

Summary
Objective
In autoimmune encephalitis the etiologic role of neuronal cell‐surface antibodies is clear; patients diagnosed and treated early have better outcomes. Neuronal antibodies have also been described in patients with pediatric epilepsy without encephalitis. The aim was to assess whether antibody presence had any effect on long‐term outcomes in these patients.
Methods
Patients (n = 178) were recruited between 1988 and 1992 as part of the prospective Dutch Study of Epilepsy in Childhood; none received immunotherapy. Healthy age‐matched bone‐marrow donors served as controls (n = 112). All sera were tested for serum N‐methyl‐d‐aspartate receptor (NMDAR), alpha amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor, leucine rich glioma inactivated 1, contactin associated protein like 2 (CASPR2), contactin‐2, glutamic acid decarboxylase, and voltage gated potassium channel (VGKC)‐complex antibodies by standard techniques. No cerebrospinal fluid (CSF) samples were available. Results were correlated with clinical data collected over 15 years.
Results
Seventeen patients (9.5%) were positive for VGKC complex (n = 3), NMDAR (n = 7), CASPR2 (n = 4), and contactin‐2 (n = 3), compared to three (3/112; 2.6%) healthy controls (VGKC complex [n = 1], NMDAR [n = 2]; p = 0.03; Fisher's exact test). Titers were relatively low (≤1:100 for cell‐surface antibodies), but 8 (47%) of the 17 positive samples bound to the surface of live hippocampal neurons consistent with a potential pathogenic antibody. Preexisting cognitive impairment was more frequent in antibody‐positive patients (9/17 vs. 33/161; p = 0.01). Fourteen antibody‐positive patients were treated with standard antiepileptic drugs (AEDs); three (17%) became intractable but this was not different from the 16 (10%) of 161 antibody‐negative patients. In 96 patients with available follow‐up samples at 6 and/or 12 months, 6 of 7 positive antibodies had disappeared and, conversely, antibodies had appeared for the first time in a further 7 patients.
Significance
Neuronal antibodies were found at low levels in 9.5% of patients with new‐onset pediatric epilepsy but did not necessarily persist over time, and the development of antibodies de novo in later samples suggests they could be due to a secondary response to neuronal damage or inflammation. Moreover, as the response to standard AEDs and the long‐term outcome did not differ from those of antibody‐negative pediatric patients, these findings suggest that routine neuronal antibody testing is unlikely to be helpful in pediatric epilepsy. However, the higher incidence of preexisting cognitive problems in the antibody‐positive group, the CASPR2 and contactin‐2 antibodies in 7 of 17 patients, and the binding of 8 of 17 of serum samples to live hippocampal neurons suggest that neuronal antibodies, even if secondary, could contribute to the comorbidities of pediatric epilepsy.
Keywords: Autoantibodies, Pediatric epilepsy, Voltage‐gated potassium channel complex, NMDA receptor
Key Points.
Low levels of neuronal antibodies are present in ˜10% of patients with pediatric epilepsy at onset but are not associated with poor long‐term outcomes or treatment intractability
Antibodies can develop during the course of epilepsy and are not likely to be the sole cause of epilepsy in pediatric patients
However, if associated with clinical features suggestive of autoimmune encephalitis, this “secondary inflammation” may be immunotherapy responsive as seen in other antibody‐mediated diseases
In adults, autoantibodies to essential neuronal proteins such as the N‐methyl‐d‐aspartate receptor (NMDAR) and the voltage gated potassium channel (VGKC)‐complex antigen, leucine rich glioma inactivated 1 (leucine rich glioma inactivated 1 (LGI1), are now widely recognized as an important treatable cause of encephalitis.1, 2 Patients present with memory loss, confusion, and seizures in limbic encephalitis with predominantly LGI1 antibodies3, 4 and neuropsychiatric features, movement, and autonomic symptoms in NMDAR‐Ab (antibody) encephalitis.5, 6 However, the recent characterization of faciobrachial dystonic seizures (FBDS) in patients with LGI1 antibodies has widened the phenotype to include patients presenting with seizure predominance.7, 8 Recognition of each of these diseases is important, as they are responsive to immunotherapies.
