

Abstract
The gamma‐aminobutyric acid type A receptor β3 gene (GABRB3) encodes the β3‐subunit of the gamma‐aminobutyric acid type A (GABAA) receptor, which mediates inhibitory signalling within the central nervous system. Recently, GABRB3 mutations have been identified in a few patients with infantile spasms and Lennox–Gastaut syndrome. We report the clinical and electrographic features of a novel case of GABRB3‐related early‐onset epileptic encephalopathy. Our patient presented with neonatal hypotonia and feeding difficulties, then developed pharmacoresistant epileptic encephalopathy, characterized by multiple seizure types from 3 months of age. Electroencephalography demonstrated ictal generalized and interictal multifocal epileptiform abnormalities. Using a SureSelectXT custom multiple gene panel covering 48 early infantile epileptic encephalopathy/developmental delay genes, a novel de novo GABRB3 heterozygous missense mutation, c.860C>T (p.Thr287Ile), was identified and confirmed on Sanger sequencing. GABRB3 is an emerging cause of early‐onset epilepsy. Novel genetic technologies, such as whole‐exome/genome sequencing and multiple gene panels, will undoubtedly identify further cases, allowing more detailed electroclinical delineation of the GABRB3‐related genotypic and phenotypic spectra.
Short abstract
This article is commented on by Pearl on pages 330–331 of this issue.
Abbreviations
- GABAA
Gamma‐aminobutyric acid type A
- GABRB3
Gamma‐aminobutyric acid type A receptor β3 gene
What this paper adds.
A report of a novel GABRB3 mutation associated with early infantile epileptic encephalopathy.
A detailed description of the clinical and electrographic features of the case.
A review of the few other reported cases of GABRB3‐related epileptic encephalopathy.
A discussion of this gene's association with other neurodevelopmental disorder phenotypes.
Increased clinical awareness of the role of multiple gene panels in the diagnosis of early infantile epilepsy.
Gamma‐aminobutyric acid type A (GABAA) receptors are ligand‐gated chloride channels that act as the primary mediators of fast inhibitory synaptic transmission in the central nervous system. They belong to the Cys‐loop superfamily, and are formed by pentameric assemblies of different subunit subtypes: α1–α6, β1–β3, γ1–γ3, δ, ε, π, θ, and ρ1–ρ3.1 Most GABAA receptors contain two α‐subunits, two β‐subunits, and another, most commonly a γ‐subunit.2, 3 These subunits have four transmembrane domains, of which the second transmembrane domain forms a central ion pore with the other four subunits4 (Fig. 1a). When GABA, the physiological ligand of GABAA receptors binds to the receptor, the ion pore opens, facilitating chloride influx or efflux. The direction of chloride flux depends on intracellular chloride concentration, regulated by the potassium‐chloride KCC2 and sodium–potassium–chloride NKCC1 co‐transporters. In developing brains, the predominance of NKCC1 leads to increased intracellular chloride, resulting in GABA‐mediated chloride efflux which depolarizes the cell membrane, thus leading to neuronal excitation.5 Mature neuronal cell bodies have low intracellular concentrations of chloride due to high levels of KCC2 expression, leading to GABA‐mediated chloride influx that hyperpolarizes the cell membrane leading to neuronal inhibition.6
Figure 1.
