

Abstract
SUCLA2 encodes for a subunit of succinyl-coenzyme A synthase, the enzyme that reversibly synthesises succinyl-coenzyme A and ATP from succinate, coenzyme A and ADP in the Krebs cycle. Disruption of SUCLA2 function can lead to mitochondrial DNA depletion. Patients with a SUCLA2 mutation present with a rare but distinctive deafness-dystonia syndrome. Additionally, they exhibit elevated levels of the characteristic biochemical markers: methylmalonate, C4-dicarboxylic carnitine and lactate are increased in both plasma and urine. Thus far, eight different disease-causing SUCLA2 mutations, of which six missense mutations and two splice site mutations, have been described in the literature. Here, we present the first patient with an intragenic deletion in SUCLA2 and review the patients described in literature.
Keywords: 3-Methylglutaconic aciduria, Hearing impairment, Mitochondrial DNA depletion, Movement disorder, SUCLG1
Introduction
Mitochondrial diseases are a heterogeneous group of disorders, caused by mutations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA). nDNA mutations can cause instability or a decreased quantity of mtDNA, or changed functioning of mitochondrial proteins. This will lead to dysfunction of the respiratory chain in mitochondria, where ATP is synthesised. Tissues highly dependent on oxidative energy supply (central nervous system, sensory organs, skeletal muscles and heart) are therefore most commonly affected in mitochondrial disease (Menezes et al. 2014).
Infantile mitochondrial encephalomyopathic depletion syndrome, which is associated with methylmalonic aciduria, has been connected to mutations in SUCLA2 (MIM#612073, (Carrozzo et al. 2007; Ostergaard et al. 2007b)) and SUCLG1 (MIM*611224, (Ostergaard et al. 2007a)). These genes encode for subunits of the adenosine diphosphate (ADP)-dependent isoforms of succinyl-coenzyme A synthase (SCS-A). SCS-A is a mitochondrial matrix enzyme that reversibly synthesises succinyl-coenzyme A from succinate and coenzyme A in the Krebs cycle. The hypothesis is that SCS-A forms a complex with mitochondrial nucleoside diphosphate kinase, an enzyme important in the dNTP salvage pathway in mtDNA replications. Disruption of SCS-A could thereby lead to impaired mtDNA synthesis and thus mtDNA depletion (Elpeleg et al. 2005).
SCS-A is a heterodimer that occurs in a G-SUCL and an A-SUCL form. They share an invariant alpha-subunit encoded by SUCLG1. The variable beta-subunit is encoded by SUCLG2 or SUCLA2 and determines the enzymatic nucleotide specificity (Johnson et al. 1998). The G-SUCL form initiates the reversible conversion of succinyl-CoA and GDP to succinate and GTP. A-SUCL reversibly converts succinyl-CoA and ADP to succinate and ATP in the Krebs cycle. A-SUCL is mainly expressed in testis, brain and skeletal muscle tissue, whereas G-SUCL is expressed in liver and anabolic tissues (Johnson et al. 1998; Lambeth 2006; Lambeth et al. 2004; Miller et al. 2011).
Mutations in SUCLA2 give rise to a typical but rare combination of disorders: an early onset dystonia combined with deafness (MIM#612073, (Carrozzo et al. 2007; Ostergaard et al. 2007b)). In deafness-dystonia syndromes (e.g. Mohr-Tranebjaerg syndrome (TIMM8A, MIM#304700, (Jin et al. 1996)), Woodhouse-Sakati syndrome (C2orf37, MIM#241080, (Alazami et al. 2010) and MEGDEL (SERAC1, MIM#614739, (Wortmann et al. 2012)), these features dominate the clinical picture. Other causes for the rare association of dystonia and deafness are mitochondrial disorders and organic acidurias. Perinatal hypoxic-ischemic brain injury, kernicterus, head trauma and meningoencephalitis account for a small proportion of non-genetic causes (for review, see (Kojovic, et al. 2013)).
Here, we report one new patient with a homozygous intragenic deletion of a complete exon in SUCLA2. In addition, we compared the gen- and phenotypes of all 28 patients previously reported in literature.