In adult and pediatric patients with epilepsy or seizures without encephalitis, autoantibodies are present in approximately 9–13%.9, 10, 11, 12 These patients are more likely to have been classified as “focal epilepsy of unknown cause” and show a tendency toward standard antiepileptic drug (AED) resistance.11, 12 However, in these studies, follow‐up was short, and immunologic treatments have been tried on an empirical basis at a time when the presence of an antibody was unknown. With increasing interest in the possible etiologic role of autoantibodies in epilepsy, and the recognition that early diagnosis and immunotherapy treatment improves outcome in autoimmune encephalitis, it is important for the clinician to be able to make informed decisions regarding which patients to test and whether the results will affect patient management and epilepsy outcome.13, 14
Here we studied archived samples from patients who had been sampled within a median 69 days from their first presentation to the neurologist and followed up for many years. None of the patients were given immunotherapies and some were resampled at 6 and 12 months. The results were compared with age‐ and sex‐matched healthy controls.
Methods
Patient cohort
Children (aged 1 month to 16 years) were enrolled into the Dutch Study of Epilepsy in Childhood (DSEC) from four participating centers in The Netherlands between 1988 and 1992. Details of exclusion and inclusion criteria have been published previously.15, 16 Children with a presumed “acute symptomatic” etiology for their epilepsy (defined as seizures occurring only during the first week after the onset of acute neurologic insult, for example, stroke, head trauma, or central nervous system infection, or concurrently with an acute systemic metabolic disturbance, for example, uremia, hyponatremia, or hypoglycemia, or both.17) were excluded.
Sufficient volumes of serum were available only in 178 children at enrollment (out of the total cohort of 881 patients who were enrolled and discussed). The median period between the first seizure and first blood sampling was 69 days (range 0 days to 6.4 years). All of the serum samples were stored at −20°C from collection. A previous study by the DSEC found no significant differences in sex, etiology, or epilepsy syndrome between those children with available samples and those without.18 Follow‐up serum samples from 96 patients taken at 6 months (n = 30), 12 months (n = 34), and 6 and 12 months (n = 32) after intake were also available for testing. In addition, 112 age and sex‐matched control samples came from age‐matched sibling donors of bone marrow transplantations (BMTs), collected between 1985 and 1995 and stored under the same condition as the patients' sera.
Ethics
The DSEC was approved by the ethics committees of all involved hospitals, and informed consent was obtained in all cases before enrollment.15, 19
Epilepsy classification and definitions
At enrollment, classification of the seizures and epilepsy syndromes was made after discussion by three participating pediatric neurologists according to the 1989 International League Against Epilepsy (ILAE) criteria. This was revised after 2 years and at the end of the most recent intractability study,20 as some children proved to have neurologic brain‐related morbidities. In view of the new terminology published in the most recent reorganization of the ILAE seizure and epilepsy classification, and to facilitate interpretation of the new serologic data with the preexisting clinical data, we have provided definitions of the terms used in Table 1.
Table 1.
Definitions of terminology used from the preexisting historical clinical database of the Dutch Study of Epilepsy in Childhood
| Terminology | Definition |
|---|---|
| Idiopathic epilepsy (IE) | Epileptic syndromes with particular clinical characteristics and specific EEG findings. Unknown origin but presumed genetic etiology |
| Remote symptomatic epilepsy (RSE) | Epilepsies considered the consequence of a known or suspected disorder of the central nervous system resulting in a static encephalopathy. All children with mental retardation (MR) with epilepsy of unknown cause were classified as RSE |
| Cryptogenic epilepsy (CE) | Epilepsies of unknown origin that do not conform to the criteria for IE or RSE |
| Terminal remission | Interval between the very last seizure and the end of follow‐up |
| Fast response to medication | 6 Months of remission starting within 2 months after initiation of AED |
| Intractability | No remission exceeding 3 months (at least one seizure per 3 months) during a minimum period of 1 year of observation despite adequate treatment.16 (Arts et al). Early onset intractability: onset of intractability within the first 5 years of follow‐up. Late onset intractability: onset after the first 5 years of follow‐up |
EEG, electroencephalogram; AED, antiepileptic drug.