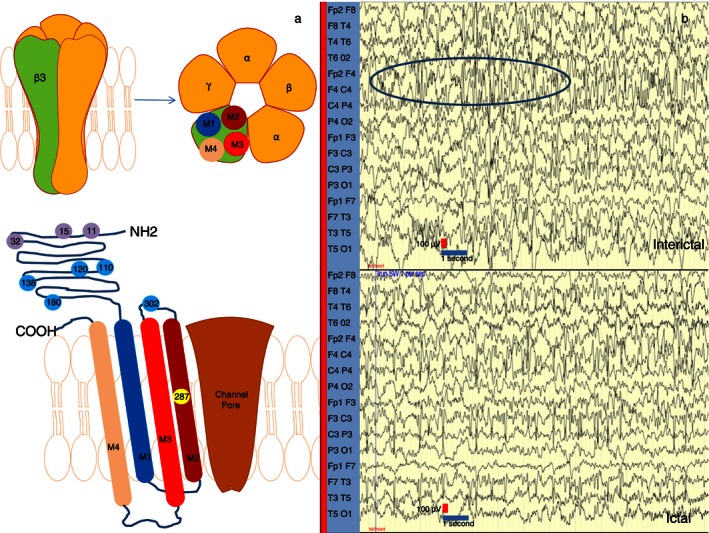
(a) Top: Schematic diagram of the gamma‐aminobutyric acid type A (GABAA) receptor and the gamma‐aminobutyric acid type A receptor β3 (GABRB3) subunit (depicted on the left). Most physiological heteromeric GABAA receptors are thought to include two α‐, two β‐, and one other (most frequently a γ‐) subunits. Bottom: Each β3‐subunit consists of an extracellular domain, an α‐helical M1–M4 transmembrane bundle, and an M3–M4 intracellular loop. The M2 segments line the ion channel pore that tapers as it traverses towards the intracellular side of the membrane. The de novo heterozygous mutation in GABRB3 in our proband is located in amino acid position 287. Thr287 is located in the M2 segment. Previously reported mutations in patients with epileptic encephalopathies are located in amino acid positions 110, 120, 138, 180, and 302. Polymorphisms and mutations implicated in childhood absence epilepsy are in positions 11, 15, and 32, clustered closer to the N terminus. All above mutations and variants are depicted as circles with corresponding numbers. (b) Interictal electroencephalogram (EEG) recording is seen above, showing high‐amplitude multifocal discharges, particularly in the right anterior region (circle). Ictal EEG is depicted below, demonstrating generalized fast activity, more prominent over the frontal regions.
Given the central inhibitory role of GABAA receptors, it is not surprising that, to date, several genetic epilepsy syndromes have been associated with variants in GABAA receptor subunit genes including GABRA1, GABRB3, GABRD, and GABRG2.7 GABRB3, located on chromosome 15q11.2‐q12, encodes the β3‐subunit of the GABAA receptor. Reduced GABRB3 expression has been postulated in the pathogenesis of absence seizures, abnormal sensory processing, and other neurodevelopmental disorder phenotypes such as Angelman syndrome, autism spectrum disorders, and intellectual disability.8, 9, 10 Furthermore, single nucleotide polymorphisms and missense mutations in GABRB3 have previously been implicated in childhood absence epilepsy.11, 12
In 2013, GABRB3 mutations were identified in patients with infantile spasms and Lennox–Gastaut syndrome.13 To date, few cases of GABRB3 epileptic encephalopathy have been described in the literature. We report a case of early‐onset epilepsy with a de novo GABRB3 mutation identified on a diagnostic multiple gene panel, and delineate the electroclinical phenotype in our patient.
Case Report
A male, first‐born to unrelated white parents, was delivered after induction of labour, by forceps delivery at 42 weeks' gestation. The antenatal period was unremarkable and he was born in good condition. His maternal grandfather reported having seizures in childhood provoked by startling, but these resolved spontaneously and there were no further neurodevelopmental concerns. Family history was otherwise non‐contributory. The patient presented with neonatal hypotonia, feeding difficulties, and failure to thrive in early infancy. At 3 months of age, he developed clusters of short seizures characterized by eye deviation, eyelid flickering, tonic arm extension, and back arching. Ictal electroencephalography (EEG) revealed generalized fast activity, more prominent over the frontal regions, and interictal recordings showed high‐amplitude delta/theta multifocal (but predominantly right anterior) discharges (Fig. 1b). Initially, complete cessation of seizures was achieved in response to therapeutic doses of the GABA transaminase inhibitor vigabatrin. However, severe hypotonia, sedation, and respiratory difficulties ensued, hence the patient was weaned off vigabatrin and it was discontinued. Seizures recurred at 5months of age, and, despite multiple antiepileptic therapies (initially carbamazepine 14 mg/kg/day; then levetiracetam 40 mg/kg/day, topiramate 4 mg/kg/day, and sodium valproate 25 mg/kg/day in combination; and finally ketogenic diet with levetiracetam up to 50 mg/kg/day), he continued to have ongoing intractable epilepsy. At the time of last review, age 3 years 2 months, he had 10 to 20 epileptic seizures per day. Seizure semiology was varied, with episodic behavioural arrest, focal motor events, myoclonic jerks, and brief tonic seizures with lower‐limb extension. Parents also reported frequent paroxysms of laughter of undetermined origin, but these episodes have not been captured on EEG.
