

Abstract
Classical neonatal-onset glutaric aciduria type 2 (MAD deficiency) is a severe disorder of mitochondrial fatty acid oxidation associated with poor survival. Secondary dysfunction of acyl-CoA dehydrogenases may result from deficiency for riboflavin transporters, leading to severe disorders that, nevertheless, are treatable by riboflavin supplementation. In the last 10 years, we identified nine newborns with biochemical features consistent with MAD deficiency, only four of whom survived past the neonatal period. A likely iatrogenic cause of riboflavin deficiency was found in two premature newborns having parenteral nutrition, one of whom recovered upon multivitamin supplementation, whereas the other died before diagnosis. Four other patients had demonstrated mutations involving ETF or ETF-DH flavoproteins, whereas the remaining three patients presumably had secondary deficiencies of unknown mechanism. Interestingly, six newborns among the seven tested for plasma amino acids had pronounced hyperprolinemia. In one case, because the initial diagnostic workup did not include organic acids and acylcarnitine profiling, clinical presentation and hyperprolinemia suggested the diagnosis. Analysis of our full cohort of >50,000 samples from >30,000 patients suggests that the proline/alanine ratio may be a good marker of MAD deficiency and could contribute to a more effective management of the treatable forms.
Introduction
Multiple acyl-CoA dehydrogenase deficiency (MADD), also known as glutaric aciduria type 2 (GA2, OMIM 231680), is a rare autosomal recessive metabolic disease, caused by defects in the mitochondrial electron transfer flavoprotein (ETF) or, in its electron acceptor counterpart, the electron transfer flavoprotein dehydrogenase (ETF-DH or ETF-QO) (Frerman and Goodman 1985). Neonatal-onset forms are characterized by nonketotic hypoglycemia, metabolic acidosis, hepatomegaly, and hypotonia, usually with fatal outcome during the neonatal period or the first months of life (Frerman and Goodman 2001). A few of these patients have congenital anomalies, such as renal cystic dysplasia, facial dysmorphism, rocker bottom feet, and abnormalities of external genitalia (Frerman and Goodman 2001), while the others often develop severe cardiomyopathy during the neonatal period. Cases of secondary MADD related to riboflavin deficiency have been described in neonates of mothers carrying heterozygous mutations in a riboflavin transporter (deficiency for GPR172B/SLC52A1) and showed a good response to transient riboflavin supplementation (Chiong et al. 2007; Harpey et al. 1983; Ho et al. 2011). A different entity with later presentation, Brown–Vialetto–Van Laere syndrome, is caused by mutations in two related riboflavin transporters SLC52A2 and SLC52A3 (Bosch et al. 2011; Green et al. 2010; Johnson et al. 2012).
Important biochemical anomalies guide the diagnosis of primary or secondary MADD. Plasma acylcarnitine profiles show an increase of all chain-length acylcarnitines, while urine organic acid profiles display high concentrations of glutaric, 2-hydroxyglutaric, ethylmalonic and other dicarboxylic acids, and several glycine derivatives (Frerman and Goodman 2001). Early studies reported that a marked increase of plasma proline is common in neonatal-onset patients (Frerman and Goodman 2001; Goodman et al. 1983; Przyrembel et al. 1976; Sweetman et al. 1980), but plasma amino acid analysis does not usually play a part in the diagnostic workup.
In this report, we have identified nine newborns with biochemical features consistent with primary or secondary MAD deficiency in our local cohort and investigated the potential clinical interest of amino acid analyses in this context by comparison to a large cohort of patients.
Subjects and Methods
Between 2003 and 2014, we have identified four neonates with primary MAD deficiency and five others with suspected secondary MAD deficiency. Of these, 1/4 and 3/5 patients, respectively, survived past the neonatal period. Table 1 summarizes the relevant clinical and biochemical features and further details are presented in this section.
Table 1.