Antibody testing of serum samples
Antibodies to the VGKC complex and to the intracellular enzyme glutamic acid decarboxylase (GAD) were measured by radioimmunoassays. To avoid reporting results of low specificity,21, 22 VGKC‐complex antibody positivity was set at >400 pm and GAD positivity at >100 units/ml.11 Cell‐based assays (CBAs) were used to detect antibodies to the NMDAR, alpha amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPAR), LGI1, CASPR2, and contactin‐2. These tests were scored on a visual scale: 0, no binding; 1, low but specific binding; to 4, strong binding to all transfected cells by two independent observers as previously described3, 5 and in use for routine diagnostics. All positive samples were confirmed, and then tested blind by LW as part of a routine antibody service, with dilutions to assess the titer; the positives samples were also tested for binding to the surface of live hippocampal neurons in culture, prepared from P0 Sprague‐Dawley rat pups as described previously.23, 24
Statistical analysis
Descriptive statistics were used to summarize patient data. Fisher's exact test was used to compare categorical data. Data were analyzed using GraphPad Prism 6.0.
Results
Autoantibody testing
Seventeen patients (17/178; 9.5%) were positive for one antibody (VGKC complex [n = 3]; NMDAR [n = 7], CASPR2 [n = 4], and contactin‐2 [n = 3], compared to 3 [2.6%] of the 112 healthy controls (VGKC complex [n = 1], NMDAR [n = 2]; p = 0.03; Fisher's exact test; see Fig. 1, Table 2). Antibodies to LGI1, AMPAR, or GAD were not identified in any patients or controls. Although antibodies binding to the cell‐surface antigens were not high (scoring 1 at dilution 1:20 in 5 and at dilution 1:100 in 9), eight of these samples bound to the surface of live hippocampal neurons (see Table 2), which suggests potential clinical relevance.
Figure 1.
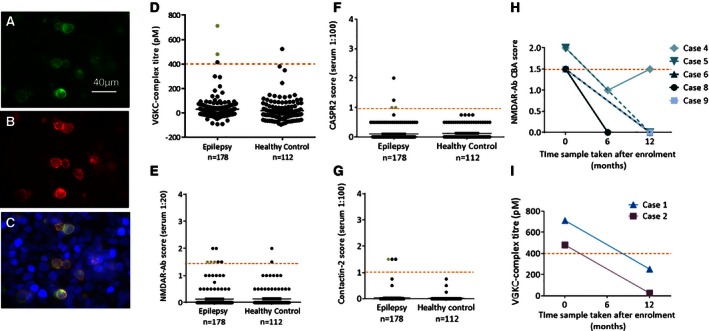
Autoantibody testing results of the epilepsy and healthy control cohorts. The transfected CASPR2‐EGFP tagged transfected HEK cells (green) are shown (A). Serum from patient 16 binds to the surface of the CASPR2 transfected cells, seen with anti‐human IgG labelling (red, B).The transfected cells (A) and anti‐human immunoglobulin (IgG)–labelled cells (B) colocalize indicating a positive result for this patient (C). The scatter diagrams show the titers and CBA scores of positive tests for each antigen tested at the onset of epilepsy compared with healthy controls; VGKC complex (D), NMDAR (E), CASPR2 (F), and contactin‐2 (G). The red dashed line indicates the positive cut‐off used for each assay. The serum samples highlighted by green dots were positive on surface hippocampal staining in vitro. When available, follow‐up samples were tested; four of five NMDAR‐Ab positive patients and both VGKC‐complex antibody‐positive patients were negative at either 6 or 12 months after intake (H, I). Patient 4 showed an initial reduction in antibody levels then increase over time. These fluctuating antibody levels did not correlate with developmental regression or seizure activity.
Table 2.