Over time, he developed severe global developmental delay with signs of slow progress but no obvious evidence of regression. At 3 years 2 months, head circumference plotted on the 0.4th centile. A few subtle dysmorphic features were evident, including mild prominence of his forehead with long eyelashes, tented mouth appearance, high‐arched palate, and bilateral undescended testes. There was also marked axial hypotonia and severe head lag. An overall paucity of limb movements was observed, although some antigravity movements were evident. Peripheral tone was low/normal, deep tendon reflexes were brisk throughout, and plantar responses up‐going bilaterally. There was no evidence of ankle clonus. He could make some vocalizations and was feeding orally with only occasional drooling, and no episodes of choking or swallowing difficulties. Extensive neurometabolic investigations were essentially unremarkable (Table SI, online supporting information), including a normal MRI brain (age 14 mo) and comparative genomic hybridization microarray analysis for genomic copy number variants.
Molecular Genetic Investigation
Further diagnostic testing was undertaken using a multiple gene panel covering 48 genes (Table SII, online supporting information) causing early infantile epileptic encephalopathy. A custom SureSelect library was created (Agilent's SureDesign tool, https://earray.chem.agilent.com/suredesign/). Libraries were made following the SureSelectXT Custom Capture protocol (Agilent Technologies, Santa Clara, CA, USA) and sequenced in‐house on an Illumina MiSeq. Of targeted regions (target genes and their intron–exon boundaries), 99.7% were covered at ≥30×. A single heterozygous missense mutation of GABRB3 (c.860C>T, p.Thr287Ile) was identified (Fig. 1a). Subsequent Sanger sequencing of both proband and parents confirmed these findings, establishing that the mutation was absent in the parents and likely to have occurred de novo (Fig. S1, online supporting information). The variant identified is classified as ‘likely pathogenic’ according to the American College of Medical Genetics/Genomics Standards and Guidelines,14 which infers greater than 90% certainty of being disease‐causing. This variant occurred de novo, providing strong evidence of pathogenicity. It was absent in control population databases including ExAC (http://exac.broadinstitute.org/), 1000 Genomes (http://browser.1000genomes.org/index.html), and Exome Variant Server (http://evs.gs.washington.edu/EVS/) (moderate evidence). Furthermore, the amino acid change occurs in a transmembrane domain lining the ion channel pore4 (moderate evidence). In silico analysis provided further evidence of pathogenicity, with Polyphen2 (score 1.000, sensitivity 0.00, specificity 1.00), SIFT (score 0.000), and PROVEAN (score −5.65) predicting the variant to be probably damaging, damaging, and deleterious, respectively.
Discussion
We report a novel mutation in GABRB3 in a patient with severe intractable early infantile epileptic encephalopathy. Our report highlights the recent finding that mutations in GABRB3 are an emerging cause of early‐onset epilepsy syndromes.
To date, five other cases of GABRB3 early infantile epileptic encephalopathy have been reported,10, 13 to our knowledge (Table 1). We postulate that there are several features common in all these cases, including: (1) seizure onset in infancy, <10months of age, (2) multiple seizure types including infantile spasms, (3) variable EEG abnormalities, and (4) associated comorbidities such as neurodevelopmental delay, attention‐deficit–hyperactivity disorder, autistic features, and intellectual disability. Similar to previous reported cases, our patient manifested features of a non‐specific early infantile epileptic encephalopathy disorder, with multiple seizure types and significant delay in development.
Table 1.
