectal malformations (Loening-Baucke and Kimura 1999). Meconium ileus is known to be associated with cystic fibrosis (van der Doef et al. 2011). In the case presented, fetal hyperechogenic bowel, along with meconium ileus, was associated with TALDO deficiency. Shortly after birth, the bowel obstruction caused severe deterioration. We can now postulate that the liver malfunction may explain the meconium plug – as malsecretion of bile acids could lead to the creation of sticky and viscous stool (Harries 1978). In addition, the postnatal comatose event lasting 3 days postoperatively may be related to the impaired liver function and its failure to effectively eliminate anesthetic drugs. As described, no other cause for this event was found. The severe hypothyroidism described in this case is unusual. Triiodothyronine in treating infantile hypothyroidism is rarely needed (Wartofsky 2013) and can probably be explained by disrupted pentose phosphate pathway, known to be important for thyroid function (Wartofsky 2013). It is possible that this neonate harbors a variation in a modifier gene, which may cause a severe form of clinical presentation. Also, this could be the result of a second mutated gene. These possibilities can be explored in whole-genome sequencing or whole-exome sequencing, which were not part of this report.
In the paper by Engelke et al. (2010), six patients with TALDO deficiency were described, all reported with high levels of erythronic acid (EA) compared with age-matched controls, including one patient at the age of 1 month. The mechanism responsible for the elevation of EA is not clear. EA is a product of the pentose phosphate pathway, produced from d-erythrose-4-phosphate, which, in turn, is a product of active transaldolase. However, more research is needed to evaluate the role of EA as a biomarker in TALDO deficiency.
Figure 1c outlines the distribution qof the published TALDO1 mutations showing that as of now all mutations are clustered in exons 5 through 7. There is no clear genotype-phenotype correlation. The severe presentation of TALDO deficiency may of course be ascribed to variations in additional genes serving as phenotype modifiers. There is also the possibility of coexistence of a second disease. This can be further explored using whole-exome or whole-genome sequencing which was not performed in this case. It is also possible that environmental factors, such as anesthetic drugs used in the perinatal operation, had a role in the severe deterioration of the disease described.
In conclusion, the severe disease caused by this specific nonsense TALDO1 mutation further delineates the variable presentation of this important-to-recognize disease.
Synopsis
This article presents a new case of transaldolase deficiency with symptoms previously undescribed, diagnosed with nmr spectrometry and cgh array technology.
Compliance with Ethical Guidelines
Conflict of Interest
Ehud Banne, Vardiella Meiner, Avraham Shaag, Rachel Katz-Brull, Ayelet Gamliel, Stanley Korman, Horowitz Cederboim, Morasha Plesser Duvdevani, Ayala Frumkin, Amir Zilkha, Vadim Kapuller, Dan Arbell, Elite Cohen, and Smadar Eventov-Friedman declare that they have no conflict of interest.
Informed Consent
All procedures described in this manuscript were performed in accordance with the ethical standards of the responsible Institutional Review Committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the patient’s family for blood and urine tests.