Demographic, clinical, and paraclinical features, and long‐term outcomes of antibody‐positive epilepsy patients
| Case no./timing of sample (from onset of epilepsy, days) | Age and sex | Seizure type at onset | Type of epilepsy/etiology | Associated clinical features | Treatment history first 5 years/at end of follow‐up (FU, years) | Epilepsy course/seizure type at end of 5 year follow‐up/outcome (TR, years) | Ab positivity (pm/titers) |
|---|---|---|---|---|---|---|---|
| 1 (33) | 4 F | CPS | Localization related symptomatic/remote symptomatic | Right hemiplegia Learning difficulties |
4 AEDS: no fast response/off AED at final FU (14.6) | Improving course SPS, SE, unclear seizures TR = 10.2 |
VGKC (712 pm)a 252 pm at 12 months |
| 2 (0) | 1.7 M | TC | Localization‐related symptomatic/cryptogenic | Mental retardation, not progressive | 3 AEDs: no fast response/on AED at final contact (14.7) |
Poor course TC, SPS, CPS, unclear seizures TR < 1 year, intractable |
VGKC (480 pm)a 25 pm at 12 months |
| 3 (2,333) | 7.4 F | TC | Generalized idiopathic/idiopathic | Mild developmental delay | No AEDs; not on AED at final contact (15.3) |
Improving course TC, unclear seizures TR = 1.5 |
VGKC (414 pm) |
| 4 (365) | 7.4 M | Absences | Generalized idiopathic childhood absence epilepsy/idiopathic | – | 2 AEDs: fast response/off AED at final contact (15.7) |
Good course absences TR = 15.6 |
NMDAR (1 in 100) 1 in 20 at 12 months |
| 5 (106) | 1.5 F | Minor motor and absences | Generalized idiopathic childhood absence epilepsy/idiopathic | – | 2 AEDs: no response (bad compliance)/on AED at final contact (13.5) |
Improving course Minor motor and absences TR = 1.7 |
NMDAR (1 in 100) Negative at 12 months |
| 6 (4) | 12.6 F | TC | Generalized idiopathic with photosensitivity/remote symptomatic | Mild learning difficulties (LD) –attended regular school | 1 AED: fast response/on AED at final contact (13.1) |
Good course TC TR = 13.1 |
NMDAR (1 in 20) Negative at 12 months |
| 7 (0) | 12 M | SE | Localization related symptomatic/remote symptomatic |
Learning difficulties, IQ < 50 Tetraparesis |
2 AEDs: no fast response/on AED at death (died 1.4 years after enrollment) |
SE, unclear small seizures TR = 1 |
NMDAR (1 in 20)a |
| 8 (11) | 3.6 F | Atonic seizures | Generalized Lennox‐Gastaut syndrome/remote symptomatic | Mild global delay | 2 AEDs: no fast response/on AED at final FU (12.7) |
Deteriorating course Atonic seizures, TC TR = 0.5 |
NMDAR (1 in 20)a
Negative at 6 months |
| 9 (89) | 6.6 F | Absences | CAE/idiopathic | – | 1 AED: fast response/off AED at final FU (14.8) |
Good course Absences TR = 14.8 |
NMDAR (1 in 20)a
Negative at 12 months |
| 10 (0) | 15.5 F | TC | IGE/idiopathic | – | No AED/off AED at final FU (2) |
Lost to follow‐up after 2 years TC TR = 2 |
NMDAR (1 in 20) |
| 11 (46) | 4.1 M | SE | Localization related symptomatic/RS including MR | Mild learning difficulties, autism spectrum disorders | 1 AED: fast response/off AED at final FU (15.9) |
Good course SE TR 12.1 |
Contactin‐2 (1 in 100)a |
| 12 (124) | 4.2 M | Unclear seizures | Remote symptomatic including MR | Global mental retardation, spasticity, visual problems | 1 AED: fast response/on AED at final FU (5) |
Improving course TC with focal onset TR > 2 |
Contactin‐2 (1 in 100) |
| 13 (271) | 8.8 F | Atonic, astatic | BECTS/idiopathic | – | 3 AED: no fast response/off AED at final FU (5) |
Improving course SPS with generalization TR > 2 |
Contactin‐2 (1 in 100) |
| 14 (31) | 12.5 M | SPS | Benign partial epilepsy/idiopathic | – | Never used AED |
Good course SPS with generalization TR > 5 |
CASPR2 (1 in 100)a |
| 15 (49) | 8.9 M | TCS | IGE/idiopathic | – | 1 AED: fast response/no AED at final FU (14) |
Good course TCS TR > 5 |
CASPR2 (1 in 100)a |
| 16 (1) | 10.5 M | PS with secondary generalization | Localization related cryptogenic/cryptogenic | – | 3 AED: no fast response, polytherapy/on AED at final FU (13.3) |
Poor course, intractable Clustered PS with gen leading to hospitalization TR < 0.1 |
CASPR2 (1 in 100) |
| 17 (130) | 0.6 M | CPS, myoclonic, atonic | Secondary generalized multifocal with atonic and atypical absence seizures/RS including MR |
Severe mental retardation Febrile seizures during FU |
4 AED: no fast response/on AED at final FU (16.5) |
Improving course. Intractable at 5 years – bad compliance. TCS TR > 5 |
CASPR2 (>1 in 100) |
AED, antiepileptic drug; BECTS, benign epilepsy with centrotemporal spikes; CAE, childhood absence epilepsy; CPS, complex partial seizures; CT, computed tomography; EEG, electroencephalography; FU, follow‐up; IGE, idiopathic generalized epilepsy; MJ, myoclonic jerks; PS, partial seizure; RS, remote symptomatic; SPS, simple partial seizures; SWD, spike‐wave discharge; TC, tonic–clonic; TCS, tonic–clonic seizure; TR, terminal remission; SE, status epileptic.