Reported patients with early‐onset epileptic encephalopathy due to GABRB3 mutations
| Mutation | De novo/inherited | Age at onset (mo) | Seizure semiology | EEG | Seizure evolution and treatment response if reported | Other features | Intellectual disability | References |
|---|---|---|---|---|---|---|---|---|
|
c.328A>G; p.Asn110Asp |
De novo | 5 |
Infantile spasms Myoclonic seizures |
Hypsarrhythmia | No follow‐up data | None reported | None at presentation; no follow‐up data | Allen et al.13 |
|
c.358G>A; p.Asn120Asp |
De novo | 10 | Infantile spasms | Generalized 2Hz bursts | Lennox–Gastaut syndrome |
ADHD Impulsivity |
Severe | Allen et al.13 |
|
c.413_415dupACC; p.Asn138_Arg139insHis |
De novo | 2 |
Myoclonic Focal seizures Atonic head nods |
Multifocal/modified burst suppression | Responsive to levetiracetam and topiramate | ‘Mild’ autistic features | Severe | Hamdan et al.10 |
|
c.539A>G; p.Glu180Gly |
De novo | 10 | Infantile spasms | Generalized 2Hz bursts | Lennox–Gastaut syndrome |
ADHD Impulsivity Sleeping difficulties |
Severe | Allen et al.13 |
|
c.905A>G; p.Tyr302Cys |
De novo | 10 |
Focal dyscognitive seizures Behavioural arrests |
Slow, some left temporal features | Lennox–Gastaut syndrome | None reported | Severe: 20 words at 4y | Allen et al.13 |
|
c.860C>T; p.Thr287Ile |
De novo | 3 |
Focal motor seizures Tonic seizures |
Generalized fast activity. Interictal delta/theta multifocal discharges |
Ongoing seizures with behavioural arrests, focal motor, myoclonic, tonic seizures | None currently | Severe | This paper |
ADHD, attention‐deficit–hyperactivity disorder; EEG, electroencephalography.
Our patient harboured a mutation affecting a highly conserved protein domain located in the second transmembrane loop lining the ion channel (Fig. 1a). It is difficult to postulate what the exact pathogenic mechanism of this variant is without any in vitro or in vivo functional work; however, it is probably due to loss of function and haploinsufficiency. It is possible that the position of the amino acid change within the receptor pore could negatively impact on receptor function, for example by inhibiting the pore's opening and subsequent chloride flux. Other mechanisms including reduced GABRB3 protein expression and incorrect cellular trafficking of GABRB3 could also play a role. This is the first reported mutation affecting this region and, to date, most other reported GABRB3 mutations causing epileptic encephalopathies target the extracellular protein domains, near the amino (N)′ terminus13 (Fig. 1a).
GABRB3 has also been implicated in other neurodevelopmental disorders such as autism, childhood absence epilepsy, and Angelman syndrome. A single nucleotide polymorphism at the promoter region of GABRB3 (T>C substitution in position −897, numbering with respect to the initiator methionine of exon 1a), which leads to reduction of the promoter's transcriptional ability, has been associated with childhood absence epilepsy in previous studies.12 Other researchers have reported that a rare GABRB3 single nucleotide polymorphism (c.31C>T; p.Pro11Ser)15 and other variants (c.44C>T; p.Ser15Phe, and c.94G>A; p.Gly32Arg) (NM_021912.4) are linked with childhood absence epilepsy phenotypes associated with eyelid myoclonus and generalized tonic–clonic seizures.6, 11 Probands had typical generalized 3Hz spike and wave discharges during childhood, with clinical symptoms later remitting, without neurological sequelae. The identified mutations were also present in other family members who never had epileptic seizures, suggesting variable penetrance. The same mutations were not found in 630 healthy ethnically and sex‐matched comparison individuals in one study. However, p.Pro11Ser was later described in an asymptomatic individual, suggesting it may be a rare single nucleotide polymorphism.6, 15 Interestingly, genetic variants implicated in childhood absence epilepsy are clustered much closer to the N‐terminal domain of the protein than those identified in GABRB3 early infantile epileptic encephalopathy (Fig. 1a).
In Angelman syndrome, deletions encompassing GABRB3 have been reported.16 Mice with Gabrb3 knockout have epilepsy and other abnormalities that show some similarities to patients with Angelman syndrome.17 The extent to which GABRB3 accounts for the clinical phenotype of Angelman syndrome is currently unclear. Other genes including GABRG3 and GABRA5 are also often contained within deletions causing Angelman syndrome.16 The more severe phenotype associated with such deletions might indicate an additive role for these genes in disease pathogenesis. EEG recordings in Angelman syndrome often demonstrate specific rhythmic patterns that are less prominent in patients with deletions encompassing GABRB3. Indeed, no such rhythmicity was evident in our participant.