Footnotes
Competing interests: None declared
Contributor Information
Ehud Banne, Email: ehud.banne@gmail.com.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Al-Kouatly HB, Chasen ST, Streltzoff J, et al. The clinical significance of fetal echogenic bowel. Am J Obstet Gynecol. 2001;185:1035–1038. doi: 10.1067/mob.2001.117671. [DOI] [PubMed] [Google Scholar]
- Al-Shamsi AM, Ben-Salem S, Hertecant J, et al. Transaldolase deficiency caused by the homozygous p.R192C mutation of the TALDO1 gene in four Emirati patients with considerable phenotypic variability. Eur J Pediatr. 2015;174:661–668. doi: 10.1007/s00431-014-2449-5. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam S, Wamelink MM, Ngu LH, et al. Novel heterozygous mutations in TALDO1 gene causing transaldolase deficiency and early infantile liver failure. J Pediatr Gastroenterol Nutr. 2011;52:113–116. doi: 10.1097/MPG.0b013e3181f50388. [DOI] [PubMed] [Google Scholar]
- Engelke UF, Zijlstra FS, Mochel F, et al. Mitochondrial involvement and erythronic acid as a novel biomarker in transaldolase deficiency. Biochim Biophys Acta. 2010;1802:1028–1035. doi: 10.1016/j.bbadis.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyaid W, Al Harbi T, Anazi S, et al. Transaldolase deficiency: report of 12 new cases and further delineation of the phenotype. J Inherit Metab Dis. 2013;46:997–1004. doi: 10.1007/s10545-012-9577-8. [DOI] [PubMed] [Google Scholar]
- Harries JT. Meconium in health and disease. Br Med Bull. 1978;34:75–78. doi: 10.1093/oxfordjournals.bmb.a071462. [DOI] [PubMed] [Google Scholar]
- Leduc C, Crouch EE, Wilson A, et al. Novel association of early onset hepatocellular carcinoma with transaldolase deficiency. JIMD Rep. 2014;12:121–127. doi: 10.1007/8904_2013_254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loening-Baucke V, Kimura K. Failure to pass meconium: diagnosing neonatal intestinal obstruction. Am Fam Physician. 1999;60:2043–2050. [PubMed] [Google Scholar]
- Mohamed M, Kouwenberg D, Gardeitchik T, et al. Metabolic cutis laxa syndromes. J Inherit Metab Dis. 2011;34:907–916. doi: 10.1007/s10545-011-9305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A. The pathogenesis of transaldolase deficiency. IUBMB Life. 2007;59:365–373. doi: 10.1080/15216540701387188. [DOI] [PubMed] [Google Scholar]
- Perl A, Hanczko R, Telarico T, et al. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med. 2011;17:395–403. doi: 10.1016/j.molmed.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylki-Szymanska A, Stradomska TJ, Wamelink MM, et al. Transaldolase deficiency in two new patients with a relative mild phenotype. Mol Genet Metab. 2009;97:15–17. doi: 10.1016/j.ymgme.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Tylki-Szymanska A, Wamelink MM, Stradomska TJ, et al. Clinical and molecular characteristics of two transaldolase-deficient patients. Eur J Pediatr. 2014;173:1679–1682. doi: 10.1007/s00431-014-2261-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valayannopoulos V, Verhoeven NM, Mention K, et al. Transaldolase deficiency: a new cause of hydrops fetalis and neonatal multi-organ disease. J Pediatr. 2006;149:713–717. doi: 10.1016/j.jpeds.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Van der Doef HP, Kokke FT, van der Ent CK, et al. Intestinal obstruction syndromes in cystic fibrosis: meconium ileus, distal intestinal obstruction syndrome, and constipation. Curr Gastroenterol Rep. 2011;13:265–270. doi: 10.1007/s11894-011-0185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven NM, Huck JH, Roos B, et al. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001;68:1086–1092. doi: 10.1086/320108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven NM, Wallot M, Huck JH, et al. A newborn with severe liver failure, cardiomyopathy and transaldolase deficiency. J Inherit Metab Dis. 2005;28:169–179. doi: 10.1007/s10545-005-5261-6. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Smith DE, Jansen EE, et al. Detection of transaldolase deficieny by quantification of novel seven-carbon chain carbohydrate biomarkers in urine. J Inherit Metab Dis. 2007;30:735–742. doi: 10.1007/s10545-007-0590-2. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Salomons GS, et al. Transaldolase deficiency in a two-year-old boy with cirrhosis. Mol Genet Metab. 2008;94:255–258. doi: 10.1016/j.ymgme.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Jacobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. J Inherit Metab Dis. 2008;31:703–717. doi: 10.1007/s10545-008-1015-6. [DOI] [PubMed] [Google Scholar]
- Wartofsky L. Combination L-T3 and L-T4 therapy for hypothyroidism. Curr Opin Endocrinol Diabetes Obes. 2013;20:460–466. doi: 10.1097/01.med.0000432611.03732.49. [DOI] [PubMed] [Google Scholar]