Positive serum staining on the surface of hippocampal neurons in vitro.
Clinical features of antibody positive patients
The clinical and paraclinical features, treatment responses, and outcomes of the antibody‐positive patients are listed in Table 2. All three patients with VGKC complex Abs had cognitive impairment/learning difficulties (this was not present in the VGKC complex antibody‐positive healthy control). The two patients (cases 1 and 2; Table 2) with the highest titers (712, 480 pm) had focal epilepsy that was difficult to treat, needing at least three AEDs for seizure control initially. Although there were no antibodies detected to the VGKC‐complex‐associated proteins, LGI1 and CASPR2 in these samples, both bound to the surface of hippocampal neurons suggesting potential clinical relevance.
Three of the seven NMDAR‐Ab‐positive patients had learning difficulties before the onset of epilepsy (cases 6–8), including one severely affected (case 7), and required AEDs until the end of follow‐up. Three others (cases 4, 5, and 9) had childhood absence epilepsy with 3‐Hz generalized spike‐wave discharge on electroencephalography (EEG). Three patients were positive for contactin‐2 antibodies (cases 11–13); two were known to be on the autistic spectrum and one had pharmacoresistant benign epilepsy with centrotemporal spikes (BECTS) requiring three AEDs for seizure control. Four patients were positive for CASPR2 antibodies; two (cases 16, 17) had focal epilepsy with periods of resistance to AEDs.
Comparison of clinical features and long‐term outcomes between antibody‐positive and antibody‐negative patients
There were no differences in the sex distribution or age at onset of epilepsy between antibody‐positive and antibody‐negative patients. Seizure semiology and frequency also showed no long‐term difference, and computed tomography (CT) and EEG findings were comparable (Table 3). Overall, however, there was a significantly higher rate of cognitive impairment/developmental delay in the antibody‐positive patient group (9/17 vs. 33/161; p = 0.01). These features were all present before the onset of epilepsy (and hence antibody testing), and included patients with structural brain abnormalities, mild autism, and severe global development delay.
Table 3.
Comparison of clinical features and outcomes of new onset epilepsy antibody positive and negative patients
| Characteristic | Antibody positive (n = 17) | Antibody negative (n = 161) | p‐Value | ||
|---|---|---|---|---|---|
| Sex | M:F – 9:8 | M:F – 72:89 | |||
| Median age of presentation | 5.7 years (range 0.9–15.5) | 6.2 (range 0.2–15.8) | |||
| Type of epilepsy at enrollment | Generalized | 9 (53%) | Generalized | 75 (47%) | 0.8 |
| Focal | 6 (35%) | Focal | 82 (51%) | 0.3 | |
| Other | 2 (12%) | Other | 4 (2%) | 0.1 | |
| Frequency of seizures within first 6 months | 1–3 | 6 (35%) | 1–3 | 71 (44%) | 0.6 |
| 4–25 | 4 (24%) | 4–25 | 34 (21%) | 0.8 | |
| Uncountable | 7 (41%) | Uncountable | 56 (35%) | 0.6 | |
| Etiology at onset | Idiopathic | 8 (47%) | Idiopathic | 87 (54%) | 0.6 |
| RS incl MR | 7 (41%) | RS incl MR | 43 (27%) | 0.3 | |
| Cryptogenic | 2 (12%) | Cryptogenic | 31 (19%) | 0.7 | |
| Preexisting neurologic signs/abnormal neurologic examination | 3 (17.6%) | 17 (10.6%) | 0.4 | ||
| Mental retardation/cognitive impairment at intake | 9 (52.9%) | 33 (20.4%) | 0.01a | ||
| History of febrile seizures before or after intake | 1(5.8%) | 32 (19.8%) | 0.2 | ||
| Family history | 2 (11.7%) | 20 (12.4%) | 1 | ||
| Status epilepticus as presenting feature | 2 (11.7%) | 9 (5.6%) | 0.2 | ||
| Abnormal EEG at intake | 14 (82%) | 126 (78%) | 1 | ||
| CT at intake | Normal | 8 (47%) | Normal | 85 (53%) | 0.8 |
| Abnormal | 4 (24%) | Abnormal | 29 (18%) | 0.5 | |
| Not done | 5 (29%) | Not done | 48 (30%) | 1 | |
| Polytherapy during FU, range (2–16 years) | 4/14 (28.5%) | 22/143 (15.4%) | 0.3 | ||
| On AED at final contact, range (2–16 years) | 8/14 (57%) | 44/143 (30.7%) | 0.07 | ||
| Intractable at last contact | 3 (2 with late onset) (17.6%) | 16 (8 with late onset) (9.9%) | 0.4 | ||
AED, antiepileptic drug; MR, mental retardation; RS, remote symptomatic.