Methods
Patients
We report one new patient who was under care of one of the authors (ADM) and performed a literature review on PubMed accessed on December 2014. We used the search term “SUCLA2” and had 44 hits. After reading the abstracts, eight relevant articles remained (Carrozzo et al. 2007; Elpeleg et al. 2005; Jaberi et al. 2013; Lamperti et al. 2012; Matilainen et al. 2014; Morava et al. 2009; Nogueira et al. 2015; Ostergaard et al. 2007b). No additional articles were found in the citation lists of used articles. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
DNA Sequence Analysis
DNA was extracted from peripheral venous blood samples using standard procedures. The complete coding region of SUCLA2 (GenBank accession#NM_003850.2, chromosome 13q12.2-q13.3, 11 exons) was sequenced as described previously (Carrozzo et al. 2007).
Multiplex Ligation-Dependent Probe Amplification
MLPA (multiplex ligation-dependent probe amplification) to screen for deletions and duplications of exons 1, 2, 6, 9, 10 and 11 of SUCLA2 was performed using the SALSA P089 probemix (MRC-Holland, The Netherlands), following the manufacturer’s procedures.
Results
The patients’ findings are summarised in Tables 1 and 2 and Fig. 1.
Table 1.
Clinical, radiological and metabolic findings in patients with SUCLA2 deficiency
| Symptoms | Patients reported in the literature (n = 28)a | This study | Total (n = 29) |
|---|---|---|---|
| Signs and symptoms | |||
| Gender | 19 males, 9 females | Male | 20 males |
| Age of onset; birth–6 months | 22/24 | + | 23/25 |
| Age of death (12/28) | 6 months–21 years | Alive 6 years | 6 months–21 years |
| Muscle hypotonia | 27/28 | + | 28/29 |
| Delayed motor development | 27/28 | + | 28/29 |
| Sensorineural hearing loss | 25/28 | + | 26/29 |
| Dystonia/hyperkinesia | 24/28 | + | 25/29 |
| Absent speech | 17/20 | + | 18/21 |
| Feeding problems | 22/28 | + | 23/29 |
| Failure to thrive | 20/27 | + | 21/28 |
| Progressive spasticity | 15/22 | − | 15/23 |
| Ophthalmoplegia/strabismus/ptosis | 17/26 | − | 17/27 |
| Hyperhidrosis | 4/14 | NA | 4/14 |
| Epilepsy | 4/21 | − | 4/22 |
| Ataxia | 1/1 | − | 1/2 |
| Athetosis | 2/2 | − | 2/3 |
| Increased deep tendon reflexes | 1/1 | − | 1/2 |
| MRI | |||
| Cerebral atrophy on MRI | 12/24 | + | 13/25 |
| Cerebellar atrophy on MRI | 4/24 | − | 4/25 |
| Basal ganglia lesions on MRI | 18/24 | − | 18/25 |
| Metabolic investigations | |||
| Elevated serum lactate | 17/19 (range 0.9–7.5; N < 2.2 mmol/l) | + | 18/19 |
| Elevated urinary lactate | 2/2 (N < 150 μmol/mmol creatinine) | + | 3/3 |
| Elevated plasma methylmalonate | 7/7 (range 0.8–33; N < 0.33 μmol/l) | + | 8/8 |
| Elevated urinary methylmalonate | 17/18 (range “marginal”-212; N < 5 μmol/mmol creatinine) | + | 18/19 |
| Elevated plasma C4-dicarboxylic carnitine | 4/4 (range 0.23–2.2; N <0.6 μmol/l) | + | 5/5 |
| Elevated urinary C4-dicarboxylic carnitine | 3/3 (N 0.04–0.5 μmol/mmol creatinine) | + | 4/4 |
Table 2.
Genetic findings in patients with SUCLA2 deficiency
| Mutation | Predicted effect of the mutation on protein level | Type of mutation | Reference | Country of origin |
|---|---|---|---|---|
| c.308C>A | p.(Ala103Asp) | Missense | P I, II (Lamperti et al. 2012) | Italy |
| c.352G>A | p.(Gly118Arg) | Missense | P 2 (Carrozzo et al. 2007) | Italy |
| c.534+1G>A | p.(Pro125Valfs*4) | Splice | P 1–16 (Morava et al. 2009; Ostergaard et al. 2007b) | Faroe Islands |
| c.751G>A | p.(Asp251Asn) | Missense | P 1,2 (Jaberi et al. 2013) | Iran |
| c.789_802+29delinsATAAA | p.(Tyr222Lysfs*51) | Splice | P II6, II7 (Elpeleg et al. 2005) | “Israelic muslim” |
| c.850C>T | p.(Arg284Cys) | Missense | P 1–3 (Carrozzo et al. 2007) | Italy |
| c.985A>G | p.(Met329Val) | Missense | P 1 (Nogueira et al. 2015) | Portugal |
| c.998A>G | p.(Asp333Gly) | Missense | P 1,2 (Matilainen et al. 2014)a | Finland |
| Deletion of exon 6 | p.(Tyr222Lysfs*51) | Deletion | This study | Turkey |
aHeterozygous mutation with additional deletion of 13q14 in P2
Fig. 1.