Summary of clinical and biochemical findings in patients with MADD or MADD-like presentation
| Patient n/sex | Age of onset | Survival | Clinical picture | Glutaric acid (mmol/mol of creatinine) in urine | Other anomaliesa | Proline/alanine (μmol/L) in plasma | Diagnosis |
|---|---|---|---|---|---|---|---|
| 1/F | 34 h | Deceased at 8 days | Hypoglycemia, feeding difficulties, hypotonia, mild hyperammonemia, hyperlactatemia, convulsion, hepatomegaly | 7,080 | Urine: EMA, IBG, BG, HG, 2-hydroxyglutaric acid, adipic acid, suberic acid | 369/130 | Primary MADD (ETF-DH deficiency) |
| 2/M | 1 h | Deceased at 4 days | Hypoglycemia, metabolic acidosis, mild hyperammonemia, lethargic coma, hypotonia, hepatomegaly, subependymal hemorrhage, PAH | 6,507 | Urine: EMA, 2-hydroxyglutaric acid, adipic acid, suberic acid, sebacic acid, HG, IBG, BG, IVG | 622/126 | Primary MADD (ETF-DH deficiency) |
| 3/F | Antenatal diagnosis | Deceased at 38 days | Macrocephaly, dysmorphia, hepatomegaly, metabolic acidosis, hyperlactatemia, mild hyperammonemia, hypotonia | 5,280 | Urine: EMA, adipic acid, 2-hydroxyglutaric acid, IBG, IVG | NR/NR | Primary MADD (ETF-DH deficiency) |
| 4/F | 1 day | Alive | Feeding difficulties, hypotonia, hypothermia, hepatomegaly, hypoglycemia, hyperammonemia, hyperlactatemia, renal insufficiency | 1,669 | Urine: EMA, adipic acid, suberic acid, sebacic acid, HG, IBG, BG, IVG Plasma acylcarnitines: C4–C16:1 |
272/905 | Primary MADD (ETFA deficiency) |
| 5/F | 12 h | Alive | Hypoglycemia, hyperlactatemia, pulmonary distress, metabolic acidosis, subependymal hemorrhage, | 175 | Urine: EMA, 2-hydroxyglutaric, IBG, IVG, HG, adipic acid, suberic acid, sebacic acid | 1,152/1,134 | Suspected secondary MADD |
| 6/M | 2 days | Alive after the neonatal period lost to follow-up | Hypoglycemia, hypotonia, feeding difficulties, hyperammonemia | 2,272 | Urine: adipic acid, suberic acid, sebacic acid, HG, SG, PG, BG, IVG | NR/NR | Suspected secondary MADD |
| 7/F | 1 day | Deceased at 3 days | Cardiorespiratory arrest, metabolic acidosis with hyperlactatemia, hepatomegaly, dilated left ventricle, PAH, liver dysfunction, pulmonary hemorrhage, multiorgan failure | 2.87b | Plasma: 2-hydroxyglutaric acid, EMA, adipic acid, suberic acid | 1,457/429 | Suspected secondary MADD |
| Plasma acylcarnitines: C16-OH, C14-DC, C18-DC, glutarylcarnitine | |||||||
| 8/M | 35 days | Alive | Metabolic acidosis, mild hyperlactatemia, pancytopenia, lethargy, hypotonia, myoclonia, hepatomegaly | 535 | Urine: EMA, 2-hydroxyglutaric, adipic acid, suberic acid, HG, SG, BG, IVG | 1,181/247 | Iatrogenic riboflavin deficiency |
| Plasma acylcarnitines: C3-C14 | |||||||
| 9/M | 13 days | Deceased at 17 days | Abnormal movements, anemia, thrombopenia, and severe metabolic acidosis | 1,018 | Urine: EMA, 2-hydroxyglutaric, adipic acid, suberic acid, HG, SG, BG, IVG | 1,230/599 | Iatrogenic riboflavin deficiency |
| Plasma acylcarnitines: C4-C14 | |||||||
| Control | <14 | 83–281/166–514 |
EMA ethylmalonic, BG butyrylglycine, HG hexanoylglycine, IVG isovalerylglycine, SG suberylglycine, PG propionylglycine, IBG isobutyrylglycine, PAH pulmonary arterial hypertension, NR not reported
aIncrease of the reported metabolites
bPlasma value in μmol/L (normal values 0.53 μmol/L)
We observed primary neonatal MADD in four newborns (patients 1–4). Patients 1 and 2 showed typical neonatal MADD onset with hypoglycemia, metabolic acidosis with hyperlactatemia, mild hyperammonemia, hepatomegaly and hypotonia, and classical biochemical profiles associating very high glutaric levels (7,080 and 5,280 mmol/mol creatinine, respectively) with increased ethylmalonic acid, 2-hydroxyglutaric acid, dicarboxylic acids, and multiple acylglycines in urine, along with multiple acylcarnitines in plasma. Both died at a few days of life. Patient 1 showed compound heterozygosity for ETFDH mutations (c.50dup/p.His17GlnfsX6 in exon 2 and c.313A>G/p.Lys105Glu in exon 3). Patient 2 had a complete loss of ETF-DH activity and a homozygous mutation involving ETFDH (c.34+5G>C, donor splice site of intron 1). Patient 3 was the sister of patient 2 and MADD diagnosis was obtained during pregnancy. The child died at 38 days of life of respiratory failure following aspiration pneumonia. Patient 4 made good progress under treatment (Table 1), yet primary MADD was confirmed by molecular genetic testing (homozygous known pathogenic mutations in ETFA: c.797C>T, p.Thr266Met, in exon 9). She is now 9 years old. Of note, levels of glutaric acid (1,669 vs 5,280–7,980 mmol/mol creatinine) and other markers in urine were lower for patient 4 than patients 1–3.