Analyzed by Fisher's exact test.
At 5‐year follow‐up, 65% (11/17) of patients in the antibody‐positive and 78% (125/161) in the antibody negative group had been seizure‐free for >12 months (not significant). At final contact, 57% (8/14) of the antibody‐positive patients were taking AEDs compared to 31% (44/140) of antibody‐negative patients (p = 0.07, Table 3), but the rate of intractability was not different between the two groups (p = 0.18, Fisher's exact test).
Testing of follow‐up samples
Of the 17 patients who were antibody positive at intake, further samples at 6 and 12 months were available for antibody testing in 7 (Fig. 1H,I). Changes in these short‐term antibody levels (Fig. 1H,I) did not correlate with changes in seizure frequency or cognitive development (Table 2). Moreover, 7 (7.7%) of 89 sera from patients who had been antibody negative (n = 161) were found to be positive at follow‐up (NMDAR‐Abs [n = 3], CASPR2‐Abs [n = 2], CASPR2‐ and NMDAR‐Abs [n = 1], and contactin‐2‐Abs [n = 1]). Two of these patients became intractable in the long‐term (cases 27 and 64). The antibodies and clinical features of these patients are listed in Table S1.
Discussion
Autoantibodies to neuronal surface antigens have been reported in adults and children with epilepsy and could indicate an immune basis with obvious management implications.11, 12, 13 However, these studies have been complicated by some use of immunotherapies, and long‐term treatment and outcome data of untreated antibody‐positive patients have not been reported. We were able to test a large, historical cohort of patients with pediatric epilepsy for neuronal surface antibodies and relate the findings to their epilepsy course over time. The frequency of antibodies were similar to those reported previously, but were mainly transient and occurred sporadically; despite lack of immunotherapies, most autoantibody positive patients had a good outcome and responded to standard AEDs. The results suggest, therefore, that routine antibody testing is not necessarily helpful in children with epilepsy and should be restricted to those with evidence of neuroinflammatory disease. Nevertheless, the number of patients (n = 11) who had or developed antibodies to CASPR2 or contactin‐2, proteins that are linked to genetic forms of epilepsy or neurodevelopmental disorders, is an intriguing finding that deserves further study.
The frequency of antibody positivity in this study cohort (9.5% compared with 2.6% in controls) was similar to that published previously for both adult and pediatric epilepsies (10–16% in patients and <5% in controls). Patients with acute symptomatic epilepsy had been excluded and, importantly, the patients did not have any associated clinical features of encephalitis (e.g., confusion, memory loss) at the time of sampling, and none had received immunotherapies. All cell‐based assay–positive samples obtained at onset contained relatively low antibody titers, 8/14 bound to the surface of hippocampal neurons in vitro, and most available follow‐up samples had normalized by 12 months, whereas antibodies had appeared de novo in seven patients. Although samples were taken as close to seizure onset as possible, the median sampling time of 69 days means that some were taken after the establishment of epilepsy and the results could, therefore, reflect the consequences of the epileptogenic process rather than being the primary cause.