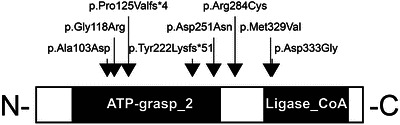
The positions of all mutations identified in human
Clinical Report
The male patient was born at term as the 4th child of consanguineous healthy Turkish parents (first cousins) after an uneventful pregnancy and delivery. An older sister died due to cardiac failure at the age of 1.5 years; no more details have been documented. At the age of 2 months, generalised muscular hypotonia was noticed, followed by a severe delay in motor development. At the age of 5 months, hearing loss became apparent, and sensorineural hearing loss was proven with brainstem evoked potentials (BAEP). Cranial MRI findings at the age of 6 months revealed unspecific enlargement of ventricular system and delayed myelinisation. Echocardiography, electrocardiogram as well as electroencephalogram showed no abnormalities. At the age of 22 months, the patient was unable to sit independently or to turn himself from the prone to supine position. Proximal muscular hypotonia with reduced deep tendon reflexes and hyperkinetic-dystonic movements of his extremities and facial dyskinesia were noted. Facial features included high-arched palate, elongate facies and large ears. Eye movements were free in all directions, and the patient was able to follow objects. According to his parents, he was able to pronounce “mum”, but no active speech was observed during clinical follow-up until 22 months. Feeding difficulties lead to failure to thrive with metric data for weight and head circumference on the 3rd percentile and length on the 25th percentile. The parents refused further academic care and follow-up, and we only know that the patient is alive at the age of 6 years but do not have further details.
Metabolic Findings
Serum lactate was elevated (4.4 mmol/l, normal <2), the serum amino-acid profile was within the normal range, and in particular alanine was not elevated. The urinary C4-dicarboxylic carnitine (C4DC) excretion was strongly increased, as was urinary lactate (314 μmol/mmol creatinine, normal <150) and urine methylmalonate (47 μmol/mmol creatinine, normal <5). Homocysteine, vitamin B12 and folate in blood were all within normal limits.
Genetic Investigations
Sanger sequencing of the entire coding region of SUCLA2 did not reveal any mutations, with the exception of exon 6 for which no PCR product could be obtained. Therefore, an MLPA analysis was performed, which confirmed the presence of a homozygous deletion of exon 6.
Review of Patients Reported in the Literature
A total of 28 patients were reported in literature (Carrozzo et al. 2007; Elpeleg et al. 2005; Jaberi et al. 2013; Lamperti et al. 2012; Matilainen et al. 2014; Morava et al. 2009; Nogueira et al. 2015; Ostergaard et al. 2007b) of whom nine were female.
In these patients, eight different disease-causing variants have been described in SUCLA2, two of which are splice site mutations and six missense mutations (Table 2). All are found in homozygous state in the affected patients except for one patient described with compound heterozygosity (Carrozzo et al. 2007) and one patient with a mutation of one of the alleles and a deletion of 13q14 (Matilainen et al. 2014).
Twenty-two of 24 patients presented symptoms within the first 6 months of life. Life span varies greatly, from 6 months to 21 years. Most prevalent clinical symptoms are muscle hypotonia and delayed motor development (27/28), sensorineural hearing loss (25/28), dystonia/hyperkinesia (24/28), absent speech (17/20), feeding problems (22/28), and failure to thrive (20/27). On MRI, basal ganglia lesions have been reported in 18 of 24 cases. Cerebral (12/24) or cerebellar (4/24) atrophy can also be part of the phenotype. The typical metabolic findings in SUCLA2 deficiency encompass increased plasma (17/19) and urinary (2/2) lactate, increased plasma (7/7) and urinary (17/18) methylmalonic acid as well as increased plasma (4/4) and urinary (3/3) C4DC.