The remaining five patients with biochemical features of MADD were suspected of having secondary MADD.
Riboflavin deficiency linked to riboflavin deficiency in the mothers was suspected for three of these patients (referred to as 5–7). Their clinical presentations were comparable to neonatal MADD but biochemical abnormalities were milder. Notably, urine glutaric acid levels were between 175 and 2,272 mmol/mol creatinine (patients 5 and 6). For patients 5 and 6, vitamin supplementation completely normalized clinical and biochemical features. The patients did not have further episodes of decompensation under riboflavin supplementation and eventually without any therapy. Normalization of biochemical parameters and uneventful evolution after discontinuation of therapy are not consistent with the diagnosis of primary MADD. In addition, for patient 5, fatty acid oxydation flux analysis was normal in fibroblasts, and for patient 6, we did not detect mutations of ETFA, ETFB, or ETFDH. Patient 7 died at 3 days of life, and we suspect a secondary MADD deficiency, because only moderate abnormalities were detected for acylcarnitines and organic acids in plasma (urine analysis unavailable).
Our last two cases (patients 8 and 9) likely suffered from iatrogenic riboflavin deficiency. Both were premature newborns (32 and 31 weeks gestation, respectively), fed by parenteral nutrition for over 2 weeks. Clinical signs and metabolic acidosis appeared after 2 and 3 weeks, respectively. They showed milder but suggestive biochemical profiles of MADD. Patient 8 recovered upon multivitamin supplementation while patient 9 died before diagnosis.
All procedures were in accordance with the ethical standards of the local committees on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Amino Acid Analyses and Statistics
Quantitative plasma amino acid analysis was performed by ion-exchange chromatography: 40-μL deproteinized samples were injected into a JEOL AminoTac JLC-500/V amino acid analyzer calibrated according to standards of known concentration. Amino acid identification was based on retention time and quantification was obtained by integration. Statistical analyses were performed in R (cran.r-project.org). In particular, the ellipse of Fig. 2 was obtained by robust covariance estimation (covRob module).
Fig. 2.
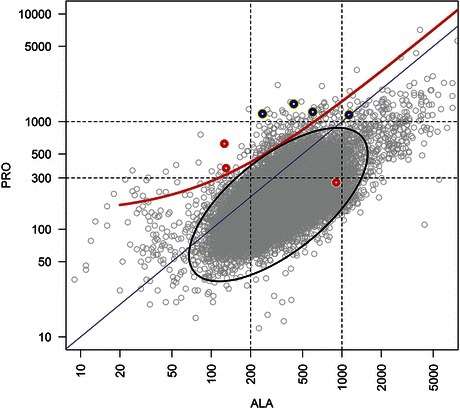
The plasma levels of alanine (“ALA”, x-axis) and proline (“PRO”, y-axis) in micromoles/liter are shown for 53,338 samples corresponding to our complete cohort (gray). The samples collected at presentation from the patients reported in this study are shown as red (newborns with primary MADD) or blue circles (newborn with suspected secondary MADD). The equality line is blue, indicating that average reference alanine levels are greater than proline. Useful cutoffs are shown by black dashed lines and a proposed continuous cutoff by a red line (corresponding to the equation PRO = 1.4 × ALA + 140) (see text). The black ellipse represents the estimated bivariate normal distribution at alpha = 10−5
Results
We first investigated individual amino acid profiles to assess the degree of hyperprolinemia relative to reference intervals. Of the seven cases tested for plasma amino acids, six patients showed hyperprolinemia (Table 1). Two of four cases of primary MADD had only mild hyperprolinemia, yet contrasting with a paradoxical, moderate decrease of alanine concentration below the normal/reference interval. The only primary MADD patient who survived (patient 4) never showed marked hyperprolinemia at diagnosis or during follow-up. All the informative cases suspected of secondary MADD (patients 5, 7, 8, 9) showed much more pronounced hyperprolinemia (1,152–1,457 vs 272–622 μmol/L), whereas alanine levels were only moderately increased. For patient 7, as the initial diagnostic workup did not include organic acid or acylcarnitine profiling, clinical presentation and hyperprolinemia suggested to complete biochemical investigations leading to the diagnosis.