Indeed, low‐titer antibodies to VGKC complex or NMDARs have been found in some patients without autoimmune neurologic diseases,21, 25 and the fact that NMDARs were found in some children with generalized absence epilepsy makes them unlikely to be pathogenic in these cases. However, the binding to the surface of live hippocampal neurons, present in 8 of 17 sera, suggested that the antibodies could be pathogenic if they reach the brain parenchyma. In the remaining patients whose antibodies did not bind to the neurons or to LGI1, the VGKC‐complex Abs may be markers for a neuroinflammatory process rather than for an antibody‐mediated syndrome, as recently discussed.12, 22, 26 Surprisingly, therefore, antibodies to CASPR2 or contactin‐2, the other known components of the VGKC complex, were found in 11 pediatric epilepsy patients (seven at onset, four during follow‐up), but not in controls. This finding draws attention to the growing area of shared targets and partially overlapping phenotypes between antibody and genetic forms of pediatric neurologic disease.12, 27
Despite the limitations of a retrospective study with no access to cerebrospinal fluid (CSF) samples for testing, this observational study enabled us to investigate the long‐term outcome of antibody positivity in a cohort of patients with pediatric epilepsy in whom immunotherapies were not used. Because most patients responded well to standard AEDs and had a good long‐term outcome, this questions the pathologic relevance of these low positive antibodies particularly as, in most cases, the antibodies were transient and disappeared by 6 or 12 months follow‐up. It seems likely that the antibodies may have been a secondary response to neuronal damage before the seizures came under control, rather than the primary pathogenic agent. Nevertheless, this is not unprecedented and the development of high levels of NMDAR‐Abs following herpes simplex virus encephalitis (HSVE) with neurologic deterioration (movement disorder, behavioral change, seizures, and worsening of brain lesions on magnetic resonance imaging [MRI]), in patients with no evidence of reactivation of the herpes simplex virus,28 and their response to immunotherapy,29 demonstrates that this “secondary inflammation” may still be pathogenic and respond to immunotherapy.
From these results, a “mono‐immunogenic” cause for pediatric epilepsy is unlikely, with neuronal antibodies forming only part of the complex etiologic framework that includes inflammation, structural abnormalities, and genetic susceptibility; similar diverse factors may explain the final outcomes. Future studies investigating predictive biomarkers in epilepsy need to include all these factors to indicate the likely progression of disease and treatment response. This would be invaluable to both patients and clinicians,30 particularly in drug‐resistant refractory cases with associated comorbidities, commonly seen in pediatric epilepsy. This group of patients may benefit most from further studies into antibody presence, antigenic targets, relevance, and immunotherapy treatment trials.
Disclosure of Conflict of Interest
AV, BL, PW, and the Nuffield Department of Clinical Neurosciences in Oxford receive royalties and payments for antibody assays. The remaining authors have no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1. Clinical features of latent antibody‐positive epilepsy patients.
Acknowledgments
S. Wright was funded by an Oxford University/Wellcome Trust Clinical Research Training Fellowship. Work in the Oxford laboratory is supported by the NIHR Oxford Biomedical Research Centre.
Biography
Dr. Sukhvir Wright is a pediatric neurology specialist registrar researching autoimmune epileptic encephalopathy.

References
- 1. Irani SR, Gelfand JM, Al‐Diwani A, et al. Cell‐surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014;76:168–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann N Y Acad Sci 2014;1338:94–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel‐complex proteins leucine‐rich, glioma inactivated 1 protein and contactin‐associated protein‐2 in limbic encephalitis. Morvan's syndrome and acquired neuromyotonia. Brain 2010;133:2734–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010;9:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irani SR, Bera K, Waters P, et al. N‐methyl‐d‐aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non‐paraneoplastic disorder of both sexes. Brain 2010;133:1655–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti‐NMDA‐receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011;69:892–900. [DOI] [PubMed] [Google Scholar]