Discussion
Dystonia deafness syndromes are a rare and heterogeneous group of disorders. Known genetic causes include Mohr-Tranebjaerg syndrome (TIMM8A, MIM#304700, (Jin et al. 1996)), Woodhouse-Sakati syndrome (C2orf37, MIM#241080, (Alazami et al. 2010)) and mitochondrial disorders, such as MEGDEL syndrome (SERAC, MIM#614739,(Alexoudi and Schneider 2012)) and SUCLA2 mutations (MIM#612073, Carrozzo et al. 2007; for review see (Kojovic, et al. 2013)). We show here that the dystonia deafness syndrome caused by SUCLA2 dysfunction can also be caused by an intragenic deletion of SUCLA2.
Our patient presented with the same characteristic symptoms as other patients with a mutation of this gene. Most prevalent symptoms of this syndrome, apart from the dystonia (25/29 patients, 86%) and deafness (26/29, 90%), are failure to thrive (21/28, 75%), delayed motor development (28/29, 97%), muscle hypotonia (28/29, 97%), progressive spasticity (15/23, 65%) and ophthalmoplegia (17/27, 63%). Almost all patients present symptoms within the first 6 months of their lives (22/24, 92%), and in our patient, this was not different. Because follow-up of our patient was not possible beyond the age of nearly 4 years, a description of progressiveness in this specific intragenic SUCLA2 deletion in comparison to other SUCLA2 mutations cannot be given. Review of the previously described cases shows there are no patients with a mild disease presentation and/or course of disease reported to date. In the patient described in this paper, there was only one MRI made at the age of 6 months. It showed widening of the ventricle system, which is fairly unspecific and can be seen in early stages of many neurodegenerative diseases. MRIs of patients with SUCLA2 mutations can show no abnormalities or mild cerebral atrophy with widened ventricle system and subarachnoid spaces in the first year. As patients grow older, Leigh-like lesions of the basal ganglia appear, starting in the putamen and caudate nucleus being the cause of dystonia development.
Our patient had increased lactate in urine and plasma, as well as elevated urinary excretion of methylmalonate and C4DC. Also the biochemical parameters are strikingly similar amongst patients. The most important indicators for a SUCLA2 deficiency are a mild increase of methylmalonate in plasma and urine in combination with an abnormal profile of carnitine esters. C4DC in plasma and especially in urine is elevated. Because of the TCA cycle defect caused by these mutations, lactate builds up as well.
In conclusion, mutations in SUCLA2 cause one of the rare but distinctive dystonia deafness syndromes. The clinical phenotype together with the metabolic findings in blood and urine is so specific that careful description without further investigations (e.g. brain MRI) would be enough to classify patients. Mutation analysis of the SUCLA2 gene will be necessary to confirm the diagnosis. Both mutations and intragenic deletions of this gene can cause this disorder. Both Sanger sequencing and MLPA (or another method to detect single exon deletions and duplication) of the SUCLA2 gene is subsequently needed to confirm the diagnosis.
Synopsis
A newly reported SUCLA2 deletion gives rise to a distinct deafness-dystonia syndrome.
Compliance with Ethics Guidelines
Competing Interests
Adela Della Marina, Arjan P.M. de Brouwer, Richard J Rodenburg, Roeltje R. Maas, Ron A. Wevers and Saskia B. Wortmann declare that they have no competing interests.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Authors’ Contribution
ADM provided the clinical data. AB, RW, RR and SW were involved in obtaining the biochemical data and genetic investigation, the data acquisition and analysis. RM and SW wrote the manuscript; all authors read and approved the final manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Saskia B. Wortmann, Email: saskia-wortmann@gmx.de
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Alazami AM, Schneider SA, Bonneau D, et al. C2orf37 mutational spectrum in Woodhouse–Sakati syndrome patients. Clin Genet. 2010;78(6):585–590. doi: 10.1111/j.1399-0004.2010.01441.x. [DOI] [PubMed] [Google Scholar]