To evaluate the relevance of the association between hyperprolinemia and biochemical features of MADD, we noticed that alanine showed the minimum median levels across all the patients compared to 20 additional amino acids (Fig. 1). Because of the observed dissociation between proline and alanine levels, we reasoned that the ratio or the difference between the levels of these two amino acids could be of diagnostic relevance. Figure 2 plots proline against alanine in our entire cohort of plasma amino acid profiles archived since 1995 including patients with known metabolic disorders (N = 53,338). Figure 2 shows a clear correlation between alanine and proline, with on average higher concentrations of the former amino acid. We observed that some MADD-like profiles clearly deviate from this trend, characterized by high proline concentrations relative to alanine.
Fig. 1.
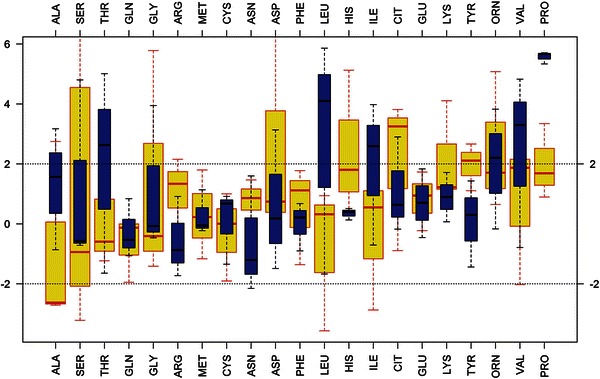
Box plots of age-normalized levels of 21 plasma amino acids for samples from patients with primary (yellow) or presumably secondary (blue) MAD deficiency at presentation. Y-axis: age-normalized standard deviations from a hospital reference population. Amino acids are ordered from the lowest (alanine, ALA) to the highest median levels (proline, PRO) across all samples. The boxes cover the ±25th percentile from the median (horizontal line in the boxes); the dotted lines cover the ±95th percentile from the median
The plot shows that 3/4 tested patients with suspected secondary MADD were very unusual in having very high proline levels (>1,000 μM) contrasting with lower alanine levels (<1,000 μM). Two of three tested patients with primary MADD were also unusual in having moderate increases of proline (>300 μM) contrasting with reduced alanine (<200 μM). The plot also suggests that a potentially useful, continuous cutoff could be plasma proline at levels greater than 1.4 × (alanine concentration) + 140 (red line in Fig. 2; in micromoles/liter). Only 153 of the 53,338 samples show proline values above such cutoff and would be regarded as consistent with MADD, with a p-value <0.003 (the same p-value is obtained by restricting the analysis to the first sample of each patient in the cohort, which yields 35 positive out of 34,525 patients). This cutoff is a tangent to the ellipse of Fig. 2, corresponding to a bivariate reference distribution, at alpha error of 10−5, thus supporting the conclusion that the values associated with most neonatal MADD or MADD-like samples are highly deviant.
These analyses showed an overrepresentation of MADD-like cases among patients with hyperprolinemia, confirming the relevance of the association between these biochemical features. In addition, we suggest that hyperprolinemia with dissociated alanine levels can be useful to suggest a MADD-like disorder. Of note, other etiologies with proline much greater than alanine were primary hyperprolinemia, E3 (DLD) deficiency, lysinuric protein intolerance, and peri-mortem conditions.
Discussion
Early reports pointed to hyperprolinemia as a common feature of MADD, yet plasma proline is currently not regarded as a marker of the disease. Our study indicates that it is a frequent finding both in primary and in suspected cases of secondary early-onset forms of MADD. Our uniquely large cohort of >50,000 plasma amino acid samples also indicates that the simple dissociation between high proline and lower alanine levels may be suggestive of the diagnosis, by using simple cutoffs such as proline levels greater than 140% of alanine levels plus 140 μM. This is of particular interest considering that secondary MADD readily responds to riboflavin supplementation, provided that the diagnosis is suggested promptly. In addition, a small fraction of primary MADD cases can also respond to therapy involving riboflavin, carnitine, and appropriate diet.