At present, there are too few reported cases of GABRB3‐related epilepsy for clear phenotype–genotype correlations or comparisons with Angelman syndrome due to chromosome deletions with GABRB3 haploinsufficiency. It is currently unknown whether mutation type and location affect clinical disease presentation, or whether other environmental or epigenetic factors may play a role. Increasing availability of novel genetic technologies, such as multiple gene panels and whole‐exome sequencing will undoubtedly accelerate the identification of further cases, allowing more detailed delineation of the spectrum of GABRB3‐related disorders.
Recent studies suggest that GABRB3 mutations cause attenuated chloride currents and channel activity impairment through subunit hyperglycosylation.11 The mechanisms through which GABRR3 mutations affect neuronal networks giving rise to epilepsy and neurodevelopmental delay are yet to be fully elucidated. Further research is needed, not only to understand the underlying disease pathogenesis but also to identify novel therapies, which may also have wider implications for GABAA modulation in other neurological disorders.
Supporting information
Figure S1: Top panel: De novo heterozygous mutation in GABRB3, c.860C>T (p.Thr287Ile) detected by panel and confirmed by Sanger sequencing (red rectangle).
Table SI: Neurometabolic investigations undertaken in patient.
Table SII: Epilepsy and severe delay gene panel result.
Acknowledgements
The authors have stated that they had no interests that might be perceived as posing a conflict or bias.
References
- 1. Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci 1994; 17: 569–602. [DOI] [PubMed] [Google Scholar]
- 2. Baumann SW, Baur R, Sigel E. Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J Biol Chem 2002; 277: 46020–25. [DOI] [PubMed] [Google Scholar]
- 3. Sigel E, Steinmann ME. Structure, function, and modulation of GABAA receptors. J Biol Chem 2012; 287: 40224–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature 2014; 512: 270–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fukuda A. Diuretic soothes seizures in newborns. Nat Med 2005; 11: 1153–54. [DOI] [PubMed] [Google Scholar]
- 6. Hirose S. Mutant GABAA receptor subunits in genetic (idiopathic) epilepsy. Prog Brain Res 2014; 213: 55–85. [DOI] [PubMed] [Google Scholar]
- 7. Macdonald RL, Kang JQ, Gallagher MJ. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol 2010; 588: 1861–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanaka M, DeLorey TM, Delgado‐Escueta AV, Olsen RW. GABRB3, epilepsy, and neurodevelopment. Epilepsia 2010; 51: 77. [PubMed] [Google Scholar]
- 9. Delahanty RJ, Kang JQ, Brune CW, et al. Maternal transmission of a rare GABRB3 signal peptide variant is associated with autism. Mol Psychiatry 2011; 16: 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hamdan FF, Srour M, Capo‐Chichi JM, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet 2014; 10: e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka M, Olsen RW, Medina MT, et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet 2008; 82: 1249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Urak L, Feucht M, Fathi N, Hornik K, Fuchs K. A GABRB3 promoter haplotype associated with childhood absence epilepsy impairs transcriptional activity. Hum Mol Genet 2006; 15: 2533–41. [DOI] [PubMed] [Google Scholar]
- 13. Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lachance‐Touchette P, Martin C, Poulin C, Gravel M, Carmant L, Cossette P. Screening of GABRB3 in French‐Canadian families with idiopathic generalized epilepsy. Epilepsia 2010; 51: 1894–97. [DOI] [PubMed] [Google Scholar]
- 16. Dan B, Boyd SG. Angelman syndrome reviewed from a neurophysiological perspective. The UBE3A‐GABRB3 hypothesis. Neuropediatrics 2003; 34: 169–76. [DOI] [PubMed] [Google Scholar]
- 17. DeLorey TM, Handforth A, Anagnostaras SG, et al. Mice lacking the β3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 1998; 18: 8505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Top panel: De novo heterozygous mutation in GABRB3, c.860C>T (p.Thr287Ile) detected by panel and confirmed by Sanger sequencing (red rectangle).
Table SI: Neurometabolic investigations undertaken in patient.
Table SII: Epilepsy and severe delay gene panel result.