- Alexoudi A, Schneider SA. Mutations in the phospholipid remodeling gene SERAC1 cause MEGDEL syndrome. Mov Disord. 2012;27(14):1738. doi: 10.1002/mds.25228. [DOI] [PubMed] [Google Scholar]
- Carrozzo R, Dionisi-Vici C, Steuerwald U, et al. SUCLA2 mutations are associated with mild methylmalonic aciduria, Leigh-like encephalomyopathy, dystonia and deafness. Brain. 2007;130(Pt 3):862–874. doi: 10.1093/brain/awl389. [DOI] [PubMed] [Google Scholar]
- Elpeleg O, Miller C, Hershkovitz E, et al. Deficiency of the ADP-forming succinyl-CoA synthase activity is associated with encephalomyopathy and mitochondrial DNA depletion. Am J Hum Genet. 2005;76(6):1081–1086. doi: 10.1086/430843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaberi E, Chitsazian F, Ali Shahidi G, et al. The novel mutation p.Asp251Asn in the beta-subunit of succinate-CoA ligase causes encephalomyopathy and elevated succinylcarnitine. J Hum Genet. 2013;58(8):526–530. doi: 10.1038/jhg.2013.45. [DOI] [PubMed] [Google Scholar]
- Jin H, May M, Tranebjaerg L, Kendall E, et al. A novel X-linked gene, DDP, shows mutations in families with deafness (DFN-1), dystonia, mental deficiency and blindness. Nat Genet. 1996;14(2):177–180. doi: 10.1038/ng1096-177. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Mehus JG, Tews K, Milavetz BI, Lambeth DO. Genetic evidence for the expression of ATP- and GTP-specific succinyl-CoA synthetases in multicellular eucaryotes. J Biol Chem. 1998;273(42):27580–27586. doi: 10.1074/jbc.273.42.27580. [DOI] [PubMed] [Google Scholar]
- Kojovic M, Parees I, Lampreia T, et al. The syndrome of deafness-dystonia: clinical and genetic heterogeneity. Mov Disord. 2013;28(6):795–803. doi: 10.1002/mds.25394. [DOI] [PubMed] [Google Scholar]
- Lambeth DO. Reconsideration of the significance of substrate-level phosphorylation in the citric acid cycle. Biochem Mol Biol Educ. 2006;34(1):21–29. doi: 10.1002/bmb.2006.49403401021. [DOI] [PubMed] [Google Scholar]
- Lambeth DO, Tews KN, Adkins S, Frohlich D, Milavetz BI. Expression of two succinyl-CoA synthetases with different nucleotide specificities in mammalian tissues. J Biol Chem. 2004;279(35):36621–36624. doi: 10.1074/jbc.M406884200. [DOI] [PubMed] [Google Scholar]
- Lamperti C, Fang M, Invernizzi F, et al. A novel homozygous mutation in SUCLA2 gene identified by exome sequencing. Mol Genet Metab. 2012;107(3):403–408. doi: 10.1016/j.ymgme.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilainen S, Isohanni P, Euro L et al (2015) Mitochondrial encephalomyopathy and retinoblastoma explained by compound heterozygosity of SUCLA2 point mutation and 13q14 deletion. Eur J Hum Genet 23:325–330 [DOI] [PMC free article] [PubMed]
- Menezes MJ, Riley LG, Christodoulou J. Mitochondrial respiratory chain disorders in childhood: insights into diagnosis and management in the new era of genomic medicine. Biochim Biophys Acta. 2014;1840(4):1368–1379. doi: 10.1016/j.bbagen.2013.12.025. [DOI] [PubMed] [Google Scholar]
- Miller C, Wang L, Ostergaard E, Dan P, Saada A. The interplay between SUCLA2, SUCLG2, and mitochondrial DNA depletion. Biochim Biophys Acta. 2011;1812(5):625–629. doi: 10.1016/j.bbadis.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Morava E, Steuerwald U, Carrozzo R, et al. Dystonia and deafness due to SUCLA2 defect; clinical course and biochemical markers in 16 children. Mitochondrion. 2009;9(6):438–442. doi: 10.1016/j.mito.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Nogueira C, Meschini MC, Nesti C, et al. A novel SUCLA2 mutation in a portuguese child associated with “mild” methylmalonic aciduria. J Child Neurol. 2015;30(2):228–232. doi: 10.1177/0883073814527158. [DOI] [PubMed] [Google Scholar]
- Ostergaard E, Christensen E, Kristensen E, et al. Deficiency of the alpha subunit of succinate-coenzyme A ligase causes fatal infantile lactic acidosis with mitochondrial DNA depletion. Am J Hum Genet. 2007;81(2):383–387. doi: 10.1086/519222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard E, Hansen FJ, Sorensen N, et al. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130(Pt 3):853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- Wortmann SB, Vaz FM, Gardeitchik T, et al. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat Genet. 2012;44(7):797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]