Proline dehydrogenase (or proline oxidase (POX)) is a flavoprotein, well characterized in microorganisms and highly conserved throughout eukaryotes and bacteria (Servet et al. 2012; Tanner 2008). POX oxidizes proline to produce Δ1-pyrroline-5-carboxylate (P5C), the first step of proline catabolism. Reduced POX may then give electrons to the electron transfer chain directly via the ubiquinone pool (Moxley et al. 2011), thus without requiring ETF (Wanduragala et al. 2010). Gene sequence comparison and absorption spectral characterization of its catalytic site have suggested that human POX features are comparable to those in microorganisms (Tallarita et al. 2012; Tanner 2008). Because POX is a flavoprotein, it is reasonable to propose that hyperprolinemia observed in riboflavin deficiency may be secondary to depletion in the FAD cofactor. The reason why hyperprolinemia is observed in MADD is still unclear. Secondary coenzyme Q10 deficiency has been observed in riboflavin-responsive MADD (Cornelius et al. 2013). It is tempting to speculate that MADD might induce alterations of the ubiquinone pool or of riboflavin metabolism leading to hyperprolinemia.
Plasma proline was most prominently elevated in the suspected cases of secondary MADD at the time of presentation and was also found in a patient with Brown–Vialetto–Van Laere syndrome during a period of cardiorespiratory distress that preceded death (data not shown). Therefore, we suggest that hyperprolinemia may be a marker of acute decompensation. Nevertheless, it was not found at any time point in a primary MADD case that survived, and the other patients with primary MADD had absolute levels of proline that were not very elevated, except if compared to alanine.
Furthermore, proline levels were higher in secondary MADD, offering the opportunity to quickly refine MADD diagnoses.
We cannot completely rule out differential diagnoses for hyperprolinemia such as medical treatment containing proline, parenteral nutrition, or gelatin administration (Illsinger et al. 2006). However, the degree of hyperprolinemia relative to other amino acids such as alanine, and its association with biochemical features of MADD in different contexts, times, and geographical areas, is not really consistent with this association being coincidental. Hyperlactatemia also induces hyperprolinemia by inhibition of proline oxidase (Kowaloff et al. 1977). However, alanine and proline are both known to increase concomitantly with increased lactate concentrations, yet alanine did not increase as much as proline or actually decreased in some cases (Fig. 2). Hyperprolinemia can also be observed in peri-mortem profiles, characterized by generalized hyperaminoacidemia with very low arginine concentrations, which was clearly different from our profiles.
Our study highlighted the clinical heterogeneity associated with biochemical profiles of MADD. A significant part of suspected neonatal MADD may be secondary forms, treatable with riboflavin. For populations at risk of riboflavin deficiency like preterm newborns under parenteral nutrition, riboflavin supplementation should be considered with particular attention. Critical care recommendations may include riboflavin supplementation as part of a multivitamin cocktail as a first-line therapy for severely ill newborns. Based on our data, in some areas, education of neonatologists may be required to ensure complete prevention of iatrogenic or other causes of secondary MADD.
As shown by our study based on >50,000 plasma amino acid profiles, biochemical profiles of MADD were overrepresented among cases of hyperprolinemia with simple cutoffs (p < 0.003), and other diagnoses with similar alanine and proline levels are associated with very different clinical pictures (see above). This observation is particularly relevant for countries like France where plasma amino acid profiles are ordered before acylcarnitines and urine organic acids profiles. Of particular interest, proline concentrations were more pronounced in secondary MADD that is treatable but can lead to death if untreated. Hyperprolinemia may be a first-line indication of the diagnosis prompting the introduction of MADD therapy.
Our study suggests that the absolute difference between proline to alanine levels in plasma may be a good marker of MAD deficiency, in particular for secondary etiologies, and could contribute to a more effective management of this disorder.
Acknowledgments
We thank Jacqueline Bardet, Odile Beaugendre, Marie Bisançon, Martine Gasquet, and Sabine Leroy for excellent technical assistance.
Synopsis
Hyperprolinemia and more precisely high proline/alanine ratio may be a good marker of MAD deficiency.