- 8. Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013;136:3151–3162. [DOI] [PubMed] [Google Scholar]
- 9. Majoie HJ, de Baets M, Renier W, et al. Antibodies to voltage‐gated potassium and calcium channels in epilepsy. Epilepsy Res 2006;71:135–141. [DOI] [PubMed] [Google Scholar]
- 10. McKnight K, Jiang Y, Hart Y, et al. Serum antibodies in epilepsy and seizure‐associated disorders. Neurology 2005;65:1730–1736. [DOI] [PubMed] [Google Scholar]
- 11. Brenner T, Sills GJ, Hart Y, et al. Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia 2013;54:1028–1035. [DOI] [PubMed] [Google Scholar]
- 12. Suleiman J, Wright S, Gill D, et al. Autoantibodies to neuronal antigens in children with new‐onset seizures classified according to the revised ILAE organization of seizures and epilepsies. Epilepsia 2013;54:2091–2100. [DOI] [PubMed] [Google Scholar]
- 13. Quek AM, Britton JW, McKeon A, et al. Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 2012;69:582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bien CG. Value of autoantibodies for prediction of treatment response in patients with autoimmune epilepsy: review of the literature and suggestions for clinical management. Epilepsia 2013;54(Suppl. 2):48–55. [DOI] [PubMed] [Google Scholar]
- 15. Arts WF, Geerts AT, Brouwer OF, et al. The early prognosis of epilepsy in childhood: the prediction of a poor outcome. The Dutch study of epilepsy in childhood. Epilepsia 1999;40:726–734. [DOI] [PubMed] [Google Scholar]
- 16. Arts WF, Brouwer OF, Peters AC, et al. Course and prognosis of childhood epilepsy: 5‐year follow‐up of the Dutch study of epilepsy in childhood. Brain 2004;127:1774–1784. [DOI] [PubMed] [Google Scholar]
- 17. Hauser WA, Anderson VE, Loewenson RB, McRoberts SM. Seizure recurrence after a first unprovoked seizure. N Engl J Med 1982;307:522–528. [DOI] [PubMed] [Google Scholar]
- 18. Callenbach PM, Jol‐Van Der Zijde CM, Geerts AT, et al. Immunoglobulins in children with epilepsy: the Dutch Study of Epilepsy in Childhood. Clin Exp Immunol 2003;132:144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stroink H, Brouwer OF, Arts WF, et al. The first unprovoked, untreated seizure in childhood: a hospital based study of the accuracy of the diagnosis, rate of recurrence, and long term outcome after recurrence. Dutch study of epilepsy in childhood. J Neurol Neurosurg Psychiatry 1998;64:595–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Geerts A, Brouwer O, Stroink H, et al. Onset of intractability and its course over time: the Dutch study of epilepsy in childhood. Epilepsia 2012;53:741–751. [DOI] [PubMed] [Google Scholar]
- 21. Paterson RW, Zandi MS, Armstrong R, et al. Clinical relevance of positive voltage‐gated potassium channel (VGKC)‐complex antibodies: experience from a tertiary referral centre. J Neurol Neurosurg Psychiatry 2014;85:625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hacohen Y, Singh R, Rossi M, et al. Clinical relevance of voltage‐gated potassium channel‐complex antibodies in children. Neurology 2015;85:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beaudoin GM III, Lee SH, Singh D, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 2012;7:1741–1754. [DOI] [PubMed] [Google Scholar]
- 24. Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc 2006;1:2406–2415. [DOI] [PubMed] [Google Scholar]
- 25. Zandi MS, Paterson RW, Ellul MA, et al. Clinical relevance of serum antibodies to extracellular N‐methyl‐d‐aspartate receptor epitopes. J Neurol Neurosurg Psychiatry 2015;86:708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hacohen Y, Wright S, Waters P, et al. Paediatric autoimmune encephalopathies: clinical features, laboratory investigations and outcomes in patients with or without antibodies to known central nervous system autoantigens. J Neurol Neurosurg Psychiatry 2013;84:748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramanathan S, Wong CH, Rahman Z, et al. Myoclonic status epilepticus as a presentation of caspr2 antibody‐associated autoimmune encephalitis. Epileptic Disord 2014;16:477–481. [DOI] [PubMed] [Google Scholar]
- 28. Hacohen Y, Deiva K, Pettingill P, et al. N‐methyl‐d‐aspartate receptor antibodies in post‐herpes simplex virus encephalitis neurological relapse. Mov Disord 2014;29:90–96. [DOI] [PubMed] [Google Scholar]
- 29. Mohammad SS, Sinclair K, Pillai S, et al. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to N‐Methyl‐d‐aspartate receptor or dopamine‐2 receptor. Mov Disord 2014;29:117–122. [DOI] [PubMed] [Google Scholar]
- 30. Hegde M, Lowenstein DH. The search for circulating epilepsy biomarkers. Biomark Med 2014;8:413–427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical features of latent antibody‐positive epilepsy patients.