Compliance with Ethics Guidelines
Conflict of Interest
Clément Pontoizeau, Florence Habarou, Anaïs Brassier, Alice Veauville-Merllié, Coraline Grisel, Jean-Baptiste Arnoux, Christine Vianey-Saban, Robert Barouki, Bernadette Chadefaux-Vekemans, Cécile Acquaviva, Pascale de Lonlay, and Chris Ottolenghi declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
AB, CG, J-BA, and PdL collected the data. CP, FH, AVM, CVS, CA, PdL, and CO analyzed the data. CP, FH, CVS, CA, PdL, and CO wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
Competing interests: None declared
C. Pontoizeau and F. Habarou equally contributed to this book.
Contributor Information
Clément Pontoizeau, Email: clement.pontoizeau@aphp.fr.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Bosch AM, Abeling NGGM, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2011;34:159–164. doi: 10.1007/s10545-010-9242-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiong MA, Sim KG, Carpenter K, et al. Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol Genet Metab. 2007;92:109–114. doi: 10.1016/j.ymgme.2007.06.017. [DOI] [PubMed] [Google Scholar]
- Cornelius N, Byron C, Hargreaves I, et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum Mol Genet. 2013;22:3819–3827. doi: 10.1093/hmg/ddt232. [DOI] [PubMed] [Google Scholar]
- Frerman FE, Goodman SI. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci U S A. 1985;82:4517–4520. doi: 10.1073/pnas.82.13.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerman FE, Goodman SI, et al. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric acidemia type II. In: Scriver CR, Sly WS, Childs B, et al., editors. The metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001. pp. 2357–2365. [Google Scholar]
- Goodman SI, Reale M, Berlow S. Glutaric acidemia type II: a form with deleterious intrauterine effects. J Pediatr. 1983;102:411–413. doi: 10.1016/S0022-3476(83)80665-9. [DOI] [PubMed] [Google Scholar]
- Green P, Wiseman M, Crow YJ, et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am J Hum Genet. 2010;86:485–489. doi: 10.1016/j.ajhg.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpey JP, Charpentier C, Goodman SI, et al. Multiple acyl-CoA dehydrogenase deficiency occurring in pregnancy and caused by a defect in riboflavin metabolism in the mother. Study of a kindred with seven deaths in infancy: value of riboflavin therapy in preventing this syndrome. J Pediatr. 1983;103:394–398. doi: 10.1016/S0022-3476(83)80410-7. [DOI] [PubMed] [Google Scholar]
- Ho G, Yonezawa A, Masuda S, et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum Mutat. 2011;32:E1976–E1984. doi: 10.1002/humu.21399. [DOI] [PubMed] [Google Scholar]
- Illsinger S, Lücke T, Offner G, et al. Status epilepticus and hyperprolinaemia following recurrent gelatine administrations in a patient on peritoneal dialysis. Nephrol Dial Transplant. 2006;21:1417–1419. doi: 10.1093/ndt/gfk046. [DOI] [PubMed] [Google Scholar]
- Johnson JO, Gibbs JR, Megarbane A, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain J Neurol. 2012;135:2875–2882. doi: 10.1093/brain/aws161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaloff EM, Phang JM, Granger AS, et al. Regulation of proline oxidase activity by lactate. Proc Natl Acad Sci U S A. 1977;74:5368–5371. doi: 10.1073/pnas.74.12.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxley MA, Tanner JJ, Becker DF. Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli. Arch Biochem Biophys. 2011;516:113–120. doi: 10.1016/j.abb.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przyrembel H, Wendel U, Becker K, et al. Glutaric aciduria type II: report on a previously undescribed metabolic disorder. Clin Chim Acta. 1976;66:227–239. doi: 10.1016/0009-8981(76)90060-7. [DOI] [PubMed] [Google Scholar]
- Servet C, Ghelis T, Richard L, et al. Proline dehydrogenase: a key enzyme in controlling cellular homeostasis. Front Biosci Landmark Ed. 2012;17:607–620. doi: 10.2741/3947. [DOI] [PubMed] [Google Scholar]
- Sweetman L, Nyhan WL, Tauner DA, et al. Glutaric aciduria type II. J Pediatr. 1980;96:1020–1026. doi: 10.1016/S0022-3476(80)80629-9. [DOI] [PubMed] [Google Scholar]
- Tallarita E, Pollegioni L, Servi S, et al. Expression in Escherichia coli of the catalytic domain of human proline oxidase. Protein Expr Purif. 2012;82:345–351. doi: 10.1016/j.pep.2012.01.021. [DOI] [PubMed] [Google Scholar]
- Tanner JJ. Structural biology of proline catabolism. Amino Acids. 2008;35:719–730. doi: 10.1007/s00726-008-0062-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanduragala S, Sanyal N, Liang X, et al. Purification and characterization of Put1p from Saccharomyces cerevisiae. Arch Biochem Biophys. 2010;498:136–142. doi: 10.1016/j.abb.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]